
Abstract
Human papillomavirus type 16 (HPV16) is associated with the development of anogenital cancers and their precursor lesions, intraepithelial neoplasia. Treatment strategies against HPV-induced intraepithelial neoplasia are not HPV specific and mostly consist of physical removal or ablation of lesions. We had previously designed an HPV-specific approach to kill HPV-infected cells by the herpes simplex virus type 1 thymidine kinase (TK) gene driven by HPV E2 binding to E2-binding sites (E2BS) in the native HPV16 long control region. E2-induced TK expression renders the cells sensitive to the prodrug ganciclovir. To optimize this therapeutic approach, we modified the native long control region by adding variable numbers of E2BS adjacent to E2BS4, resulting in greatly increased cell death in HPV-positive cell lines with variable levels of E2 protein expression and no reduction in HPV specificity. Our results showed maximum increase in TK expression and cell killing when one additional E2BS was added adjacent to E2BS. As HPV-infected patients also exhibit variable E2 expression across lesions and within a lesion, this approach may potentiate the clinical utility of the herpes simplex virus type 1 TK/ganciclovir therapeutic approach.
Introduction
Early HPV transcription is regulated by the noncoding long control region (LCR) that contains a keratinocyte-specific enhancer region involved in the regulation of early HPV transcription and viral tropism (Sen et al., 2004; Hebner and Laimins, 2006). It has binding sites for several cellular transcription factors including AP-1, Sp1, Oct-1, YY1, and NF1 (Morris et al., 1993; Tan et al., 1994; Kanaya et al., 1997) and binding sites for the viral E1 and E2 proteins, which regulate early HPV gene expression and initiation of replication (Tan et al., 1994; Frattini and Laimins, 1994).
The E2 protein is involved in the regulation of HPV gene expression and replication. The LCR of oncogenic HPV types contains four E2-binding sites (E2BS) with variable affinity toward E2 (Fig. 1A). These sites have the consensus palindromic sequence ACCN6GGT (Hegde and Androphy, 1998; Thierry, 2009). Three E2BS are located in the vicinity of the viral p97 promoter, whereas the fourth site is located at a position upstream of the LCR. E2 binds to these sites in a cooperative manner resulting in either activation or repression of transcription from the early promoter (Longworth and Laimins, 2004). Regulation of E6 and E7 oncogene expression from the LCR by E2 depends upon the E2BS that are bound and the amount of full-length E2 protein (Broker et al., 1989; Bernard and Apt, 1994; Bouvard et al., 1994; Hamid et al., 2009). Various groups have reported that the loss of expression of E2 leads to abundant expression of E6 and E7 proteins and development of cervical carcinoma (Jeon and Lambert, 1995; Corden et al., 1999). Recently it has been reported that E2 disruption is not essential for high levels of oncogene expression (Hafner et al., 2008). Data show that the majority of HPV16-positive cervical neoplastic lesions are transcriptionally active and express E2. In lesions containing integrated viral DNA, despite the E2 gene being intact and the E2 protein being expressed, LCR methylation at the E2BS may play a role in preventing E2 binding to the LCR and influencing downstream gene expression (Bhattacharjee and Sengupta, 2006; Fernandez et al., 2009). Thus, disruption of the E2 gene is not essential for cervical cancer progression. Almost all HPV16-associated lesions, regardless of grade, have at least some intact E2 genes (Arias-Pulido et al., 2006; Bhattacharjee and Sengupta, 2006; Fernandez et al., 2009). E2 expression may occur at low levels in basal layers and is required for episomal maintenance (Doorbar, 2005). E2 can interact directly with E6 and E7 proteins and modulate their activities (Grm et al., 2005; Gammoh et al., 2006). E2 can also interact with host proteins including the tumor suppressor protein p53 (Massimi et al., 1999; Brown et al., 2008; Hamid et al., 2009). This has led to suggestions that E2 can play a role in the regulation of cellular proliferation. Hence, new roles for the E2 protein are being defined now.
(
Currently, surgical removal and ablation of HPV-infected tissue are the only methods available to treat high-grade Cervical Intraepithelial Neoplasia (CIN) to prevent Invasive Cervical Cancer (ICC). As these methods do not treat HPV infection per se, they can be associated with lesion recurrence and areas of adjacent HPV-negative normal tissue may also be unnecessarily removed or ablated. An HPV-specific nonsurgical treatment approach is therefore highly desirable. We have designed a suicide gene therapy approach to specifically eliminate HPV-infected keratinocytes without significant toxicity to HPV-negative cells (Sethi and Palefsky, 2003). The E2 protein is exploited to transactivate HPV16 promoter elements within the LCR to drive expression of an exogenous cytotoxic gene, herpes simplex virus type 1 (HSV-1) thymidine kinase (TK) (Moolten, 1994; Mullen, 1994; Nishihara et al., 1998; Sawamura et al., 1999). The cells are exposed to the prodrug ganciclovir (GCV) and HSV-1 TK converts GCV into its monophosphate form. The monophosphate form is metabolized into di- and tri-phosphates by host kinases (Matthews and Boehme, 1988). GCV triphosphate is incorporated into dividing cells, inhibiting DNA polymerase and causing cell death. Cells that do not express E2 would be insensitive to GCV.
With the goal of treating a mucosal surface lesion such as CIN, a liposome-based delivery method can be used to topically apply an effective amount of therapeutic plasmid to the lesion site. Oral GCV or its equivalent, valacyclovir, would be given concurrently. As the basal layer continuously regenerates the epithelium and HPV infects the basal cell layer, killing of the infected basal and parabasal cells has the potential to lead to a clinical and virologic cure.
The number and position of the E2BS within the suicide gene plasmid to optimally drive TK expression at different levels of E2 expression are not known. As noted earlier, there are four E2BS, referred to as E2BS1–4, with the site most proximal to the p97 promoter being E2BS1 (Fig. 1A). The goal of the present study was to maximize the therapeutic benefits of this approach by manipulating the number and position of E2BS in the plasmid, thus increasing the sensitivity of the cells to GCV and minimizing nonspecific toxicity. We assessed this approach in a variety of cell lines and showed that addition of E2BS upstream of E2BS4 increased gene transcription efficiency from the p97 promoter. The addition of E2 sites led to increased expression of the HSV-1 TK gene in HPV-infected cells showing varying levels of E2 expression, with no loss of HPV specificity. Addition of a single E2BS was more efficient in increasing expression from the LCR than was addition of multiple sites.
Methods
Plasmid construction
A 620-bp DNA fragment spanning the HPV16 LCR (nucleotide 7420 to nucleotide 117) was amplified by polymerase chain reaction (PCR) from the plasmid pHPV16 (ATCC, Manassas, VA) with the following primers: primer URR1 (5′- CGGCTCGAGTGTAGCGCCAGGCCCATT-3′) and primer URR2 (5′-CGGAAGCTTGGGTCCTGAAACATTGCA-3′), as described previously (Sethi and Palefsky, 2003). The LCR sequence comprising the four E2BS and the p97 early promoter was cloned into the XhoI–HindIII site of the pGL3-basic vector (Promega, Madison, WI) upstream of the luc + cDNA encoding the modified firefly luciferase (plasmid LCR-luc).
The HSV-1 TK gene was PCR amplified as a 1.3-kb fragment from the vector pRB103 (kindly provided by E. Mocarski, Stanford University, CA) (Sethi and Palefsky, 2003), using the following primers: TK forward (5′-CGGAAGCTTCCCAGGTCCACTTCGCAT-3′) and TK reverse (5′-CGGTCTAGACATAGCGCGGGTTCCTTC-3′). The luciferase gene was excised from the plasmid LCR-luc and was replaced by the 1.3-kb PCR product containing the HSV-1 TK coding sequence, resulting in the plasmid LCR-TK, in which the HSV-1 TK is driven by the HPV16 LCR.
Constructs with additional E2 sites
The oligonucleotide RSE1 containing six E2BS (shown in bold below) was synthesized. A complimentary oligonucleotide RSE2 was synthesized and annealed to RSE1 to obtain double-stranded DNA. Restriction enzyme sites were inserted between the E2BS of the oligonucleotide to derive constructs carrying four, two, or one additional E2 site(s) after manipulation (Fig. 2A). The resulting double-stranded oligonucleotides with variable E2BS were ligated to LCR-luc or LCR-TK purified plasmid DNA fragments. Figure 2A shows all constructs. RSE1: 5′ Nhe
Nhe
Kpn
CC Eco

(
E2 expression plasmid
The E2 gene was amplified using the template plasmid pHPV16 (ATCC) with the following primers: E2KOZFOR: 5′-CGG
Cell culture and transfections
The CaSki (Pattillo et al., 1977) and 16-MT HPV16-positive immortalized foreskin keratinocyte cell lines (Turner and Palefsky, 1995) were used in the study. The HPV-negative HT-29 colon carcinoma cell line (Fogh et al., 1977) and the HPV-negative C33A (Yee et al., 1985) and HT-3 (Fogh et al., 1977) cervical cancer cell lines were studied as well. All cell lines except 16-MT were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with glutamine (25 μg/ml), antibiotics, and 10% fetal bovine serum (10% DMEM). 16-MT cells were cultured in keratinocyte growth medium-2 supplemented with DMEM.
Transfections were performed using the lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions, on a 12-well plate at a density of 4 × 105 cells per well. The cells were harvested after 24–48 hr to measure promoter activity by a standard luciferase assay (Promega). Cells were harvested after 72 hr for RT-PCR, to check gene expression levels. GCV treatments were started at 72 hr after transfection.
Transfection efficiency for the various luciferase constructs was determined by the β-galactosidase enzyme assay system (Promega). The cell lysate prepared for the reporter assay was used for measuring β-galactosidase activity by a colorimetric assay per the manufacturer's instructions.
Detection of gene expression by RT-PCR
Total RNA was extracted from the cells using the RNeasy mini kit as per manufacturer's instructions (Qiagen, Chatsworth, CA). The RNA was treated with RNase-free DNase (Promega) for 3 hr at 37°C. The RNA was again purified through the RNAeasy column per the manufacturer's instruction. Purified RNA was used for reverse transcription and PCR, using the SuperScript III First-Strand Synthesis System for RT-PCR as per manufacturer's instructions (Invitrogen).
Quantitative real-time PCR
After total RNA extraction, concentrations were determined by means of spectrophotometry; each sample yielded a minimum of 10 μg of total RNA with an A 260/A 280 ratio ranging between 1.7 and 2.0. The E2 primers used were E2RT-F2 5′-CAGAGCCAGACACCGGAAAC-3′ and E2RT-R2 5′-TCCGTCCTTTGTGTGAGCTG-3′. The TK primers used were TKRT-2F 5′-GCACGTCTTTATCCTGGATTACG-3′ and TKRT-2R 5′-TAAGTTGCAGCAGGGCGTC-3′. The level of specific mRNA expression was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The quantitative real-time SYBR Green reaction was performed in duplicate, according to recommended protocols provided by the TaqMan ABI 7900 (Applied Biosystems, Foster City, CA). The reactions were performed at the Genome Core facility, University of California–San Francisco.
Detection of luciferase gene expression by the luciferase assay
The cell lines were transfected with the LCR-luc vector or vectors containing additional E2 sites. Controls were set up with the pGL3-basic vector for all cell lines. After 24–48 hr of transfection, the cells were harvested with 100 μl of the reporter lysis buffer per well. Luciferin reagent was added to the cell lysates to measure luciferase reporter gene expression, per the manufacturer's instructions (Promega). The luciferase gene expression was measured as relative light units, using a Dynatech Microlite plate luminometer (Chantilly, VA) with signal integration for 10 sec. For cotransfection experiments, the pCMV-E2 plasmid was added in different amounts for transfection along with the LCR luciferase constructs. The luciferase activity was measured after 24–48 hr, as described previously.
In vitro effect of GCV on HSV TK+ and HSV TK− cell lines
The HPV16-positive (CaSKi, 16-MT) and HPV-negative (HT-3, HT-29, C33A) cell lines were transfected with each of the LCR-TK constructs containing additional E2 sites. Controls were set up with the pGL3-basic vector for all cell lines. Gene expression was allowed for 48–72 hr after transfection. The cells were then exposed to GCV (Invivo Gen, San Diego, CA) for 6 days at concentrations ranging from 15 to 25 μg/ml in 10% DMEM. Cell viability was measured by the MTS cell proliferation assay (Berridge and Tan, 1993) (CellTiter 96 AQueous One Solution cell proliferation assay; Promega), according to the manufacturer's instructions. The absorbance at 490 nm (A
490) is a measure of the number of living cells in the culture. Percentage of survival was calculated as follows:
PCR amplification of E2 gene in CaSki cells
The E2 gene was amplified in CaSki cells with three different sets of primers. The primer sets were designed to amplify the E2 gene from the 5′ end (A1/A2), 3′ end (C1/C2), and also the whole E2 gene (W1/W2). Primers used were as follows: W1: ATGAAAATGATAGTACAGAC; W2: CCAGTAGACACTGTAATAG A1: ATGAAAATGATAGTACAGAC; A2: TGGATAGTCTGTGTTTCTTCG C1: GTAATAGTAACACTACACCCATA; C2: GGATGCAGTATCAAGATTTGT
Western blot analysis
E2 expression in CaSki cells was analyzed after preparing cell extracts in RBS buffer (10 mmol/liter Tris at pH 7.5, 10 mmol/liter NaCl, 3 mmol/liter MgCl2) in the presence of 1% NP-40 and resuspended in SDS-PAGE loading buffer (Fernandez et al., 2009). Equal amounts of cell extracts were loaded on a 12% SDS-PAGE gel and run at 150 V. The proteins were transferred to polyvinylidene diflouride membranes. The membranes were then hybridized with HPV16 E2 antibody TVG261 (ab17185; Abcam, Cambridge, MA) at 1:100 dilution. The protein was detected by the Chemiluminescence Plus Western Blotting detection system (GE Healthcare, Piscataway, NJ).
Detection of apoptosis in transfected cell lines
The HPV-positive (CaSki, 16-MT) and HPV-negative (HT-3, HT-29, C33A) cell lines were grown on chamber slides and transfected with each of the LCR-TK vector constructs. Controls were established with the pGL3-basic vector for all cell lines. Gene expression was allowed for 48–72 hr after transfection. The cells were then exposed to GCV for 4 days in concentrations ranging from 15 to 25 μg/ml in 10% DMEM. The cells were stained with the DeadEnd colorimetric apoptosis detection system as per manufacturer's instructions (Promega). It is a modified terminal nucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay. The darkly stained cells were apoptotic. Three fields of 30–40 cells were counted for each experiment.
Results
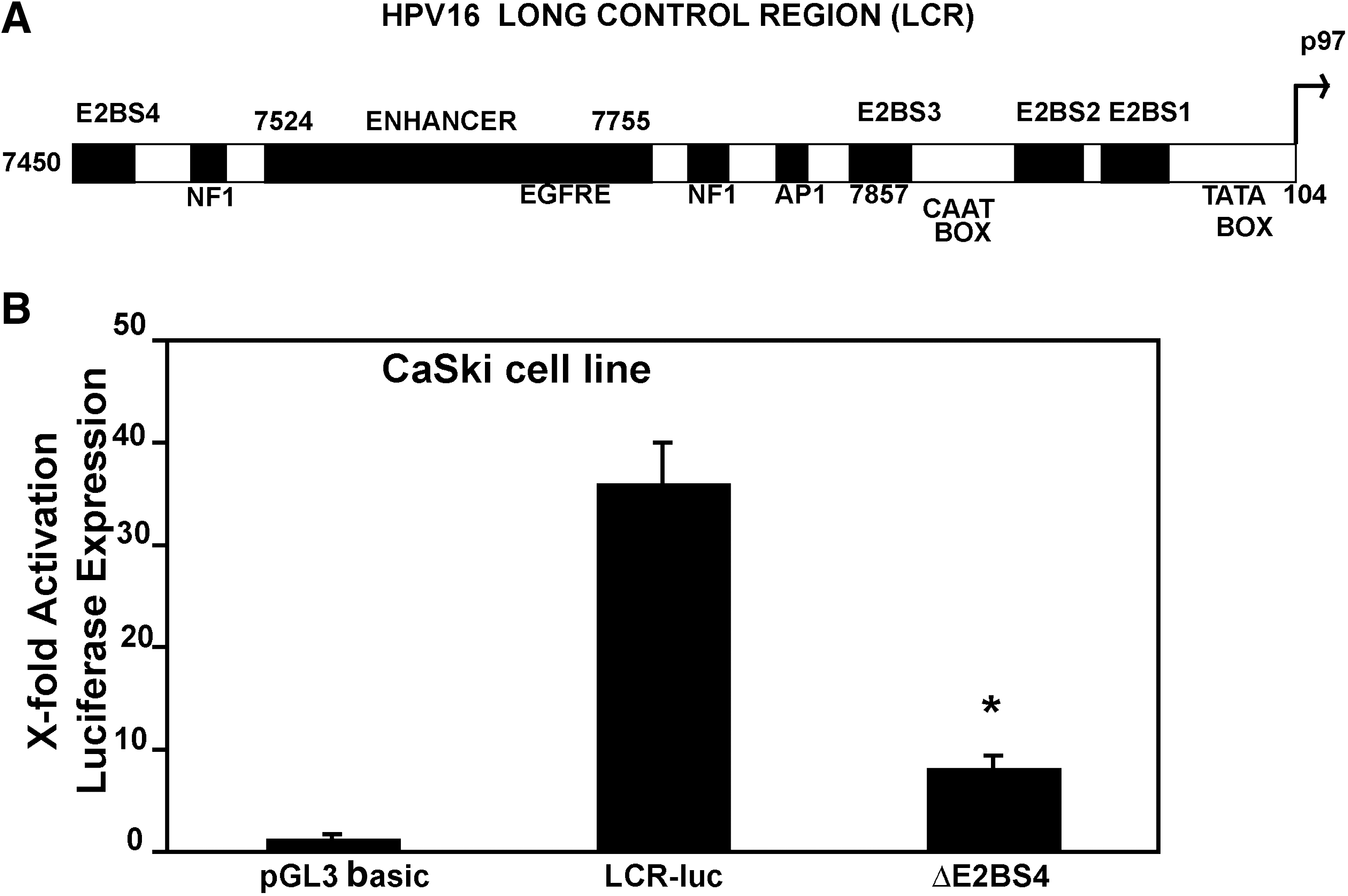
Binding of E2 protein to the E2BS4 on the HPV16 LCR upregulates gene expression from the p97 promoter
Figure 1A shows the map of the HPV16 LCR with the viral protein and cellular factor-binding sites. E2 protein binding to the LCR regulates downstream gene expression. The mutations in HPV16 E2BS4 have been reported to have little or no effect on promoter activity (Lewis et al., 1999), and also the role of E2BS4 in regulating transcription is not clearly defined for the HPV16 LCR. We therefore studied the effect of E2 protein binding to the E2BS4 on gene expression in the CaSki cell line, which expresses the E2 protein. After deletion of E2BS4 from the LCR, gene expression declined nearly fourfold in the CaSki cell line (Fig. 1B).
Addition of E2BS to the HPV16 LCR increases HPV-specific transcription from the p97 promoter
As the deletion of E2BS4 led to a decrease in gene expression, we hypothesized that addition of E2BS adjacent to E2BS4 may lead to increased gene expression compared with the native LCR. The native LCR and the LCR1, LCR2, and LCR4 plasmids containing one, two, and four additional E2BS, respectively (vector maps shown in Fig. 2A), were transfected into CaSki cells and luciferase activity was measured after 24 hr. Luciferase expression increased ∼ 90-fold for LCR1-luc and LCR2-luc compared with the 40-fold increase for the native HPV16 LCR above the luciferase expression from pGL3 basic construct. Expression increased by 60-fold for LCR4-luc (Fig. 2B). The HPV-negative C33A cell line was studied to determine the HPV specificity of this increased gene expression. In these cells, expression from the LCR-luc, LCR1-luc, LCR2-luc, and LCR4-luc plasmids remained nearly equal to pGL3 basic (Fig. 2C). The positive control pGSLV40 showed 60-fold expression above the control.
HSV-1 TK/GCV-mediated cell death of HPV16-positive cell lines increases significantly after addition of one E2BS to the HPV16 LCR
CaSki and 16-MT cells were transfected with each of the LCR-TK constructs to assess cell killing in the presence of GCV. After 72 hr of transfection, the cells were treated with GCV at concentrations of 25, 15, and 10 μg/ml for 6 days. The MTS assay was performed to assess cell viability. For CaSki and 16-MT cells, compared with the LCR-TK transfected cells, LCR1-TK, LCR2-TK, and LCR4-TK-transfected cells all showed decreased cell survival after GCV treatment at all doses used for treatment. Overall, cell killing was more efficient in CaSki cells than 16-MT cells, but for both cell lines the highest levels of cell death were seen with the LCR1 TK plasmid (Fig. 3A and B). Overall, the levels of cell death were highest at the 25 μg/ml level of GCV with the transfection of the LCR1 TK plasmid for both the cell lines.

Addition of E2BS to the HPV16 LCR enhances killing of HPV-positive cell lines, using the HSV-1 TK/ganciclovir (GCV) regimen. CaSki (
HPV specificity of HSV-1 TK/GCV-mediated gene therapy
To determine the HPV specificity of GCV-induced cell killing, we transfected the HPV-negative HT-29, HT-3, and C33A cell lines with each of the LCR-TK constructs. Seventy-two hours after transfection, the cells were treated with GCV for 6 days and cell survival was measured by the MTS assay. The results showed that the cell survival of these cell lines was not influenced by the HSV-1 TK/GCV therapy regimen (Fig. 4). There was greater than 90% cell survival in all HPV-negative cell lines at all doses and for each LCR-TK construct transfected. We also designed deletion constructs to remove the keratinocyte-specific enhancer and obtain a minimal promoter with the promoter proximal E2BS (E2BS1, E2BS2, and E2BS3) (data not shown). However, the minimal promoter was inefficient in transcribing the luciferase or TK gene cloned downstream of it after the construct was transfected into CaSki cells.
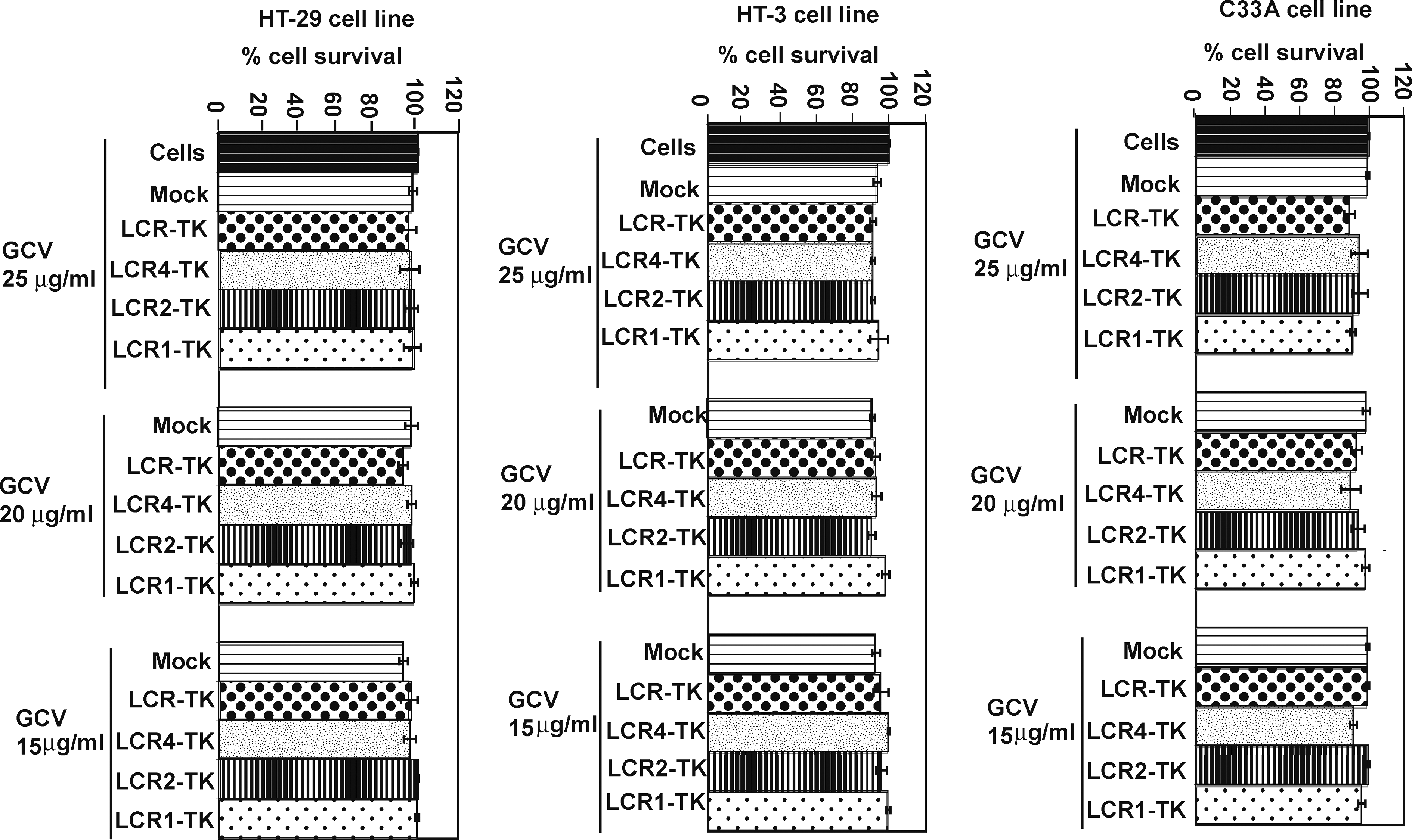
The HSV-1 TK/GCV-mediated regimen does not kill HPV-negative cell lines. The HT-29, HT-3, and C33A HPV-negative cell lines were transfected with the LCR-TK, LCR1-TK, LCR2-TK, and LCR4-TK constructs. Expression was allowed for 72 hr and the cells were then treated with GCV (25, 20, and 15 μg/ml) for 6 days. The cells were then harvested and percentage of cell survival was measured by the MTS cell proliferation assay. Untransfected/untreated cells were used as controls. The experiments were performed in triplicate and the bars represent standard deviation.
HSV-1 TK and endogenous E2 gene expression levels in the HPV16-positive cell lines
To determine the reason for the different rates of cell survival observed between the CaSki and 16-MT HPV16-positive cell lines (Fig. 3A and B), we evaluated differences in endogenous E2 gene expression levels using reverse transcriptase-PCR and real-time PCR. The presence of an intact E2 gene in the CaSki cell line was shown by PCR amplification of the 5′ and 3′ ends as well as the whole E2 gene using relevant primers (Fig. 5A). We sequenced the PCR-amplified E2 gene from CaSki cells and found the entire gene sequence to be intact and identical to that reported for the E2 gene in the CaSki cell line (Meissner, 1999). It has been reported that none of the nucleotide changes in CaSki HPV16 alter the established HPV16 splice donor or acceptor sites, polyadenylation signals, or transcription factor-binding motifs (Human Papillomaviruses, 1995; Meissner, 1999). Functional studies with the cloned E2 gene from CaSki cells will be done in future experiments.
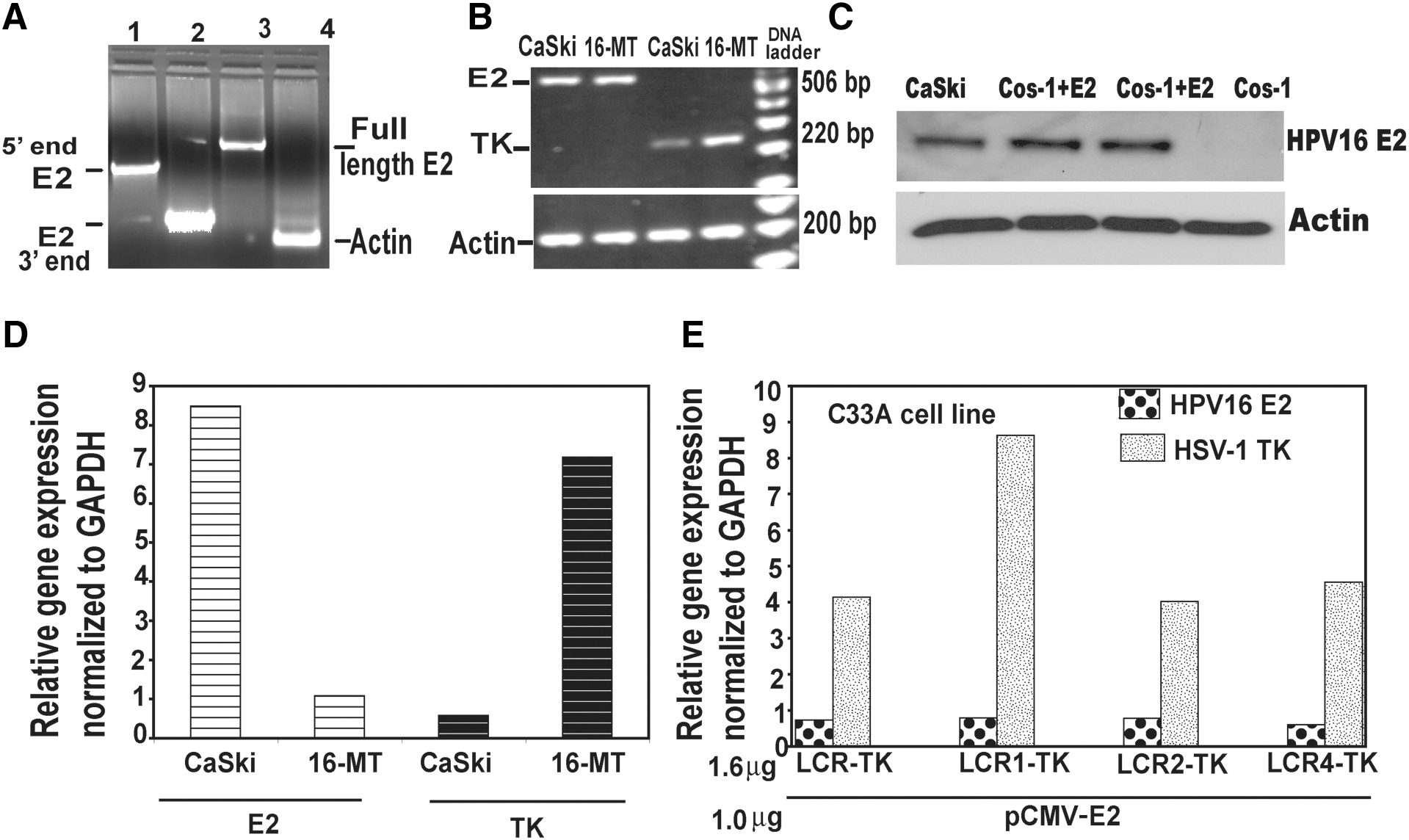
Analysis of expression of endogenous E2 expression and transfected HSV-1 TK gene expression from various LCR-TK constructs, using semiquantitative RT-polymerase chain reaction (PCR) and quantitative real-time PCR. (
Our results from RT-PCR and real-time PCR showed that CaSki cells had a higher amount of E2 gene expression than the 16-MT cells (Fig. 5B and D). To determine if the different levels of E2 expression correlated with differences in TK gene expression levels, the two cell lines were transfected with the native LCR-TK plasmid and expression was allowed for 48 hr. After normalization of relative gene expression of E2 and TK to human glyceraldehyde phosphate dehydrogenase gene expression (GAPDH), TK gene expression level was lower in CaSki cells compared with 16-MT cells (Fig. 5D). RT-PCR showed similar results for TK expression (Fig. 5B). This is consistent with our laboratory and other groups reporting repression of gene expression in the presence of high levels of E2 (Broker et al., 1989; Zur Hausen, 1991; Jeon and Lambert, 1995; Corden et al., 1999; Sethi and Palefsky, 2003). Upon cotransfection of the E2 expression vector in variable amounts in the SiHa cell line, we observed more repression of gene expression in the cells transfected with higher levels of E2 plasmid DNA (data not shown). We also performed Western blotting to show E2 protein expression in CaSki cells (Fig. 5C). E2 protein was detected in total cell lysates of CaSki cells as a 42-kDa protein after using the E2 monoclonal antibody TVG 261. Cos-1 cells were transfected with HPV16 E2 expression plasmid and E2 expression was allowed for 48 hr. These were used as positive controls and untransfected Cos-1 cells were used as negative controls. Actin was used as a housekeeping control for loading.
To further study the role of additional E2BS in TK gene expression, we cotransfected the HPV-negative C33A cell line with the HPV16 E2 expression plasmid pCMV-E2 and the various LCR-TK expression plasmids (LCR-TK, LCR1-TK, LCR2-TK, and LCR4-TK). After 72 hr, E2 and TK gene expression levels were determined by quantitative real-time PCR. Expression was normalized to human GAPDH gene expression. The data showed that in the presence of equal amounts of E2 gene expression the various LCR-TK plasmids showed different amounts of TK gene expression. TK expression was highest from the LCR1-TK construct, twofold higher than the native LCR-TK construct (Fig. 5E).
HSV-1 TK/GCV-mediated cell death is apoptotic
GCV treatment has been shown to induce apoptosis in cells expressing HSV-1 TK. To determine if apoptosis was being induced in LCR-TK-transfected cells, the various HPV-positive cell lines were transfected with the LCR-TK plasmid and TK expression was allowed for 48–72 hr. Cells were treated with GCV at a concentration of 25 μg/ml for 4 days and fixed for TUNEL staining. Untreated cells were used as controls. All the cell lines showed typical apoptotic morphology upon staining. The cells had an irregular appearance and the monolayer was disrupted. The control untreated cells showed few dead cells. We also assessed the morphology of 16-MT cells after LCR-TK transfection and GCV (25 μg/ml) treatment for 6 days in culture medium. The transfected and GCV-treated cells showed distinct apoptotic morphology with fragmented nuclei and a decrease in cell density (data not shown). A quantitative representation of the TUNEL assay is shown in Fig. 6. Consistent with the cell killing data, levels of apoptosis were higher in CaSki cells than 16-MT cells.

The HSV-1 TK/GCV regimen kills HPV-positive cell lines by apoptosis. The HPV16-positive CaSki and 16-MT cell lines were transfected with the LCR-TK plasmid. Expression was allowed for 72 hr, followed by GCV treatment (25 μg/ml) for 4 days, and cells were analyzed by TUNEL staining. The control cells were transfected with the LCR-TK plasmid but were not treated with GCV. Apoptotic cells have dark brown-stained nuclei because of fragmented DNA. The experiments were performed at least four times for all cell lines. Three fields of 30–40 cells were counted from three independent experiments for each cell line. The percentage of cells stained by TUNEL/apoptotic cells is shown for the control and GCV-treated experiments.
Discussion
There are several limitations in the current ablative approaches to treatment of CIN. One alternative is immune modulation. Prophylactic HPV vaccines have been shown to prevent HPV16- or HPV18-associated CIN (Garland et al., 2007; Markowitz et al., 2007; Paavonen et al., 2007), but these are not used for treatment of existing lesions. Recently, a therapeutic vaccine against HPV16 oncoproteins in HPV16-positive women with vulvar intraepithelial neoplasia achieved good clinical response in a subset of women (Kenter et al., 2009). However, therapeutic vaccination strategies have not been consistently effective and are not yet available for routine clinical use (Maiman, 1998; Palefsky, 1998; Palefsky et al., 1998a,b).
Another approach is to exploit the transactivation capability of the HPV E2 protein to drive expression of a suicide gene in an HPV-specific manner. The HSV-1 TK/GCV approach is the most widely used suicide gene transfer system in preclinical investigations and human clinical trials, used for brain, liver, and ovarian tumors (O'Malley, 2000; Fillat and Sangro, 2003). CIN is an excellent candidate for this approach because it is a mucosal lesion and the suicide gene plasmid could be applied topically with repeated applications, similar to treatment for candidiasis. Women would be given oral valacyclovir, and the specificity of cell killing would be conferred by expression of the HPV E2 protein.
We conducted our experiments on the CaSki and 16-MT cell lines. The CaSki cell line genome had been widely analyzed by various groups. Using PCR, it has been shown to harbor intact copies of the E2 gene (Bhattacharjee and Sengupta, 2006). We also PCR-amplified the E2 gene from CaSki cells, using primers from various regions as well as the whole gene (Fig. 5A). Several groups have also confirmed the presence of intact E2 in cervical carcinoma samples from patients (Sathish et al., 2004; Bhattacharjee and Sengupta, 2006). The integration of the E2 gene into the cellular genome is not essential for progression of cancer and E2 was found to be episomal in 52% of cervical carcinoma samples (Sathish et al., 2004).
E2 protein expression and activity in the CaSki cell line have been established (Fernandez et al., 2009). It has been shown in an HPV16 DNA methylome study using the CaSki cell line that E2 protein expression is intact but E2 is unable to bind the LCR because of methylation by mammalian methyl transferases of the E2BS upon integration of the HPV16 genome into the cellular genome (Fernandez et al., 2009). Demethylation of the HPV16 genome by agents such as 5-aza-2′-deoxycytidine in CaSki cells led to repression of E6 and E7 repression by facilitating E2 protein binding to the LCR (Fernandez et al., 2009). Notably, methylation of integrated HPV genomes would not affect our transfected HPV16 LCR-driven TK plasmid therapeutic strategy because the TK plasmid would likely remain episomal and unmethylated, and TK would be expressed in these cells in the presence of E2. This suggests that our E2-driven therapeutic approach could be potentially useful to treat that subset of patients with cervical cancer whose tumors continue to express E2 upon TK expression from the therapeutic construct and GCV treatment, leading to killing of HPV-infected cells.
As binding of the E2 protein to various E2BS on the LCR differentially regulates gene expression, we exploited this property and added a variable number of E2BS at a position adjacent to E2BS4. We obtained an increase in TK gene expression up to twofold compared with the native LCR, with the most consistent increases seen with the construct containing one additional E2BS (LCR1 TK). Although the relative increase with LCR1 compared with the native LCR was modest, the absolute increase in expression was large and the effect on cell death was substantial. Upon GCV treatment of these cells, we achieved an increase in cell death of up to 100% with constructs containing LCR1-TK compared with the native LCR-TK construct.
Different grades of CIN have been shown to express different levels of E2, and the level of E2 may vary within a given lesion (Sethi and Palefsky, 2003). We have studied the effect of variable amounts of E2 protein on gene expression from the various LCR-TK constructs (data not shown). The effect of E2 on transcriptional regulation depended on the level of the protein. Consistent with other studies (Schwarz et al., 1985; Broker et al., 1989; Stoler et al., 1992; Bouvard et al., 1994; Sethi and Palefsky, 2003), our experiments also show that the HPV16 E2 protein transactivated the native LCR at low levels and repressed transcription at progressively higher levels. For these reasons, we performed our experiments in various HPV-positive cell lines with varying HPV copy numbers and levels of E2 expression. Endogenous E2 gene expression levels as determined by quantitative real-time PCR and semiquantitative RT-PCR were found to be variable among the cell lines, with CaSki cells expressing higher amounts of E2 compared with 16-MT cells, and with higher levels of E2 expression, there was repression of TK expression. However, the CaSki cells showed higher cell killing than the 16-MT cells despite lower TK expression. Although the reasons for this are not clear, this may reflect different levels of translation or posttranscriptional modification of the TK gene or any other factors responsible for cell death within these cell lines. We did not perform experiments to directly demonstrate the transcriptional activity of the E2 protein expressed by the CaSki and other cell lines, and this will be the focus of future experiments. In addition, the optimal TK expression level required for maximum killing is not known. We speculate that cell death would be limited in cells expressing high levels of E2 because of repression of TK expression below the optimal level required for cell killing. In such cells, this therapeutic approach may not be much effective. However, as the levels of E2 expression found in the basal layers of CIN are generally thought to be low (Doorbar, 2005), targeting of basal cells infected with HPV may be possible using the suicide gene therapy approach described here, with the possibility of effecting a virologic “cure.”
The mechanism of cell death by the HSV-1 TK/GCV therapy was apoptosis. Apoptosis of 16-MT and CaSki cells may occur by p53-independent or p53-dependent mechanisms. In CaSki cells, p53 protein is low because of proteasomal degradation associated with binding to HPV E6 protein but it is still detectable (Ding et al., 2007). Our previous data with the MDCK and Vero cell lines, which have wild-type p53, showed that E2 expression was essential for the therapeutic strategy to work. There was no cell killing in the absence of E2, despite the transfection of the LCR-driven TK-expressing plasmid and GCV treatment (Sethi and Palefsky, 2003). Other groups have also demonstrated both apoptotic and necrotic cell death using the HSV-1 TK/GCV therapeutic approach (Todryk et al., 2000; Gough et al., 2001).
For HPV suicide gene therapy to be clinically useful, it must have a high therapeutic index and be nontoxic to HPV-negative cells. The HPV-negative HT-29, HT-3, and C33A cell lines did not show cell death above background when subjected to this therapy regimen. This could be due to lack of HSV-1 TK gene expression in the absence of E2 protein in these cells. Both the HPV-positive and HPV-negative cell lines had similar transfection efficiencies as determined by β-galactosidase gene expression. Thus, cell death in our system was E2 mediated and HPV specific. Besides the cervical carcinoma cell lines used, future experiments will include primary human keratinocytes that have wild-type p53 expression and in which the native HPV LCR is active.
Although we were able to show absence of cell death of HPV-negative cells using the TK suicide gene approach, killing of HPV-negative cells may occur through a different mechanism if they are in proximity to HPV-positive cells, that is, the “innocent bystander effect.” It has been observed that GCV triphosphate from TK-positive cells can induce cytotoxicity in neighboring TK-negative cells (Asklund and Almqvist, 2003; Fillat and Sangro, 2003). Toxic GCV triphosphate from HPV-infected cells may therefore diffuse to neighboring cells via gap junctions and cause cell killing. Thus, in our therapeutic approach, neighboring HPV-negative cells may also be killed because of the “bystander effect.” This would likely affect the cells adjacent to the lesion and could lead to death of some HPV-negative tissue. A “distant bystander effect” may also occur because of development of an immune response against the HPV-positive cells after HSV-1 TK transfection and GCV treatment. This can lead to induction of cytokines and heat shock proteins (hsp) including hsp70 (Todryk et al., 2000; Gough et al., 2001). However, the bystander effect, if limited, may also be advantageous. The immune response may augment clearance of HPV-infected cells, and if methods of gene delivery in vivo do not allow for high levels of transfection of basal cells of the epithelium, death of cells in the adjacent cell layer may also lead to basal cell killing with potentially beneficial effects on long-term viral clearance. However, our strategy requires DNA synthesis that is usually less active in suprabasal cells than in basal cells, but may still contribute to some additional basal cell killing. The innocent bystander effect may also produce a functional “surgical margin” around a lesion, reducing the risk of lesion recurrence after cessation of therapy.
The GCV doses used in our experiments were comparable to those used by other groups (Nishihara et al., 1998; Rubsam et al., 1998). The concentration used is also within the dose range achievable by oral administration of the oral version of GCV, valgancyclovir. GCV has been shown to induce multilog killing, which allows cells to complete one cell division after which they arrest permanently in the S-phase (Rubsam et al., 1998). Valgancyclovir could be orally administered to patients, along with topical application of the TK plasmids in a liposome-based cream. Presumably, women with CIN would apply the cream once or twice per day in a vaginal suppository for a period from several days to several weeks and would be given valgancyclovir during this period. A liposomal delivery-based gene therapy study for cystic fibrosis patients was successful when used to perform repeated applications to the nasal epithelium of patients (Hyde et al., 2000). As the cervical and anal lesions are mucosal, liposomal delivery of the therapeutic DNA may well be a feasible strategy in humans. Finally, as described earlier, functional E2 protein may be present in at least a subset of cervical cancers, and for women with those cancers, intratumoral injection of the TK construct may represent a useful adjunctive approach to standard chemotherapy and radiation therapy.
Footnotes
Acknowledgments
This work was supported by a grant from the American Cancer Society (SPRSG-03-242-01). The authors are grateful to Dr. E. Mocarski for providing the HSV-1 TK plasmid.
Author Disclosure Statement
No competing financial interests exist.