
Abstract
Manipulation of gene expression is an invaluable tool to study gene function in vitro and in vivo. The application of small inhibitory RNAs to knock down gene expression provides a relatively simple, elegant, but transient approach to study gene function in many cell types as well as in whole animals. Short hairpin structures (shRNAs) are a logical advance as they can be expressed continuously and are hence suitable for stable gene knockdown. Drug-inducible systems have now been developed; however, application of the technology has been hampered by persistent problems with low or transient expression, leakiness or poor inducibility of the short hairpin, and lack of reversibility. We have developed a robust, versatile, single lentiviral vector tool that delivers tightly regulated, fully reversible, doxycycline-responsive knockdown of target genes (FOXP3 and MYB), using single short hairpin RNAs. To demonstrate the capabilities of the vector we targeted FOXP3 because it plays a critical role in the development and function of regulatory T cells. We also targeted MYB because of its essential role in hematopoiesis and implication in breast cancer progression. The versatility of this vector is hence demonstrated by knockdown of distinct genes in two biologically separate systems.
Introduction
Lentiviral vectors are an appealing vehicle with which to deliver genes or shRNAs, particularly because they are capable of transducing most nondividing or quiescent cells (Naldini et al., 1996a). This is important for transfer into hematopoietic cells or other primary human cell types that are typically refractory to transfection and are transduced inefficiently by other types of virus (Naldini et al., 1996b; Salmon et al., 2000; Barry et al., 2001). In addition, lentiviral vectors offer several advantages over other viral vectors: they are more efficient at delivering complex gene expression cassettes compared with other retroviral vectors (May et al., 2000), they can mediate long-term gene expression (Seppen et al., 2001), and they are relatively safe.
The biological targets used to validate the vectors for this study were chosen because they are of clinical significance in autoimmunity and cancer. In the first example we focused on the transcription factor FOXP3 (forkhead box P3), the master transcription factor of regulatory T cells. Regulatory T cells (Tregs) are essential for normal immune homeostasis, balancing prevention of autoimmunity and immune pathology on the one hand, while simultaneously allowing effective immunity to infection and tumors on the other. Many now regard these cells as “master regulators” of immune responsiveness (Tang and Bluestone, 2008). The forkhead transcription factor, FOXP3, is expressed constitutively in “natural” CD4+ Tregs and has been shown to be necessary and sufficient for mouse Treg function (Bennett et al., 2001; Fontenot et al., 2003; Hori et al., 2003; Yagi et al., 2004; Chang et al., 2005). Less is known about the role human FOXP3 plays in the development and function of the Treg (reviewed by Li and Greene, 2008).
To demonstrate the robustness of the vector, we also chose a second target, the MYB oncogene, which encodes a transcription factor (MYB). MYB is associated predominantly with normal and leukemic hematopoiesis, based on its pattern of expression (Gonda et al., 1982; Westin et al., 1982), the consequences of loss of expression (Anfossi et al., 1989; Mucenski et al., 1991), and its ability to induce leukemogenesis in several species (reviewed in Ramsay and Gonda, 2008). MYB is also expressed at relatively high levels in at least two epithelial tumors: colon cancer (Torelli et al., 1987) and breast cancer (Guerin et al., 1990). Evidence of a causal role in carcinogenesis includes the correlation of expression level with poor prognosis (Biroccio et al., 2001) and the presence of mutations that are likely to dysregulate MYB expression (Thompson et al., 1997; Hugo et al., 2006).
We describe here a lentiviral system able to knock down target genes reversibly, using doxycycline (Dox) induction. This system differs from previously described vectors as it combines exquisite sensitivity to Dox with fast kinetics, efficient induction of shRNA expression, and reversibility in a single lentivector. Vectors described in prior work (Szulc et al., 2006; Aagaard et al., 2007; Pluta et al., 2007; Seibler et al., 2007; Herold et al., 2008) do not combine all of these characteristics in a single vector. We have used two biologically distinct systems to model the robust gene-silencing capability of this vector.
Materials and Methods
Vector construction
Third-generation lentiviral vectors containing the human hepatitis virus posttranscriptional regulatory element (PRE) and the HIV central polypurine tract (cPPT) element (described in Barry et al., 2001) were used for all experiments. For gene overexpression the backbone was reengineered to contain the elongation factor (EF)-1α promoter (plvEIG), a Gateway cloning site (att, CAT ccdB att), and an internal ribosome entry site (IRES) to drive the marker gene encoding green fluorescent protein (GFP). To generate the shRNAi delivery vector (plv-T-sh), this backbone was engineered to contain a Gateway cassette (att, CAT ccdB att) upstream of the Rev-responsive element into which the shRNAi cassette is recombined. The regulatory protein (T-REx) was inserted under the control of the EF-1α promoter, and the marker gene (GFP) is expressed from an IRES.
Short hairpin expression constructs
Five shRNA sequences predicted to knock down FOXP3 expression were designed, using BLOCK-iT RNAi designer (Invitrogen, Carlsbad, CA). The following FOXP3 sequences were chosen as potential shRNA targets and were numbered with respect to FOXP3 cDNA numbering: 1355: GAGTCTGCACAAGTGCTTTGT 485: GCCACATTTCATGCACCAGCT 1289: CACACGCATGTTTGCCTTCTTC 767: GCTGGCAAATGGTGTCTGCAA 981: GCACTGACCAAGGCTTCATCT
Using the preceding target sequences, shRNA constructs were generated in a sense–loop–antisense expression format, using 9-mer TTCATGAGA to form the loop. A three-step PCR procedure was used to generate the shRNA targeting sequences. PCR primers were designed to generate two PCR products: a 5′ end with the attB1 site, tH1 promoter, sense strand of the stem structure, and the loop; and a 3′ end with the loop, antisense strand of the stem structure, termination signal, and attB2 site. In step 1, a 5′ PCR product was generated with pBS-tH1, containing the tet-H1 promoter as template DNA, an attB1 forward primer, and a site-specific reverse primer (see later for primer sequences). In step 2, a 3′ PCR product was generated with a site-specific forward primer and an attB2 reverse primer (see later for primer sequences). In this reaction the forward and reverse primers overlap and no template DNA was required. Products from these two reactions were subsequently gel purified (QIAquick gel extraction kit; Qiagen, Hilden, Germany), quantitated, combined, and used as the template in a third PCR using the attB1 forward primer and the attB2 reverse primer. The final PCR product was gel purified and inserted into the pDONR entry vector as described subsequently.
Forward (5′) and reverse (3′) primers used to generate shRNAs:
attB1 forward primer: GGGGACAAGTTTGTACAAAAAAGCAGGCTT
attB2 reverse primer: GGGGACCACTTTGTACAAGAAAGCTGGGTA 1355 forward primer: TGTTTCATGAGAACAAAGCACTTGTGCAGA 1355 reverse primer: TGTTCTCATGAAACAAAGCACTTGTGCAGA 485 forward primer: GCTTTCATGAGAAGCTGGTGCATGAAATGT 485 reverse primer: GCTTCTCATGAAAGCTGGTGCATGAAATGT 1289 forward primer: GAATTCATGAGAAAGACTTCTTCCGTTTGT 1289 reverse primer: GAATCTCATGAAAAGACTTCTTCCGTTTGT 767 forward primer: CAATCTCATGAATTGCAGACACCATTTGCC 767 reverse primer: CAATTCATGAGATTGCAGACACCATTTGCC 981 forward primer: TCTTTCATGAGAAGATGAAGCCTTGGTCAG 981 reverse primer: TCTTGTCATGAAAGATGAAGCCTTGGTCAG
The Gateway technology (Invitrogen) universal cloning system was used for the transfer of short hairpin expression cassettes (FOXP3 and sh-β-catenin) and FOXP3 cDNA into expression vectors. Short hairpin cassettes were inserted into a Gateway (Invitrogen)-compatible entry vector (pDONR), using Gateway BP Clonase recombination techniques, and transferred from the entry clone to the Gateway (Invitrogen)-compatible lentiviral destination vector (plv), using Gateway LR Clonase recombination techniques. FOXP3 lentiviral expression vectors were generated in a similar fashion, inserting FOXP3 cDNA into lentiviral destination vector plvEIG. All constructs were sequenced (BigDye terminator cycle sequencing kit, version 3.1; Applied Biosystems, Foster City, CA) before use in experiments.
Cell culture
HEK293T cells were routinely cultured in Dulbecco's modified Eagle's medium (DMEM) (GIBCO; Invitrogen), 10% fetal calf serum (FCS; JRH Biosciences, Lenoxa, KA), 1% penicillin–streptomycin (Pen/Strep), 1%
The human transformed malignant breast cancer line MCF7, cultured in RPMI and transduced with the sh-Myb or sh-SCR scrambled trigger sequence, were generated according to the same protocols as described by Drabsch and colleagues (2007). GFP-positive cells were enriched by cell sorting. Transduced cells were cultured in the presence or absence of Dox (1 μg/ml) for 48 hr unless otherwise stated.
Isolation and in vitro expansion of cord blood T cell populations
Cord blood was obtained with informed maternal consent as approved by the Children's, Youth and Women's Health Service (North Adelaide, SA, Australia) Research Ethics Committee. Mononuclear cells (MNCs) were isolated from 60 to 100 ml of venous blood collected directly postpartum into preweighed blood collection bags (Blood-Pack units; Fenwal, Lake Zurich, IL) containing anticoagulant as previously described (Bresatz et al., 2007). Cord blood CD4+CD25+ cells (Tregs) and CD4+CD25− (helper T) cells were isolated from MNCs, using a Dynabeads regulatory CD4+CD25+ T cell kit (Invitrogen). The purity for each cell type was routinely greater than 90% by two-color flow cytometry for CD4 and CD25 expression. Cells were routinely expanded for 8 days in the presence of Dynabeads human T-expander CD3/CD28 (cat. no. 111-41D; Invitrogen) at a bead-to-cell ratio of 3:1, before magnetic removal of the beads. Ex vivo expansion of isolated T cell populations (1 × 106 cells per well in a 24-well plate) were performed in X-VIVO 15 medium (BioWhittaker; Lonza, Basel, Switzerland) supplemented with 20 mM HEPES (pH 7.4), 5% heat-inactivated pooled human serum (Lonza), 2 mM
Proliferation assays
MCF7 cells previously transduced with plv-T-sh-Myb or plv-T-sh-SCR were cultured in triplicate in the presence or absence of Dox for 12 days. Cell proliferation was measure by cell counting with a Coulter counter. Replicate samples were also treated to wash out the Dox from day 5. In one group of replicates of the sh-Myb-transduced cells, Dox was washed out on day 5 and added back on day 7. Western blotting was also carried out as described by Brabsch and colleagues (2007).
Lentiviral packaging
To generate lentiviral stocks, HEK293T cells were seeded at a density of 6 × 106 cells in 18 ml of medium in a 75-cm2 tissue culture flask. Cells were transfected with 12.5 μg of transfer vector (plvEIG), 7.5 μg of Gag/Pol (Δ8.2), 6.25 μg of Rev (pRSV-Rev), and 3.75 μg of Env (pCMV-VSV-G), using Lipofectamine 2000 reagent (Invitrogen) and Opti-MEM (Invitrogen) reduced serum medium, in accordance with the standard protocol. The next day, the medium was replaced with 10 ml of fresh DMEM and 48 hr later supernatant was collected, filtered (pore size, 0.45 μm), and used immediately or stored frozen (–70°C). Virus was concentrated by ultracentrifugation at 22,000 rpm for 90 min at 4°C. The supernatant was aspirated and the pellet was resuspended in X-VIVO medium (1:25, v/v) to give a 25 × concentrated stock. Virus was titered by limiting dilution, as described previously (Barry et al., 2001). Routinely we achieved titers of 5 × 105–1 × 106 transducing units per milliliter.
Lentiviral transduction
For transduction of cell lines, cells were routinely seeded at a density of 3 × 105 cells per well in a 6-well plate. Medium was replaced with 3 ml of viral supernatant (unconcentrated) containing Polybrene at a final concentration of 5 μg/ml. GFP-positive cells were sorted by flow cytometry (FACSCalibur; BD Biosciences, San Jose, CA). For transduction of primary human cells 1–3 × 105 CD4+CD25+ Tregs were transduced with lentivirus at a multiplicity of infection (MOI) of approximately 20, 24–48 hr after isolation. Cells were expanded for 8 days in the presence of Dynabeads before magnetic removal of the beads and isolation of GFP+ cells and GFP− cells by flow cytometry.
Intracellular staining
For detection of FOXP3 expression, cells (1 × 105) were washed in flow cytometry staining buffer (0.1% sodium azide, 1% FCS, in phosphate-buffered saline) and stained intracellularly with phycoerythrin (PE)-conjugated anti-human FOXP3 antibody (PCH101; eBioscience, San Diego, CA) using fixation/permeabilization and permeabilization reagents (eBioscience) and standard protocols. Cells were resuspended in fixation buffer (1% formaldehyde, 2% glucose, 0.02% sodium azide in phosphate-buffered saline) and analyzed by flow cytometry (FACSCalibur; BD Biosciences).
Purification of DNA and RNA
DNA was prepared with a QIAprep spin miniprep kit (Qiagen) and HiSpeed plasmid midi purification kit (Qiagen). RNA was prepared with an miRNeasy mini kit (Qiagen).
Analysis of RNA expression by real-time PCR
RNA was reverse transcribed, using a QuantiTect reverse transcription kit (Qiagen), and real-time PCR was performed on a Rotor-Gene 6000 (Corbett Life Science/Qiagen), using a FastStart Taq DNA polymerase kit (Roche, Indianapolis, IN) and SYBR green fluorescent dye. Transcription factor mRNA expression profiling was performed at various time points as described in the figure legends, and cells were analyzed for relative abundance of FOXP3 compared with an internal control (β-actin). FOXP3 forward: GAA ACA GCA CAT TCC CAG AGT TC FOXP3 reverse: ATG GCC CAG CGG ATG AG β-Actin forward: AAG AGC TAC GAG CTG CCT GAC β-Actin reverse: GTA GTT TCG TGG ATG CCA CAG
Stem–loop reverse transcription-polymerase chain reaction
Reverse transcriptase reactions were performed on 150 ng of total RNA. First-strand complementary DNA (cDNA) was synthesized with 0.05 μM stem–loop primer (SL-sh-1355) (5′-GTTGGCTCTGGTAGGATGCCGCTCTCAGGGCATCCTACCAGAGCCAACACAAAG-3′) and 2 μM antisense primer for the reference gene hypoxanthine phosphoribosyltransferase (Hprt-1) (5′-CAGGCAAGACGTTCAGTCCT-3′), using a SuperScript III reverse transcriptase kit (Invitrogen) according to the manufacturer's protocol. SL-sh-1355 contains a binding site for a universal reverse primer (URevP; 5′-GTAGGATGCCGCTCTCAGG-3′), a binding site for Universal ProbeLibrary (UPL) probe #21 (Roche), and a six-base 3′ overhang sequence, which is complementary to the last six bases at the 3′ end of sh-1355. Pulsed reverse transcription (RT) was carried out under previously described conditions with modifications (Chen et al., 2005; Tang et al., 2006). Briefly, RNA was initially incubated at 65°C for 5 min to uncoil the RNA. cDNA synthesis was then performed at 16°C for 30 min followed by 60 cycles of 20°C for 30 sec, 42°C for 30 sec, and 50°C for 1 sec. A final incubation at 75°C for 15 min was performed to inactivate the reaction. Additional RT-minus (i.e., without reverse transcriptase) reactions served as negative control. For quantitative PCR, a 20-μl reaction containing 0.005 × synthesized cDNA (from either an RT-positive or RT-minus reaction), 1 × LC480 master probe mix (Roche), 250 nM forward primers (sh-1355 forward primer, 5′-CGGCCGAGTCTGCACAAGTG-3′; Hprt-1 forward primer, 5′-TGACCTTGATTATTTTGCATACC-3′), 250 nM reverse primers (URevP, Hprt antisense primers) (GeneWorks, Hindmarsh, SA, Australia), 100 nM UPL probe #21 (for sh-1355), and 100 nM probe #73 (for Hprt-1) (Roche) was prepared for each sample. PCRs were performed with the LightCycler 480 system (Roche) with a predenaturing step of 95°C for 10 min, and 45 cycles of 95°C (10 sec), 60°C (30 sec), and 72°C (10 sec) followed by a cooling step at 40°C for 1 min. Semiquantitative PCR was performed with the exact reaction setup and conditions for only 34–36 cycles without the presence of UPL probe. The cycle threshold value (C t) was calculated from the amplification curve, using the second derivative maximum method (Rasmussen, 2001) described with LightCycler 480 quantification software version 1.5 (Roche). To compare the C t value with gel electrophoresis results, we normalized the value to Hprt-1, using the comparative C t method.
Dual-Luciferase reporter assays
For luciferase assays, FOXP3 and β-catenin cDNAs were inserted into the multicloning site of psiCHECK-2 (Promega, Madison, WI) vector. psiCHECK-2 is a dual-luciferase reporter construct in which the cDNA of interest is cloned into the 3′ untranslated region (UTR) of the Renilla luciferase gene. Renilla luciferase activity is normalized with respect to firefly luciferase activity, which is carried on the same vector to control for transfection efficiency. All constructs were sequenced (BigDye terminator cycle sequencing kit, version 3.1; Applied Biosystems) before use in experiments. Cells were transiently transfected as described previously, before incubation in the absence or presence of doxycycline (1 μg/ml). Subsequently, culture medium was removed and cells were lysed with Dual-Luciferase reporter assay reagents (Promega) according to the standard protocol. Samples were assayed with luciferase assay reagent II (LAR II) and Stop & Glo reagents and analyzed with a Veritas microplate luminometer (Turner Biosystems/Promega).
Results
A single lentiviral vector to conditionally knock down gene expression
A lentiviral vector was designed to achieve tightly regulated, drug-inducible knockdown of target gene expression. Short hairpin RNA expression cassettes generated by PCR were inserted into the expression vector via donor vector intermediaries, using Gateway technology. The shRNA sequence was expressed from the tet operator–H1 promoter fusion (tH1) in either donor vector for validation (pDONR), or transfer vectors for lentiviral delivery (plv-T-sh). The T-REx repressor cassette transcribed from an EF-1α promoter was included in the lentiviral backbone, thereby delivering all the components required for tetracycline-induced derepression of shRNA transcription in one vector. An internal ribosome entry site (IRES) downstream of the T-REx cassette was inserted for expression of the marker gene GFP (Fig. 1A).
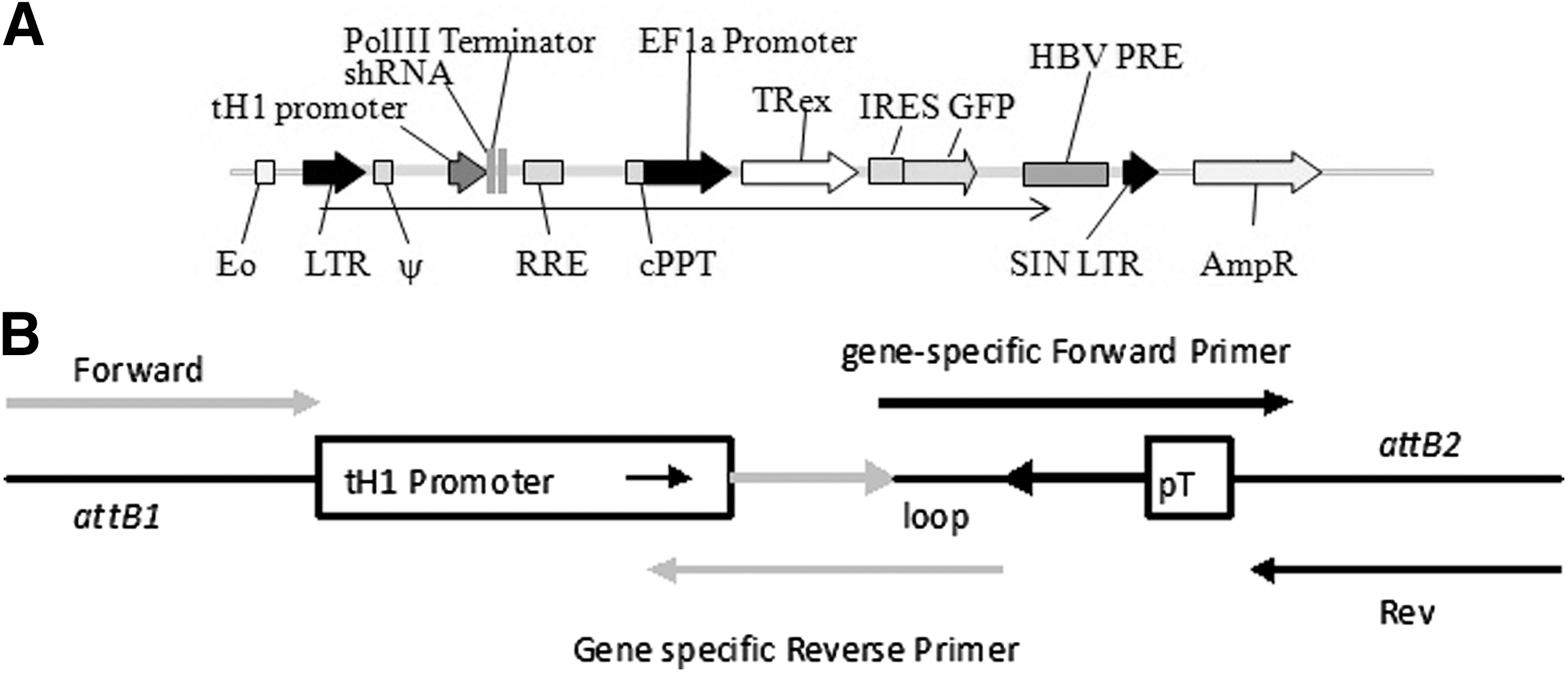
A single lentiviral expression vector for drug-inducible knockdown of FOXP3. (
Cloning and screening of short hairpin RNAs
Construction of shRNA triggers, as cassettes with the tet operator–H1 promoter fusion (tH1), facilitates the rapid screening for functional shRNA molecules in the pDONR vector before their transfer into the lentiviral backbone. A panel of five 21-mer short hairpin sequences was designed to knock down expression of human FOXP3 (BLOCK-iT RNAi designer; Invitrogen). The Gateway adaptor tailed tH1-shRNA cassettes generated by a two-step PCR (Fig. 1B) were inserted into a pDONR vector and sequenced. To screen for functional shRNA triggers, HEK293T cells were transiently cotransfected with vectors constitutively expressing the short hairpin RNAs (pDONR-shRNA) and the reporter vector psiCHECK-2 (Promega) carrying FOXP3 (psiCHECK-FOXP3). Functional shRNA triggers were then identified by a reduction in Renilla activity from the psiCHECK-FOXP3 reporter. To confirm on-target specificity of our short-hairpin sequences, a control β-catenin reporter vector (psiCHECK-β-cat) and a validated β-catenin short hairpin RNA (sh-β-cat) (van de Wetering et al., 2003) were used in all experiments.
Screening of the predicted FOXP3 shRNA targeting sequences identified one trigger, sh-1355, that substantially reduced (∼40% knockdown) Renilla activity of the psiCHECK-FOXP3 reporter compared with cells transfected with the reporter alone. No significant reduction in Renilla activity was observed in cells cotransfected with psiCHECK-FOXP3 and the sh-485, sh-767, or sh-981 expression vector, whereas sh-1289, intriguingly, led to increased FOXP3 expression (60% higher) (means of triplicate samples and representative of more than three experiments) (Fig. 2A). On-target specificity was confirmed in cells cotransfected with the β-catenin reporter vector (psiCHECK-β-catenin) and the short hairpin β-catenin (sh-β-cat), where expression was significantly reduced (80% knockdown) compared with cells transfected with psiCHECK-β-catenin alone. In contrast, no significant knockdown of Renilla activity was observed in cells cotransfected with sh-1355 and the psiCHECK-β-catenin reporter vector or in cells cotransfected with sh-β-cat and the psiCHECK-FOXP3 reporter. This screening strategy allowed rapid identification a single short hairpin RNA sequence (sh-1355) able to specifically knock down expression of FOXP3.
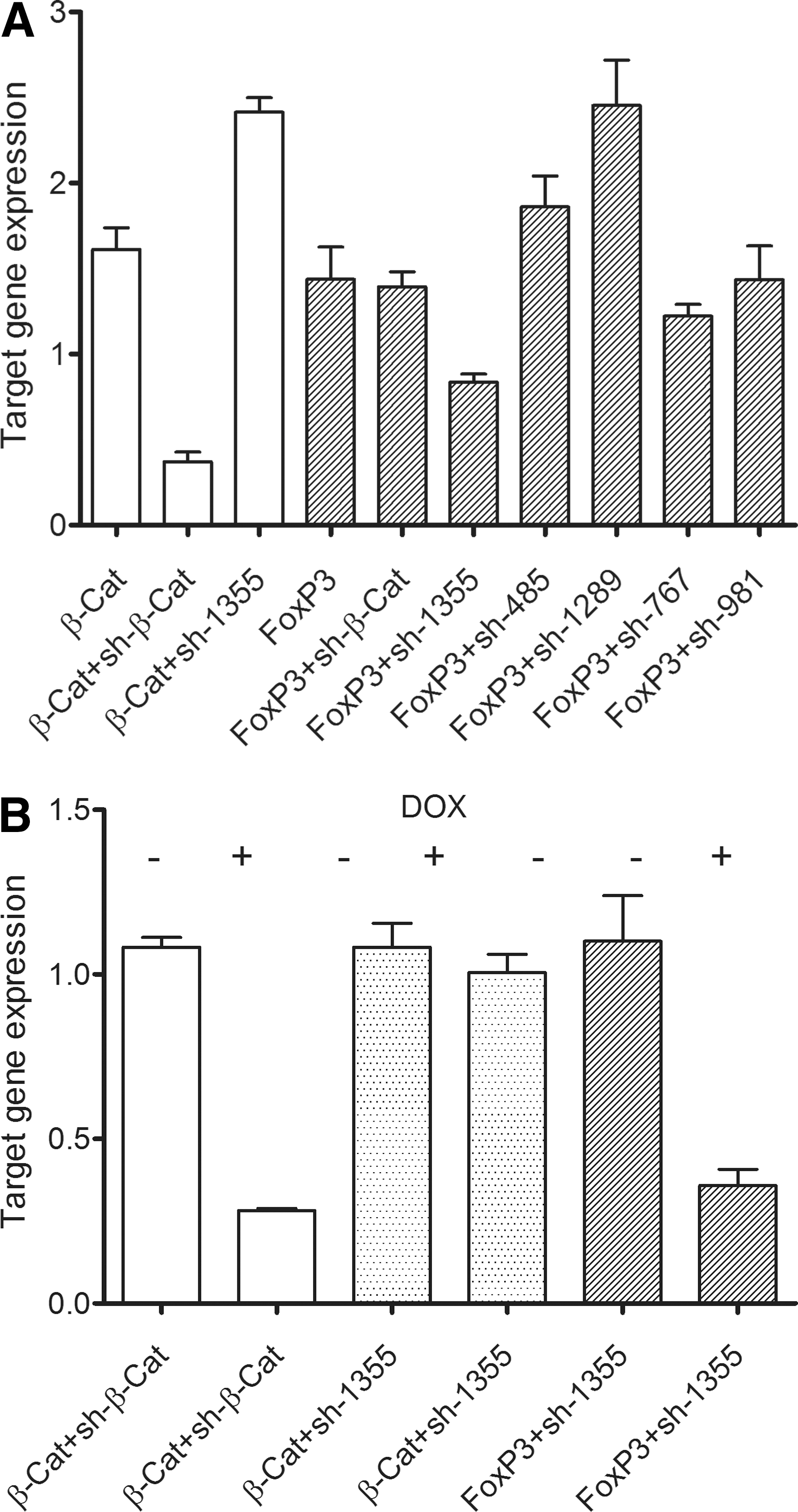
A short hairpin sequence is able to specifically and inducibly knock down expression of FOXP3. (
Conditional regulation of target genes
Next, we wished to demonstrate doxycycline-regulated knockdown of our target genes. HEK293T cells transduced with plv-T-sh-1355 and plv-T-sh-β-cat were then transiently transfected with the psiCHECK reporter construct (psiCHECK-FOXP3 or psiCHECK-β-catenin) and doxycycline (Dox) was added (1 μg/ml) to derepress shRNA transcription. Knockdown of the reporter genes was monitored by luciferase assay after 48 hr. Efficient and specific Dox-dependent knockdown of reporter gene activity was observed for both FOXP3 and β-catenin (Fig. 2B). In the absence of Dox, cells transduced with plv-T-sh-1355 virus and transfected with FOXP3 reporter vector (psiCHECK-FOXP3) showed levels of Renilla activity comparable with that in cells transfected with psiCHECK-FOXP3 alone (data not shown). However, addition of Dox (1 μg/ml) substantially reduced FOXP3 expression (>70% knockdown) (Fig. 2B). In contrast, no change in Renilla activity was observed from the β-catenin expression construct in cells transduced with plv-T-sh-1355 virus either in the absence or presence of Dox, indicating that sh-1355 activity is specific. This result also indicates that addition of Dox itself had no net effect on expression of the target gene. Target-specific and Dox-inducible knockdown was also observed with the plv-T-sh-β-cat-transduced cells transfected with psiCHECK-β-catenin. Similar results were observed in the Jurkat T cell line and NIH 3T3 cells for the sh-β-cat vector (data not shown).
Lentivirally delivered FOXP3 is conditionally regulated by sh-1355 in Jurkat T cells
The previous experiments have shown that short hairpin RNAs could downregulate Renilla activity from a hybrid mRNA containing FOXP3 or β-catenin sequences within its 3′ UTR. In this mRNA the target gene sequences are not translated. We therefore wished to determine whether sh-1355 could conditionally downregulate the intact FOXP3 mRNA. To do this Jurkat cells, which do not express endogenous FOXP3, were transduced with a lentivirus expressing FOXP3 from the EF-1α promoter (plvEIG-FOXP3) and GFP from an IRES. Cells were sorted by flow cytometry, into a population with the highest GFP+ expression (top 5%), as compared with all GFP+ cells (pooled population) (Fig. 3A). We confirmed that cells with the highest GFP fluorescence also exhibited the highest levels of FOXP3 expression by intracellular staining (Fig. 3B), suggesting that the IRES-driven reporter was a robust marker of expression of the target gene in plvEIG-FOXP3. The sorted population of plvEIG-FOXP3 cells (top 5% GFP+) was then transduced a second time (superinfected) with plv-T-sh-1355 lentivirus. As the levels of GFP in the superinfected cells were significantly higher than in the single-transduced cells (Fig. 3C), a simple gating strategy was used to analyze FOXP3 expression levels in cells with the highest mean fluorescence intensity (MFI) for GFP. Cells were treated with Dox (1 μg/ml) for 48 hr, stained intracellularly, and then analyzed by flow cytometry for FOXP3 expression. We observed that the FOXP3 level in cells transduced with both plvEIG-FOXP3 and plv-T-sh-1355 cultured in the absence of Dox was similar to that observed in cells transduced with plvEIG-FOXP3 alone (Fig. 4A). However, when Dox was added to the culture medium, FOXP3 expression in the double-transduced cells was significantly reduced, approaching the background levels of staining observed in cells not expressing FOXP3 (Fig. 4A). This result indicated that plv-T-sh-1355 downregulated plvEIG-FOXP3 in a Dox-dependent manner in Jurkat populations expressing high levels of FOXP3. Real-time PCR confirmed that FOXP3 mRNA expression was similarly downregulated (Fig. 4B). The comparable levels of FOXP3 observed in cells transduced with plvEIG-FOXP3, and in cells transduced with both plvEIG-FOXP3 and plv-T-sh-1355 cultured in the absence of Dox, is consistent with the tight regulation of sh-1355 transcription by the T-REx protein in the absence of derepression by Dox. This was confirmed by highly sensitive stem–loop RT-PCR analysis of the expression of the plv-T-sh-1355 shRNA itself, showing no product in empty Jurkat cells or Jurkat cells transduced with plv-T-sh-1355 in the absence of Dox, but significant sh-1355 product when the Jurkat cells transduced with plv-T-sh-1355 were cultured in the presence of Dox (Fig. 4C).
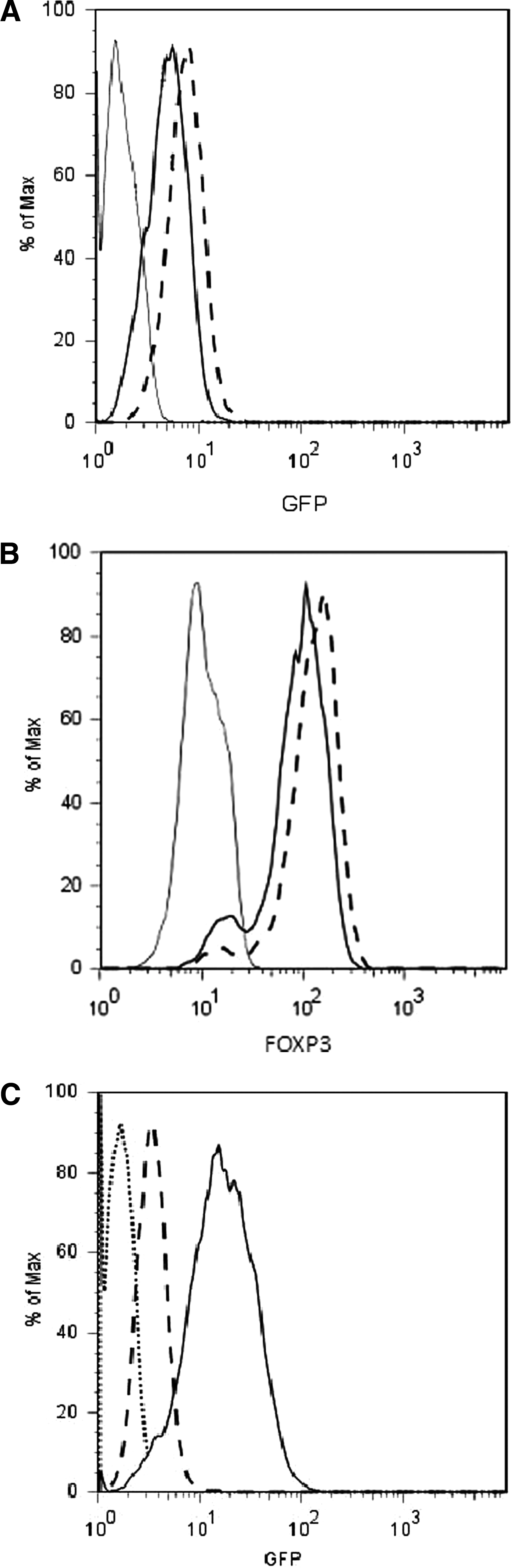
Isolation of Jurkat cell lines expressing plvEIG-FOXP3 and plv-T-sh-1135. (

Conditional regulation of FOXP3 expression in Jurkat cells and human Tregs. (
Regulation of FOXP3 in primary human Tregs
To test the broad scope of this lentiviral vector, we isolated primary human cord blood Tregs (CD4+CD25+) and transduced them with plv-T-sh-1355 viral supernatant (25 × concentrated). Transduced cells (mean transduction efficiency, 33.5%; n = 2) were then expanded ex vivo for 8 days to increase cell numbers. Cells were sorted by flow cytometry gating on GFP+ events. GFP+ and GFP− cells were then cultured in the absence or presence of Dox (1 μg/ml) for 48 hr and subsequently stained intracellularly and analyzed by flow cytometry for FOXP3 expression. We observed that FOXP3 expression in Tregs transduced with plv-T-sh-1355 and cultured in the absence of Dox was similar to FOXP3 expression in nontransduced cells in the absence or presence of Dox (data not shown). This result indicates that Dox has no nonspecific effects on FOXP3 expression in nontransduced cells and also, importantly, that plv-T-sh-1355 is not leaky in the absence of Dox. When Dox was added to the culture medium of plv-T-sh-1355-transduced Tregs, FOXP3 expression was reduced by 36.6% (MFI was reduced from 416 to 340 ± Dox, respectively) compared with plv-T-sh-1355 Tregs cultured in the absence of Dox (Fig. 4D). These results show that plv-T-sh-1355 conditionally downregulates the expression of endogenous FOXP3 in human Tregs in the presence of Dox.
Activation of short hairpin is both sensitive and rapid
A concentration curve to determine the kinetics of Dox induction of sh-1355 activity to knock down expression of FOXP3 was then generated. The results of this experiment showed that the concentration of Dox routinely used, 1 μg/ml, far exceeds that which led to maximal FOXP3 knockdown (between 20 and 40% FOXP3 remaining). At a Dox concentration of 10 ng/ml, FOXP3 expression was reduced to ∼40% of that expressed in non-Dox-treated cells (Fig. 5A), indicating the sensitivity of the system. Likewise, using SL-RT-PCR, we observed no plv-T-sh-1355 shRNA in the absence of Dox but robust expression in the presence of Dox at just 10 ng/ml (Fig. 5B), confirming the sensitivity of the system. Activation of the short hairpin was also rapid. In the experiments described here, cells were routinely treated with Dox for 48 hr before evaluating knockdown of FOXP3 expression, but we observed partial knockdown of FOXP3 as early as 12 hr after addition of Dox (68% FOXP3 remaining) and maximal knockdown of FOXP3 protein by 24 hr (22% FOXP3 remaining) compared with non-Dox-treated cells (Fig. 5C).
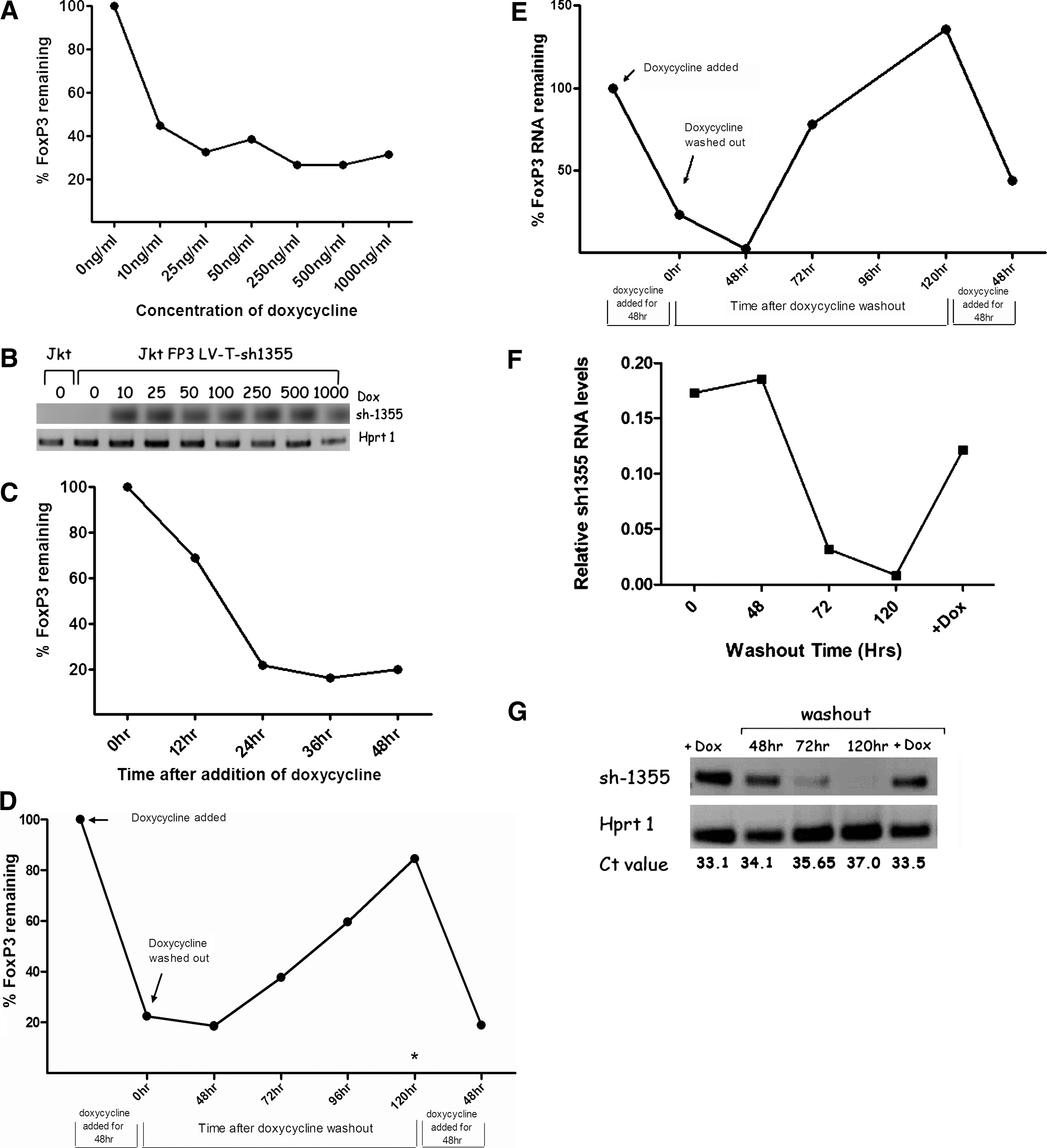
Activation of the short hairpin is sensitive, rapid, and reversible. (
Gene regulation is robust and reversible
Having shown that target gene (FOXP3) expression can be significantly downregulated in a Dox-dependent manner with lentiviral sh-1355, we next wanted to test the reversibility of the drug-induced knockdown of FOXP3 by performing a washout experiment. Jurkat cells expressing high levels of FOXP3 from plvEIG-FOXP3 and superinfected with plv-T-sh-1355 were cultured in the presence or absence of Dox (1 μg/ml) for 48 hr. After collecting an aliquot of cells for analysis, the remaining cells were washed and returned to culture. Over the next 120 hr, cells were collected every 24 or 48 hr, after which Dox was readded at 120 hr. As seen previously, FOXP3 expression was reduced to ∼20% of original levels after incubation with Dox (1 μg/ml) for 48 hr (Fig. 5D). After washout of Dox the level of FOXP3 expression remained at ∼20% for the next 48 hr, but increased at each time point thereafter until FOXP3 had returned to almost preknockdown levels (∼90%) by 120 hr after Dox removal. This result shows that drug-induced knockdown of FOXP3 is efficient and reversible. To further test the robustness of the system, after the 120-hr time point Dox (1 μg/ml) was added back and the cells were incubated for a further 48 hr. FOXP3 expression dropped back down to 20% of original levels (Fig. 5D). FOXP3 expression in cells continuously cultured in Dox for 120 hr remained low (3.8% of nontreated cells; asterisk in Fig. 5D), indicating that Dox-induced activation of the short hairpin is robust and prolonged. In all cases FOXP3 mRNA levels followed the same expression pattern (Fig. 5E). Over the course of the washout experiment, plv-T-sh-1355 shRNA expression exactly mirrored expression of FOXP3. Expression of this RNA was high after 48 hr of Dox, but had diminished to near background levels after 120 hr of washout. plv-T-sh-1355 shRNA levels increased once more when Dox was added back to the cells. These results indicate that as the Dox is washed out, plv-T-sh-1355 is concomitantly switched off and FOXP3 expression is derepressed (Fig. 5F and G).
Validation of reversible Tet-regulated gene ablation in breast cancer cells
To rigorously establish the utility and reversibility of the vector, we chose to target an endogenous cellular gene, the oncogene MYB, in the breast cancer cell line MCF7. When a prevalidated MYB-specific shRNA or a scrambled shRNA (Drabsch et al., 2007) was delivered stably into MCF7 cells, no impact on viability or growth characteristics was seen in the absence of Dox, as measured in a proliferation assay. However, when Dox was added to the culture at 1 μg/ml, a significant reduction in growth and proliferation was observed only in the plv-T-sh-Myb-transduced cells. When Dox was left with the cells for 5 days a stable gene-silencing effect was observed at the level of both protein expression and proliferation. This growth suppression was reversible, as demonstrated by washout on day 5 and restoration of growth kinetics. Furthermore, in the sh-Myb line only, the growth inhibition could be reestablished by a second Dox treatment on day 7, resulting in growth arrest for a second time in the same culture (Fig. 6A). The impact of the sh-Myb construct on MYB protein levels was confirmed by Western blot analysis of the parent cell line, the cell line transduced with sh-Myb, and two controls in which the cells were transduced with a scrambled shRNA (sh-SCR) or transduced with an empty lentiviral vector. In the plv-T-sh-Myb-transduced MCF7 cells in the absence of Dox, the levels of Myb were comparable to that of the parent cells or the two controls. MYB was significantly reduced when Dox was added, and returned to control levels after washout (Fig. 6B).

Validation of reversible gene silencing, using endogenous target gene MYB. (
Discussion
The main goal of this study was to build a simple, efficient system with which to study gene function, and we chose two genes to model this. Conditional gene knockdown has the potential to be a powerful tool, with short hairpin RNAs emerging as a highly effective and relatively straightforward mechanism to achieve this goal. However, for this technology to reach its full potential safe, efficient, and tightly regulated delivery systems need to be further developed. A system that allows regulated control of gene expression (including full reversibility) has huge advantages over systems that rely on absolute knockout or Cre-mediated excision (Ventura et al., 2004; Seidler et al., 2008), potentially overcoming issues including lethality. It also allows knockdown in the appropriate temporal- or stimulus-specific manner. Of critical importance, developing such tools allows for the controlled manipulation of gene expression in a variety of primary cell types and diverse organisms, rather than being restricted to mice. In addition, for a system to be robust, particularly for an in vivo model, manipulation of gene expression should also be enduring.
There have been many advances in the design of gene delivery systems, with lentiviruses emerging as a relatively safe system of delivery (Bauer et al., 2008). We have based the vectors described herein on the third-generation self-inactivating lentiviral vectors first described and shared by Trono and colleagues (Naldini et al., 1996a; Dull et al., 1998; Salmon and Trono, 2007) and modified by us (Barry et al., 2001). We, and others, have demonstrated that these lentiviral vectors have improved transduction efficiency, function, and specificity, particularly in primary cells (Taher et al., 2008), and are also well tolerated in vivo (Seppen et al., 2001; Yanay et al., 2003; Barry et al., 2005; Brzezinski et al., 2007), making them ideal for in vivo studies. We chose to combine the shRNA and regulatory elements into a single vector as we believed that it is less practical to perform multiple transductions or to generate clones when targeting rare primary cells or using in vivo models.
By using Gateway recombination technology we were able to rapidly screen potential short hairpins before the production of lentiviral particles. This is of potential benefit as there are still persistent problems with short hairpin design, leading to inefficient knockdown of the target gene, and thus requiring a number of targeting sequences to be tested for each gene. Screening a bank of potential short hairpin RNAs in this way revealed that one sequence out of five was able to knock down target gene expression in a highly specific way.
In this paper we describe a single lentiviral vector capable of delivering tightly regulated Dox-responsive knockdown of target genes as shown by the regulated knockdown of two different genes, FOXP3 and MYB, using single short hairpin RNAs delivered into several different cell lines (HEK293T, Jurkat, and MCF7). We also demonstrated regulated knockdown of endogenous FOXP3 in primary human Tregs. In contrast to the findings of ter Brake and Berkhout (2007), who observed a substantial loss of shRNA activity in transduced cells, we observed efficient knockdown of target genes in either transfected or transduced cells.
Throughout our experiments we observed significant knockdown of target genes (FOXP3, 60–95%; β-catenin, 80%; and MYB, ∼80% knockdown, respectively), indicating efficient expression and processing of the short hairpin cassettes. This suggests that sufficient shRNA molecules can be produced from a single copy of the tet–H1 promoter shRNA cassette in the viral backbone. We obtained high levels of FOXP3 knockdown at an MOI of 1 in cell lines (MOI of 25 in primary human Tregs), compared with others who have had to use high viral titers or isolated cell clones to obtain robust target knockdown with short hairpin RNAs (summarized in Pluta et al., 2007). This compares favorably with results using multiple copies of the short hairpin to achieve durable target gene knockdown (ter Brake and Berkhout, 2007; ter Brake et al., 2008). Significantly, the short hairpin RNA delivery vector plv-T-sh described here appears not to be susceptible to inactivating mutations (ter Brake et al., 2008) as demonstrated by the finding that MCF7 cells transduced with a lentiviral construct encoding a validated MYB shRNA (Drabsch et al., 2007) retains the ability to knock down MYB oncogene levels even after prolonged culture in the absence of Dox.
Importantly, in the absence of Dox the tet–H1 promoter appears to be tightly regulated by the repressor protein, with little evidence of leakiness. Not only was FOXP3 expression in Jurkat cells transduced with plv-T-sh-1355, when cultured in the absence of Dox, indistinguishable from that in parental cells expressing FOXP3 alone, but importantly plv-T-sh-1355 RNA was detected (using the highly sensitive SL-RT-PCR technique) only in cells treated with Dox. Similar observations were seen in MCF7 cells transduced with plv-T-sh-Myb and importantly in primary human Tregs transduced with plv-T-sh-1355 to knock down endogenous FOXP3. This has been a hurdle in the development of inducible short hairpin RNA technologies, where leakiness of shRNA expression has been observed in both transfected and transduced cells (Aagaard et al., 2007).
In experiments examining the kinetics of shRNAi induction, we show that this knockdown is both sensitive and rapid. For example, plv-T-sh-1355 RNA was induced with Dox at just 10 ng/ml and FOXP3 protein expression itself could be reduced in the presence of Dox at 10 ng/ml, 100 × lower concentrations of Dox than routinely used, with ∼60% knockdown of FOXP3 obtained and maximal repression occurring from Dox at 25 ng/ml and onward. This may be useful for in vivo experiments in which systemic administration of Dox is required. Second, after induction with Dox, derepression of the tet–H1 promoter-shRNA cassette, initiation of transcription and processing of the short hairpin must be rapid, because we observed partial knockdown of FOXP3 protein by as early as 12 hr (68% FOXP3 remaining) and maximal knockdown of FOXP3 by 24 hr (22% FOXP3 remaining) compared with non-Dox-treated cells. This compares favorably with other systems in which prolonged exposures to Dox (5 and 8 days) were required for maximal knockdown at the protein level (Szulc et al., 2006; Aagaard et al., 2007). One of the major strengths of our system is that compared with others, our system is fully and quickly reversible even when using high concentrations of Dox. In addition, washout of the Dox resulted in loss of the plv-T-sh-1355 RNA with the concomitant gain of FOXP3. Others show reversibility, but this is often incomplete or relatively slow and insensitive (Seibler et al., 2007; Herold et al., 2008).
The vector described in this paper provides a simple, single-vector system that is quickly and strongly inducible and, in addition, fully reversible. We have carried out carefully analyzed kinetics quantitating all of our results with respect to appropriate controls to show convincingly that the system described here is an advance on systems currently available. This system is valuable not only as a tool with which to study gene function, but also for modeling human disease, and may represent a potential therapeutic modality.
Footnotes
Author Disclosure Statement
No competing financial interests exist.