
Abstract
The most efficient and widely used system for generating helper-dependent adenoviral vectors (HDAds) is the Cre/loxP system developed by Graham and co-workers (Parks, R.J., Chen, L., Anton, M., Sankar, U., Rudnicki, M.A., and Graham, F.L. [1996]. Proc. Natl. Acad. Sci. U. S. A. 93, 13565–13570). Alternative systems have been developed for HDAd production, but all are limited by the technical complexity of a three-component vector production system for reproducibly generating large quantities of adenovirus with high infectivity and low helper virus (HV) contamination. Recently, these problems were addressed by Ng and co-workers (Palmer, D., and Ng, P. [2003]. Mol Ther. 8, 846–852), who developed an improved system that combines the use of a suspension-adapted producer cell line expressing high levels of Cre recombinase, a HV resistant to mutation, and a refined purification protocol. With this system, >1 × 1013 highly infectious vector particles are easily produced without vector genome rearrangements and having very low HV contamination levels. However, the Ng system incorporates a spinner flask culture system that involves considerable time, effort, and tissue culture medium to produce HDAds. We have an alternative system to obtain comparable quantities with equivalent quality to the spinner flask approach but requiring reduced labor and lower volumes of medium. This method utilizes a 10-chamber cell factory with adherent cells to produce high infectivity of HDAds with minimal HV contamination while improving yield and reducing technical complexity, effort, and medium requirements. This system is easily translatable to the production of clinical-grade HDAds for human trials.
Introduction
Materials and Methods
Plasmids, viruses, and cell lines
HDAds, HDΔ28E4LacZ coding the MCMV-LacZ expression cassette and HDΔ28E4FVIII coding the PEPCK-FVIII expression cassette, have been described in detail elsewhere (Palmer and Ng, 2003; Cerullo et al., 2007). HDΔ28E4LacZ and HDΔ28E4FVIII were amplified with 116 producer cells and the HV AdNG163 as previously described (Ng and Graham, 2002; Palmer and Ng, 2003, 2008). AdNG163 is identical to the HV AdNG163R-2 except that the viral packaging signal (ψ) is in the wild-type orientation (Ng and Graham, 2002). The physical titer of HDAds, expressed as vp/ml, was determined by absorbance at 260 nm with correction for vector genome size as previously described (Ng et al., 2002). The LacZ-mediated infectious titer, expressed as BFU/ml, of the HDAd-LacZ was determined by X-gal staining after titration on 293 cells as previously described (Ng et al., 2002). Verification of the HDAd genomic structure and determination of HV contamination were performed as described previously (Palmer and Ng, 2003).
HDAd rescue, amplification, and large-scale production
Rescue and amplification are shown in Fig. 1A. The large-scale production of HDAd is summarized in Fig. 2. HDAds (HDΔ28E4LacZ and HDΔ28E4FVIII) were rescued (serial passage 0 or P0) by transfecting a 60-mm dish of 116 cells at 60% confluency with 10 μg of PmeI-digested pΔ28E4LacZ or pΔ28E4FVIII followed, the next day, by infection with AdNG163R at an multiplicity of infection (m.o.i.) of 50 PFU/cell. HDAds were amplified by serial co-infections of 60-mm dishes of 116 cells at 90% confluency with 10% of the crude lysate from the previous passage and AdNG163 at an m.o.i. of 10 PFU/cell for serial passages 1 to 3 (P1–P3). For P4, one 150-mm dish of 116 cells at 90% confluency was co-infected with 10% of the crude serial passage 3 lysate and AdNG163 at an m.o.i. of 10 PFU/cell. For P5, four 150-mm dishes of 116 cells at 90% confluency were co-infected with the crude serial passage 4 lysate and AdNG163 at an m.o.i. of 25 PFU/cell. All serial passages were harvested 48 hr postinfection and subjected to three freeze–thaw cycles; this served as inoculum for subsequent passages. Amplification of all HDAds in 116 cells was performed under identical conditions.
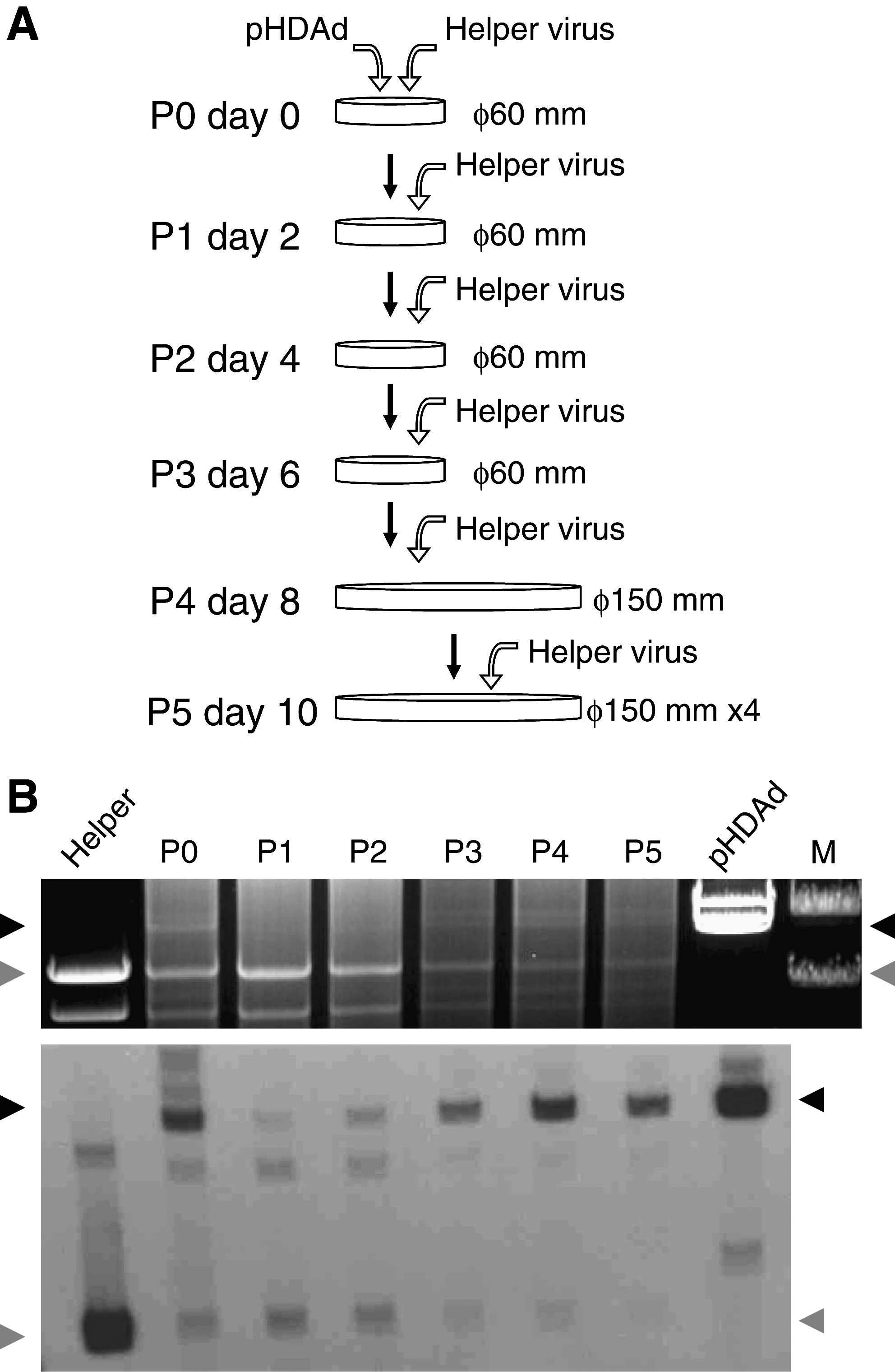
Analysis of HDΔ28E4LacZ serial amplification. (
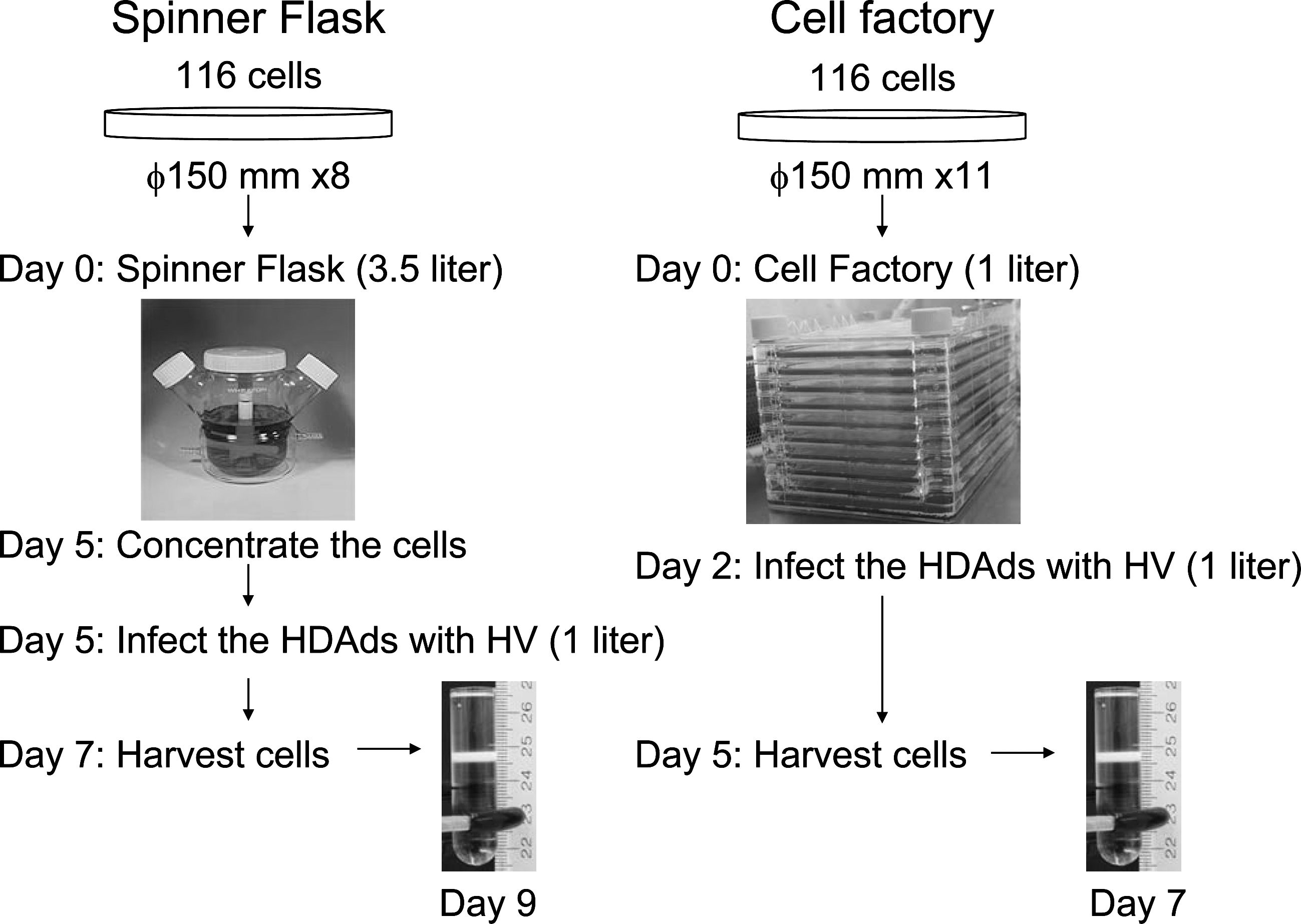
Overview of the large-scale production of helper-dependent adenoviral vectors (HDAds) with spinner flask and cell factory. The flow chart of the entire process beginning with the preparation of the spinner flask or cell factory to the final purified HDAds for large-scale production of HDAds is shown. Purification is by traditional cesium chloride ultracentrifugation.
Large-scale HDAd production was performed as follows: 3–4 × 108 of 116 cells were collected from eleven 150-mm dishes and harvested in 1 liter of medium, and then seeded in a 10-chamber cell factory (Corning, Lowell, MA). After 48 hr of incubation, medium was replaced with 1 liter of medium including 100% of the crude lysate from the 150-mm dish of serial passage 5 and AdNG163 at an m.o.i. of 25 PFU/cell. After 72 hr of incubation, co-infected cells were harvested with 1 liter of phosphate-buffered saline and centrifuged. These harvested cells were resuspended in 100 mM Tris–HCl (pH 8.0) and treated with sodium deoxycholate, DNase I, and RNase I as described in detail elsewhere (Palmer and Ng, 2008). HDAd virions were purified from cell lysate by CsCl ultracentrifugation as described in detail elsewhere (Ng et al., 2002). In an alternative simplified protocol, purified HDAds (obtained by the method described above) was used as inoculum, at a concentration of 1,000 viral particles/cell, instead of crude serial passage 5 lysate, for co-infection in a 10-chamber cell factory using the method described above. HDΔ28E4LacZ yields from each serial passage were determined by X-gal staining after titration on 293 cells expressed in BFU as described previously (Ng et al., 2002).
HDAd analysis
Physical and infectious HDAd titers were determined by OD260 and X-gal staining (HDAd-LacZ) after titration on 293 cells, as described elsewhere (Ng et al., 2002). DNA was extracted from virions obtained from CsCl gradients as described elsewhere (Ng et al., 2002) for Southern analysis and quantitative real-time polymerase chain reaction (PCR) was used to evaluate residual HV levels. DNA samples prepared from purified HDAds were subjected to quantitative real-time PCR analysis (10 min at 95°C followed by 45 cycles of 10 sec at 95°C, 7 sec at 65°C, and 30 sec at 72°C) using the Roche Light Cycler V 1.1 instrument (Roche master mix; Roche, Indianapolis, IN) with HV gene-specific primers (5′-CCCAAGAATAAAGAATCGTTTGTGTTATG-3′ and 5′- GGAGGGAGGTGGCAGGTTGAATACTA-3′). Vector copy numbers per microgram of total DNA were calculated after quantitative PCR using a standard curve generated from serial dilutions (over blank orders of magnitude) from the original plasmid DNA of HV (NG163).
Total intracellular DNA from the 293 cells infected with HDAd was extracted for measurement of vector copies in infected cells at 24 hr postinfection using DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA) according to manufacturer's protocols. DNA samples prepared from infected cells were subjected to quantitative real-time PCR analysis (10 min at 95°C followed by 45 cycles of 10 sec at 95°C, 7 sec at 60°C, and 30 sec at 72°C) using the Roche Light Cycler V 1.1 instrument (Roche master mix) and human stuffer gene-specific primers (5′-TCTGAATAATTTTGTGTTACTCATAGCGCG-3′ and 5′- CCCATAAGCTCCTTTTAACTTGTTAAAGTC-3′). Vector copy numbers per microgram of total DNA were calculated after quantitative PCR from a standard curve generated from serial dilutions (over blank orders of magnitude) with the original plasmid DNA of HDAds.
Results and Discussion
HDAd rescue and amplification
To evaluate the amplification of vector during rescue (P0) and amplification (P1–P5) steps (Fig. 1A), we extracted total intracellular DNA from the co-infected cells of each serial passage, digested it with ApaLI, and analyzed it by electrophoresis and Southern analysis (Fig. 1B). HDAds were amplified very efficiently as evidenced by their appearance and the intensity of HDAd-specific 20-kb band from passage 3 (Fig. 1B, top). Although we digested and loaded identical amounts of DNA for each sample, the intensity of HDAd-specific band gradually and significantly increased from P1 to P5, whereas that of specific HV band decreased as seen by Southern analysis (Fig. 1B, bottom). This result indicates that HDAds were significantly amplified during serial passaging.
Large-scale HDAd production
Because 116 cells can also grow in suspension (Palmer and Ng, 2003), large-scale production of HDAds is performed in a 3-liter spinner flask following the Ng protocol. With this system, >1 × 1013 highly infectious vp can be easily and reliably produced with very low HV contamination levels (0.01–0.02%), and infrequent vector genome rearrangements (Palmer and Ng, 2004). However, production of HDAds by spinner flask is labor intensive and requires a substantial quantity (4.5 liters) of medium (Fig. 2). To produce HDAds with a spinner flask, 116 cells must be supplemented with fresh media daily until the proper number of cells is obtained for infection. After producing approximately 1 × 109 cells, cells are concentrated before infection to increase infection efficiency. These steps require considerable time and increase the risk of contamination. We hypothesized that 116 cells, which are modified 293 cells (Palmer and Ng, 2003), could produce the significant quantities HDAds with a cell factory similar to FGAds, because 116 cells are able to grow as adherent cells. We thus applied cell factory technology to produce HDAds with 116 cells as adherent cells and optimized the conditions to obtain the necessary high-quality HDAds on a large scale. Specifically, we seeded the 1 liter of passage 35 116 cells at 3–4 × 105 cells/ml (approximately 3–4 × 108 cells), and co-infected with AdNG163R-2 and the crude serial passage 5 lysate from four 150-mm dishes (Fig. 1A). Seventy-two hours after co-infection, we purified HDAds by CsCl gradients as described in detail elsewhere (Palmer and Ng, 2003). Spectrophotometric analysis revealed that we obtained a total yield of 1–3 × 1013 vp corresponding for a specific yield of approximately 1 × 105 vp/cell (Table 1). The cell factory requires threefold fewer cell numbers compared with the spinner flask, and affords a total yield similar to that of the spinner flask. This result suggests that adherent 116 cells might be suitable to produce HDAds efficiently. The production of HDAds coding different transgenes by the cell factory also afforded a similar yield (Fig. 3 and Table 1). After we purified HDAds with CsCl ultracentrifugation, these purified HDAds were used as inoculum rather than crude serial passage 5 lysate. Although an additional serial passage is required, this alternative protocol is better suited for GMP large-scale production because it minimizes the handling of the large numbers of adherent cells needed to generate the inoculum, improving efficiency and greatly reducing total time, effort, and cost. HDAd preparation with inoculum of purified HDAds shows a slightly higher yield compared with that prepared from crude HDAd lysates. We also compared the yield of HDAds after infection of different m.o.i. of inoculum of HV versus purified HDAds at 500, 1,000, and 2,000 vp/cell (data not shown). HDAds produced with an inoculum of 500 vp/cell showed 10-fold less virus yield compared with 1,000 and 2,000 vp/cell. The inoculum of 2,000 vp/cell exhibited a slightly higher but not statistically significant yield compared with that infected with 1,000 vp/cell. We also hypothesized that viral production of 116 cells with the lower inoculum of 1,000 vp/cell may further reduce the magnitude of HV contamination. The infection with HV was also studied at different time points (48, 72, and 96 hr). After 48 hr, infected 116 cells were still adherent and produced lower yield of HDAds when compared harvesting after 72 hr (5.7 × 1012 vp/factory at 48 hr vs. 2.2 × 1013 vp/factory at 72 hr). The 116 cells infected for 96 hr showed complete detachment but also produced a lower yield compared with 72 hr after CsCl gradient preparation (7.2 × 1012 vp/factory at 96 hr vs. 2.2 × 1013 vp/factory at 72 hr). These data are consistent with previous observations that long-term infection of HV and HDAds might induce the disruption of infected cells and release of HDAds into medium, resulting in a lower yield of HDAds from cell lysates. There was no difference of HDAd yields among different passages of 116 cells until passage 70, indicating stability of 116 cells (in terms of HDAd yield) for at least 70 passages. Significantly, this large quantity of vector (obtained at serial passage 6) was produced in 7 days of the preparation of cell factory as shown in Fig. 2 and described in detail under Materials and Methods.
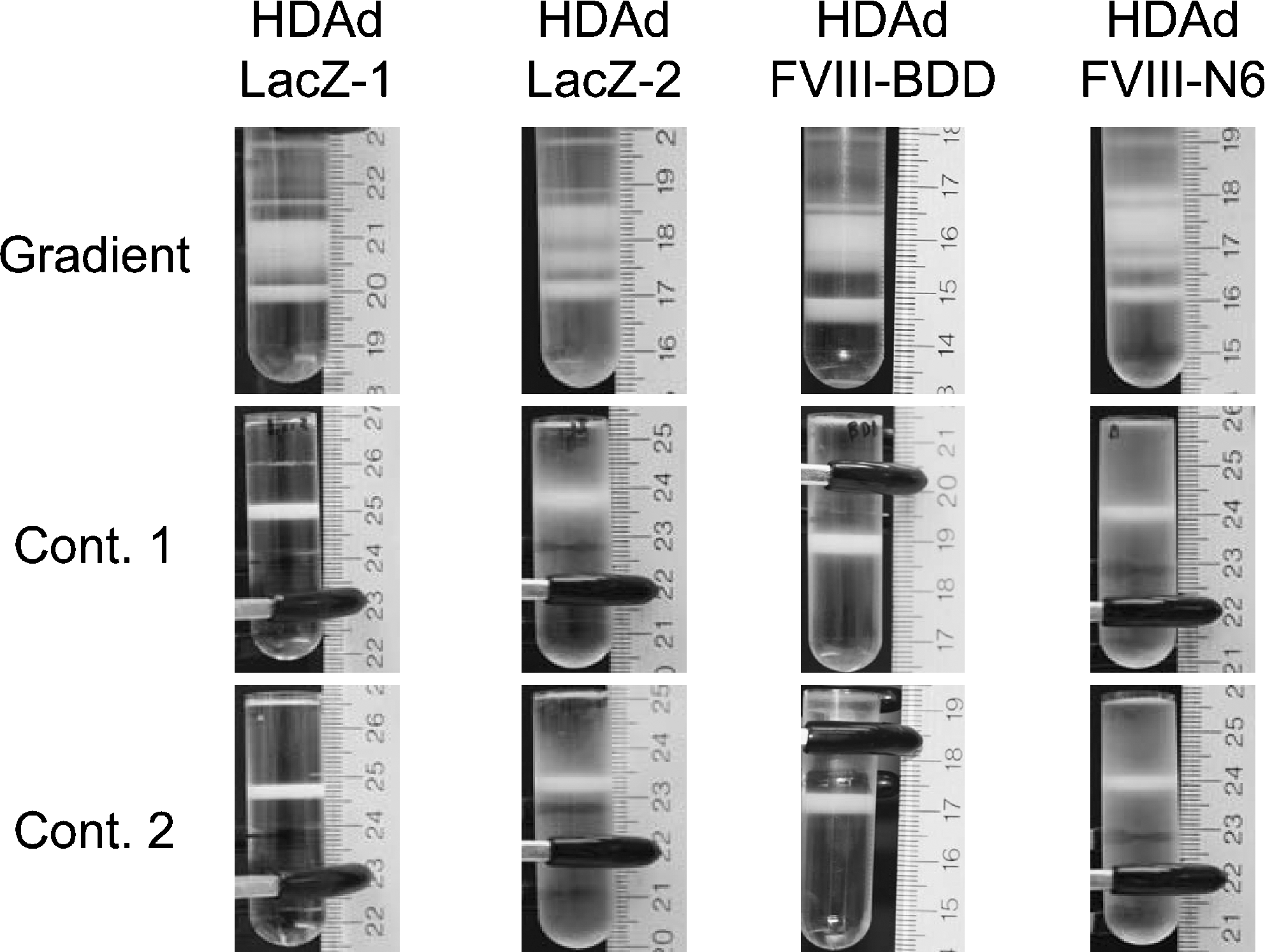
Details of helper-dependent adenoviral vector (HDAd) production with cell factory using AdNG163R-2 following the protocol shown in Fig. 2. HDAds were purified by one gradient and two continuous cesium chloride (CsCl) gradients (Cont. 1 and Cont. 2). Gradient, after first-gradient CsCl ultracentrifugation; Cont. 1, after first continuous CsCl ultracentrifugation; Cont. 2, after second continuous CsCl ultracentrifugation. HDAd-LacZ: HDΔ28E4LacZ coding LacZ expression cassette (Palmer and Ng, 2003). HDAd-FVIII BDD, HDAd-FVIII N6: HDΔ28E4FVIII BDD and HDΔ28E4FVIII N6 coding FVIII variants expression cassette (Cerullo et al., 2007). Total viral particle yield, infectivity (particle-to-infectious unit ratio expressed as vector particles:BFU) of HDAd-LacZ, and the ratio of helper virus contamination in these purified HDAds are shown in Table 1.
N/A, not applicable.
HDAd structure and yields
HDAds were evaluated, after extraction of virion DNA from the each of two continuous CsCl gradients, by digestion with ApaLI (Fig. 4A). Agarose gel electrophoresis analysis revealed that the genomic structure of HDAds was indistinguishable from that of the parental plasmids, confirming the absence of gross vector DNA rearrangement (Fig. 4B, top). To determine the level of HV contamination, we performed Southern blot analysis probing with ψ to compare the intensities of the 6.7-kb HDAd-specific band and a 1.5-kb AdNG163R-2-specific band (Fig. 4B, bottom). HDAds produced by the cell factory showed the 6.7-kb HDAd-specific band, whereas the 1.5-kb AdNG163R-2-specific band was undetectable in the virion DNA produced with cell factory (a result equivalent to the spinner flask technique). We also quantified the contamination level of HV in HDAds purified from cell factory preparations by quantitative real-time PCR (Table 1). Although the cell factory requires a fivefold greater inoculum (1,000 vp/cell) compared with the inoculum for spinner flasks (200 vp/cell), HDAds prepared by cell factory exhibited low levels of HV level (0.03–0.05%) in purified HDAds after CsCl gradient centrifugation similar to that prepared by spinner flask (Palmer and Ng, 2005). There is no difference in HV levels among HDAds prepared by cell factory (Table 1). These results confirm the ability of the 116 cell to express sufficient Cre recombinase to effectively eliminate HV packaging and the effectiveness of the two continuous CsCl gradient purifications in reducing contamination of HV to levels of those purified from spinner flask preparations.
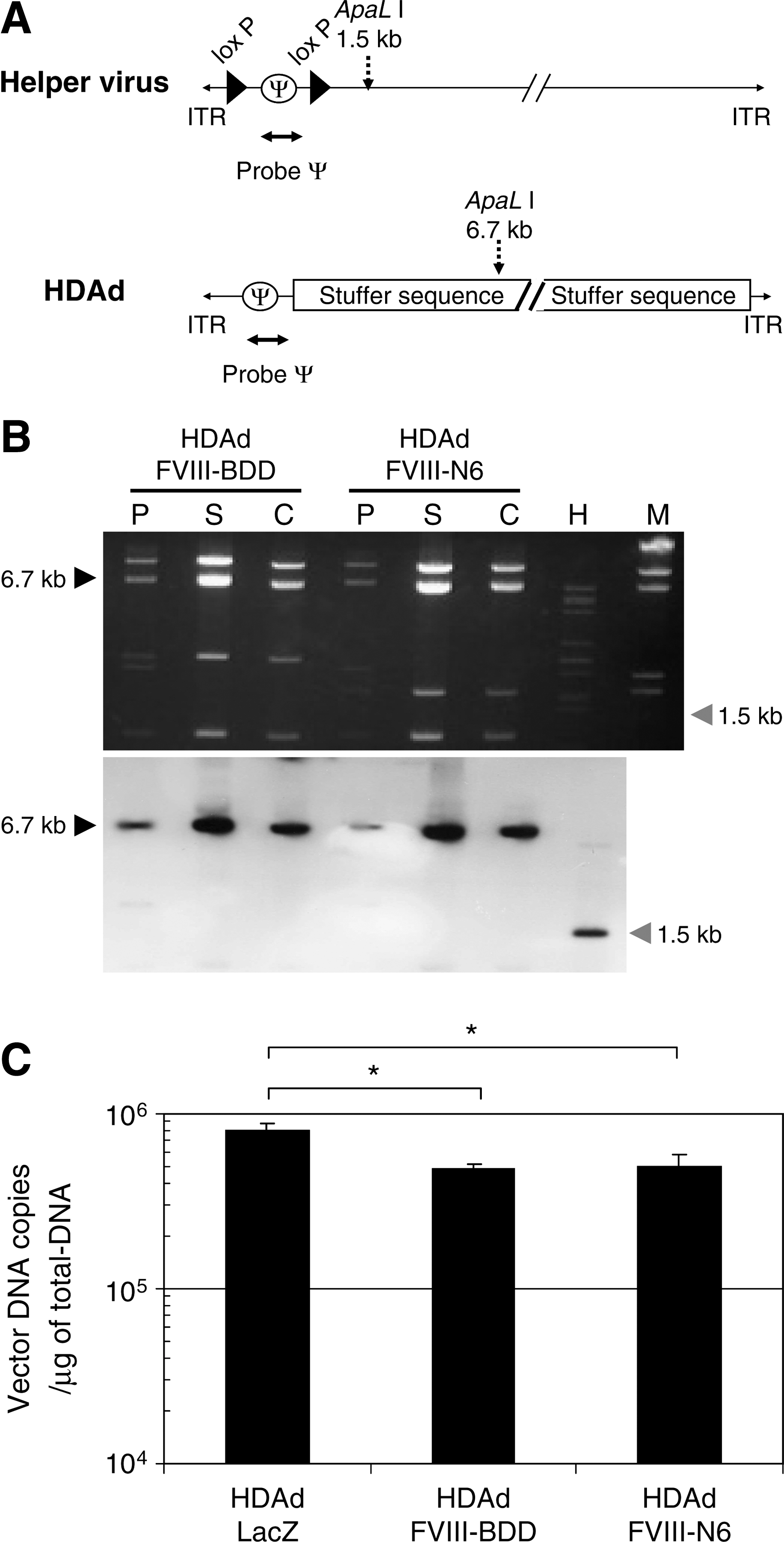
Analysis of helper-dependent adenoviral vectors (HDAds) produced from the cell factory. (
Different preparations of HDAd-LacZ produced by the cell factory technique and purified from the CsCl gradients showed a particle-to-infectious unit ratio (vp:BFU) ranging from 5:1 to 30:1 as determined by X-gal staining after titration on 293 cells (Table 1). The particle-to-infectious unit ratio (vp:IU) represents the infectivity of the vector and, therefore, the proportion of the total particles that are infectious. By definition, the higher vp:IU ratio corresponds to lower infectivity. This is an important parameter for meaningful comparisons between different vector preparations (or between different preparations of the same vector), because erroneous conclusions will be drawn if different infectivities are obtained. Moreover, because the viral particle itself may mediate dose-dependent acute toxicity (Schnell et al., 2001; ZhaNg et al., 2001; Brunetti-Pierri et al., 2004), noninfectious particles (reflected in a high vp:IU ratio) may increase risk and decrease benefit of any treatment. Therefore, the FDA has recommended that the infectivity of clinical-grade vectors be <30:1 to address this concern (Palmer and Ng, 2004). HDAd-LacZ produced with cell factories exhibits infectivity equivalent to that of the spinner flask preparations. To test whether HDAds coding different transgenes also show similarly infectivity of HDAd-LacZ, total DNA was isolated from 293 cells infected with same virus particle numbers of HDAds after 24 hr postinfection. These DNA samples were subjected to quantitative PCR to compare vector copy numbers in infected cells (Fig. 4B). There was slight but significant difference in vector copies among these HDAds. We also compared the vector copies among different preparations of same HDAds (data not shown). There was slight different of vector copies among these different preparations. These results indicated that the infection efficiency of HDAds is dependent on different transgene and different preparations. We estimated the particle-to-infectious unit ratio based on the result of quantitative PCR of HDAd-LacZ. These ratios were still within the FDA-approved ratio (under 1:30). These results confirmed that HDAds coding different transgenes and HDAds produced with different preparations exhibit acceptable infectivity.
Culture of 116 cells in a cell factory requires amounts of serum similar to spinner HDAd production. This, however, is not considered problematic given that the use of serum is permitted by the FDA in the manufacturing of Ad vectors for human use as long as it is demonstrated to be free of adventitious viruses and bovine spongiform encephalopathy agent. However, if necessary, the 116 cells could be modified to produce HDAds in serum-free medium with the cell factory for large scale of production.
Our system for producing HDAds with the cell factory represents an improvement over previous spinner methods in terms of simplicity and efficiency as well as vector yield while maintaining a high level of purity. We have (to date) produced over a dozen different HDAds using this system with consistent yields and purities, allowing us to evaluate the safety and efficacy of these vectors in meaningful preclinical studies in small and large animal models with the aim of translating this method for production in human clinical studies.
Footnotes
Acknowledgments
This work was supported by the National Institutes of Health DK56787 and HL87836 to B. Lee. The authors thank Philip Ng and Donna J. Palmer for the 116 cells and HV, AdNG163R-2.
Author Disclosure Statement
No competing financial interests exist.