
Abstract
Ischemia–reperfusion (IR) injury is an important cause of primary graft failure in lung transplantation. In this study, viral interleukin-10 (vIL-10)-engineered mesenchymal stem cells (MSCs) were tested for their ability to prevent lung IR injury. Bone marrow-derived MSCs were transduced with rvIL-10-retrovirus. After 120 min of warm left lung ischemia, rats received ∼15 × 106 vIL-10-engineered MSCs (MSC-vIL-10), empty vector-engineered MSCs (MSC-vec), or saline intravenously. Mean blood oxygenation (PaO2/FiO2 ratio, mmHg) was measured at 4 hr, 24 hr, 72 hr, and 7 days. As early as 4 hr post-IR injury with MSC-vIL-10 treatment, blood oxygenation was significantly (p < 0.05) improved (319 ± 94; n = 7) compared with untreated (saline) controls (63 ± 19; n = 6). At 24 hr post-IR injury, in the MSC-vIL-10-treated group there was a further increase in blood oxygenation (353 ± 105; n = 10) compared with the MSC-vec group (138 ± 86; n = 9) and saline group (87 ± 39; n = 10). By 72 hr, oxygenation reached normal (475 ± 55; n = 9) in the MSC-vIL-10-treated group but not in the saline-treated and MSC-vec-treated groups. At 4 hr after IR injury, lungs with MSC-vIL10 treatment had a lower (p < 0.05) injury score (0.9 ± 0.4) compared with lungs of the untreated (saline) group (2.5 ± 1.4) or MSC-vec-treated group (2 ± 0.4). Lung microvascular permeability and wet-to-dry weight ratios were markedly lower in the MSC-vIL10 group compared with untreated (saline) controls. ISOL (in situ oligonucleotide ligation for DNA fragmentation detection) and caspase-3 staining demonstrated significantly (p < 0.05) fewer apoptotic cells in MSC-vIL10-treated lungs. Animals that received MSC-vIL10 therapy had fewer (p < 0.05) CD4+ and CD8+ T cells in bronchoalveolar lavage fluid compared with untreated control animals. A therapeutic strategy using vIL-10-engineered MSCs to prevent IR injury in lung transplantation seems promising.
Introduction
IR injury in the lung is thought to be due to, in large part, the collateral damage caused by an inflammatory response to an ischemic event. Previous studies have demonstrated that neutrophil infiltration into lung tissue can cause damage to the surrounding parenchyma in the early postoperative period (Haydock et al., 1992; Eppinger et al., 1997; Ross et al., 1999). In addition, IR injury after lung transplantation has been shown to be associated with the upregulation of inflammatory cytokines (Bando et al., 1995; Snell et al., 1996). Prevention of IR injury is critical to reduce the lung graft dysfunction and improve long-term survival among lung recipients.
The ability to transfer genes into stem cells has raised hopes of using gene therapy to provide long-term therapeutic impacts (Bianco and Robey, 2001; Vollweiler et al., 2003). The unique biology of stem cells with their vast clinical potential is emerging rapidly (Rezai et al., 2004). The bone marrow (BM) is often used as a source of stem cells for gene therapy approaches. Both hematopoietic stem cells (HSCs) and mesenchymal stem cells (MSCs) can undergo self-renewal and maintain their stem cell phenotype. There is increasing interest in the use of MSCs in both cell-based and gene-based therapies (Jorgensen et al., 2003; Prockop, 2003; Hamada et al., 2005). MSCs overexpressing interleukin (IL)-10 have been shown to attenuate collagen-induced arthritis in mice (Choi et al., 2008).
MSCs as a vehicle to deliver genes are ideal because they are self-renewable, easily expandable ex vivo, and can engraft well. The mechanisms that allow MSCs to escape from host allogeneic responses are particularly interesting. MSCs express MHC class I antigens, but lack MHC class II, and costimulatory molecules (CD40, CD80, and CD86); this suggests that they can stimulate alloreactive T cells but cannot engage in secondary signaling as they lack costimulatory molecules. In the absence of costimulation T cells become anergic and are unable to reject the allograft. MSCs also appear to modulate host dendritic cell and T cell function, by promoting induction of suppressor or regulatory T cells. These effects are complemented by the production of soluble immunomodulatory factors, including IL-10, transforming growth factor-β (TGF-β), prostaglandin E2 (PGE2), and hepatocyte growth factor (HGF) (Togel et al., 2007; Caplan, 2008; Patel et al., 2008). In addition, MSCs express the enzyme indoleamine 2,3-dioxygenase (IDO), which creates a tryptophan-depleted milieu, which in turn promotes immunosuppression via activated T cell apoptosis (Plumas et al., 2005; Stagg, 2007). MSCs can be potentially used to induce immunosuppression and reduce inflammation in a transplant setting.
We demonstrated that delivery of viral interleukin-10 (vIL-10) via HSCs significantly prolonged cardiac allograft survival among mice (Salgar et al., 2004). A therapeutic effect of viral and cellular IL-10 gene transfer has been shown in experimental transplantation (Qin et al., 1996; David et al., 2000), and in autoimmune disease, such as experimental arthritis (Apparailly et al., 1998; Ma et al., 1998), and diabetes in nonobese diabetic (NOD) mice (Pennline et al., 1994; Zhang et al., 2003). Viral IL-10, which is highly homologous to mouse and human IL-10, is encoded by the Epstein–Barr virus (EBV) gene BCRF1 (Hsu et al., 1990; Moore et al., 1990; Vieira et al., 1991). Viral IL-10 impairs the function of antigen-presenting cells (APCs) by downregulating the expression of MHC class II, costimulatory molecules, and adhesion molecules (intercellular adhesion molecule-1). Similar to human and murine IL-10, vIL-10 has the ability to inhibit cytokine synthesis (de Waal Malefyt et al., 1991). It has been shown that vIL-10 can downregulate the expression of helper T cell type 1 (Th1) cytokines, particularly interferon (IFN)-γ and IL-2 (Fiorentino et al., 1991a,b; Taga and Tosato, 1992). These aforementioned properties constitute important components of the antiinflammatory and immunosuppressive effects of IL-10. Unlike mammalian IL-10, vIL-10 does not exhibit stimulatory effects on thymocytes, B cells, or mast cells (Hsu et al., 1990; de Waal Malefyt et al., 1991; Thompson-Snipes et al., 1991). This property makes vIL-10 gene therapy a better therapeutic strategy than cellular IL-10. In the present study vIL-10-engineered MSCs were used with an expectation to harness their immunosuppressive, antiinflammatory, and regenerative properties to prevent lung IR injury. The results demonstrate significant improvement in lung function after MSC-vIL10 therapy in a rat lung IR injury model.
Materials and Methods
Recombinant vIL-10 vector construction
The recombinant vIL-10 vector was constructed as described previously (Salgar et al., 2004). Briefly, vIL-10 cDNA (pcDSRa-BCRF; Hsu et al., 1990) was digested with EcoRI enzyme and the vIL-10 coding sequence (0.5 kb) was isolated and cloned into a retroviral expression vector, pMSCVneo (Clontech Laboratories, Palo Alto, CA). The clone with an intact vIL-10 gene sequence in the correct orientation (5′ to 3′) was further expanded for large-scale vIL-10-vector preparation.
vIL-10 vector production
Mammalian cell transfection
Cell transfection was performed by the calcium phosphate method (CalPhos mammalian transfection kit; Clontech Laboratories) as previously described (Salgar et al., 2004). RetroPack PT67 (Clontech Laboratories), a fibroblast (NIH/3T3)-derived cell line designed for stable production of high-titer retrovirus, was used. Virus produced by RetroPack PT67 cells expresses a dual-tropic envelope, 10A1, that recognizes receptors on mouse, rat, human, hamster, mink, cat, dog, and monkey cells. A broad mammalian host range is caused by the fact that the virus can enter target cells via two surface molecules, the amphotropic retrovirus receptor RAM1 (Pit2) and the GALV (Pit1) receptor, such that if one receptor is not abundantly expressed by a given species or cell type, the alternative receptor may still allow viral entry (Miller and Miller, 1994; Miller, 1996). Briefly, PT67 packaging cells (1 × 106) were plated in 25-cm2 flasks 12–24 hr before transfection; when the cells became 50–80% confluent transfection was performed. One to 2 hr before transfection, 25 μM chloroquine replacement medium was exchanged to increase transfection efficiency. The calcium phosphate method (CalPhos mammalian transfection kit; Clontech Laboratories) was used for transfection as described previously (Salgar et al., 2004). The cells were washed twice with phosphate-buffered saline (PBS) to remove calcium phosphate and then fed with 5 ml of fresh complete growth medium and incubated at 37°C until needed for assay. Stable transformants were selected 24–72 hr posttransfection by growing in medium containing neomycin (Geneticin 418 [G418]) at 1 mg/ml for 7 days.
Viral titer determination
The viral titer was determined as described previously (Salgar et al., 2004). Briefly, NIH/3T3 cells (CRL-1658; American Type Culture Collection [ATCC], Manassas, VA) were plated in 6-well plates (105 cells per well). After 12–24 hr, 1-ml aliquots of 10-fold serial dilutions of the viral supernatant (collected from the virus-packaging PT67 cells) were added to each well. After 48 hr the cells were selected by culturing for 1 week in medium containing antibiotic G418 (1 mg/ml). Viral titers corresponded to the number of colonies present at the highest dilution (number of colonies × the dilution). The viral supernatants had a titer of 5 × 106 particles/ml and were used to transduce MSCs.
Animals
Eight- to 10-week-old inbred male Lewis (RT1l) rats, weighing ∼200 g, were purchased from Harlan Sprague Dawley (Indianapolis, IN). MSCs isolated from Lewis rats were engineered with vIL-10 vector or empty vector (control). Empty vector-engineered MSCs (MSC-vec), or vIL-10 vector-engineered MSCs (MSC-vIL10) were administered to Lewis rats intravenously soon after inducing left lung ischemia. All experiments were conducted as per National Institutes of Health (Bethesda, MD) and institutional guidelines with the approval of the University of Miami (Miami, FL) Animal Care and Use Committee.
Mesenchymal stem cell isolation, expansion, and characterization
Rats were killed by cervical dislocation under general anesthesia. Bone marrow cells (BMCs) were isolated from long bones (femur and tibia) and suspended at 10 × 106 cells/ml in complete medium (Iscove's Dulbecco's medium containing 10% fetal bovine serum, 2 mM
MSC engineering
MSC engineering with vIL-10 was performed similar to the protocol described previously for hematopoietic stem cells (Salgar et al., 2004). Briefly, MSCs isolated from rat bone marrow and expanded ex vivo (passages 3 to 6) were plated (106 cells per 25-cm2 plate) and cultured in complete medium. After 24–48 hr of culture, 75% of the medium was replaced by adding viral supernatants derived from vIL-10 vector- or vector DNA-engineered PT67 cell cultures. Infection was allowed for 6 hr in the presence of Polybrene (8 μg/ml of medium) and the supernatant was removed and replaced with fresh complete medium. The transduction was repeated for the second time on the next day in the same manner. After transduction (48–72 hr), the supernatants were assayed for vIL-10 production by enzyme-linked immunosorbent assay (ELISA) to confirm the transduction of MSCs. A human IL-10 detection kit (555157; BD Biosciences), which recognizes vIL-10, was used as per the manufacturer's recommendations. Stable transfectants were selected by culturing transduced cells in G418 (1 mg/ml) for 7 days. MSCs that produced vIL-10 were then injected into animals.
Experimental design
The following experimental groups were included in this study: group A received syngeneic MSC-vIL10, group B received empty vector-MSCs, group C received saline (no treatment control), and group D received nothing (sham control). Groups A through C underwent 120 min of left lung ischemia before MSC (15 × 106 cells per animal) or saline administration. No ischemia was introduced in group D. Left lung ischemia was induced by clamping the left pulmonary artery/vein/bronchus at end inspiration as described later. At 4 hr, 24 hr, 72 hr, and 7 days after ischemia–reperfusion animals were anesthetized and a midline incision from the pubis to the neck was created to gain access to the heart–lung block. Blood from the left and right pulmonary veins was collected for blood gas analysis. Subsequently, the animals were killed by exsanguination. The heart–lung block was rapidly excised, and the pulmonary circulation was flushed via the right ventricle with 20 ml of normal saline. The left lung was then separated, cut into three equal portions (cranial to caudal end), and stored in buffered formalin, liquid nitrogen (snap frozen), and O.C.T. compound (−80°C) for molecular, cellular, and histological analyses. Serum specimens were stored at −80°C for further analyses. Functional assessments of lungs were done by analyzing blood gases (arterial oxygen pressure [PaO2]/fraction of inspired oxygen [FiO2], mmHg) with a Stat Profile pHOx Plus L autoanalyzer (Nova Biomedical, Waltham, MA).
MSC transplantation
Ex vivo-expanded MSCs (long-term passages, >25) engineered with vIL-10 vector (MSC-vIL-10) or empty vector (MSC-empty vector) were intravenously injected via the dorsal penile vein into rats (∼15 × 106 cells per animal). Harvested MSCs were first filtered through a 40-μm (pore size) filter to obtain single-cell suspensions and kept cold in calcium- and magnesium-free PBS (to prevent cell aggregation) until injected. We occasionally observed the occurrence of pulmonary emboli leading to animal death (1 of 25 animals) with the intravenous MSC injections. vIL-10-MSCs was confirmed for vIL-10 production before injection by analyzing cell supernatants by ELISA. Also, negative controls (MSC-empty vector) were confirmed for the absence of vIL-10 production before injection.
Lung ischemia–reperfusion injury
Male Lewis rats (body weight, 200–250 g) were anesthetized with inhalant isoflurane (Baxter, Deerfield, IL) in a drop jar. After intramuscular injection of 0.04 mg of atropine (American Reagent, Shirley, NY) the animals were endotracheally intubated with a 14-gauge angiocatheter and placed on a rodent ventilator (Harvard Apparatus, Holliston, MA) with a standardized inspired oxygen flow of 0.2 liter/min, 75 to 85 breaths/min, and a positive end-expiratory pressure of 2 cmH2O. Anesthesia was maintained with isoflurane (IsoVet; Abbot Laboratories, Abbot Park, IL), using a multistation anesthetic vaporizer at a concentration of 1 to 2%. Animals were positioned on their right side on a warming pad to maintain body temperature between 37 and 38°C. Temperature was monitored continuously with a rectal probe. Blood pressure was measured with the XBP1000 noninvasive tail blood pressure system (Kent Scientific, Torrington, CT) after induction of anesthesia and continuously throughout the experiment. A left posterior lateral thoracotomy through the fifth intercostal space was then performed. The left lung was mobilized atraumatically, and 50 units of heparin (dissolved in normal saline solution to obtain a total volume of 0.5 ml) was administered via the dorsal penile vein. Five minutes after heparin administration, the left pulmonary artery, left pulmonary vein, and left mainstem bronchus were clamped with a noncrushing microvascular clamp at end inspiration. The lungs were covered with moist gauze, and kept moist by periodic application of warm (37°C) saline solution. The left lung was kept ischemic for 120 min. After 120 min of warm ischemia (van der Kaaij et al., 2005) the microvascular clamp was removed, the lung was ventilated and reperfused, and the thoracotomy wound was closed.
Lung vascular permeability
Lung vascular permeability was measured by measuring the lung wet-to-dry weight ratio and Evans blue dye extravasation.
Lung wet-to-dry weight ratio
The lung wet-to-dry (W/D) ratio is a reflection of lung edema that is increased during lung injury. The left lungs were harvested, the specimens were weighed (wet weight) before drying at 50°C for 72 hr, and then reweighed (dry weight) to calculate the wet-to-dry ratio (Kozower et al., 2002).
Evans blue dye extravasation
Pulmonary microvascular permeability was estimated as described previously (Kabay et al., 2007). Briefly, Evans blue dye (30 mg/kg) was injected into the tail vein just before reperfusion of the lung after 120 min of ischemia. At 4 or 24 hr after the injection of Evans blue dye animals were anesthetized and perfused with saline to remove extra dye that was lodged in the vasculature, and then the left lung was harvested. A small portion of the lung was homogenized and the Evans blue dye concentration in the lung (mg/g of tissue) was measured spectrophotometrically (620 nm) by comparison with the standard curve.
Myeloperoxidase activity
Quantitative myeloperoxidase activity was measured on the basis of the chlorination reaction and peroxidation reaction (Mohanty et al., 1997; Zhou et al., 1997) as described in the myeloperoxidase (MPO) chlorination/peroxidation detection kit (FLAPF100-2; Cell Technology, Mountain View, CA).
Chlorination assay protocol
Briefly, 50 μl of sample or standards (for standard curve) and 50 μl of chlorination reaction cocktail were added to black clear-bottom 96-well plates and incubated at room temperature in the dark for 15–30 min. The fluorescence was read with a fluorescence plate reader (excitation, 499 nm; emission, 515–530 nm).
Peroxidation assay protocol
The Fluoro MPO detection kit (Cell Technology) uses a nonfluorescent detection reagent, which is oxidized in the presence of hydrogen peroxide and MPO to produce its fluorescent analog. Briefly, 50 μl of sample (or standard) was mixed with 50 μl of reaction cocktail and incubated at room temperature in the dark for 30–60 min. Fluorescence was measured by excitation at 530–570 nm and emission at 590–600 nm.
Reverse transcription-polymerase chain reaction to detect vIL-10 mRNA
Total RNA isolated from lung tissue was used for RT-PCR amplification of vIL-10 mRNA. Using sequence-specific primers, vIL-10 transcripts (370- and 460-bp amplicons) were amplified. The following primer pairs were used: (1) 370 bp: forward primer (5′-GTGCTGCTTTACCTGGCACCTGAG-3′) and reverse primer (5′-TTATCTGTTCCACAGCTTTACTCT-3′) (Invitrogen, Carlsbad, CA); (2) 460 bp: forward primer (5′-CGAAGGTTAGTGGTCACTCTGCAG-3′) and reverse primer (5′-TGTCAAATTCACTCATGGCTTTGT-3′) (Invitrogen); and (3) 207-bp rat β-actin (a housekeeping gene) amplification served as an internal control, using the PPRO6570A-200 RT2 PCR primer set obtained from SABiosciences (Frederick, MD).
Histopathology
Histopathological examination was performed on 4-μm paraffin-embedded sections obtained from formalin-fixed lung tissues. Hematoxylin- and eosin-stained sections were used for the determination of ischemia–reperfusion injury score, based on previously established criteria (Kabay et al., 2007). Acute lung injury was scored on the basis of a five-point scale according to combined assessments of alveolar congestion, hemorrhage, edema, and infiltration of inflammatory cells in the airspace or vessel wall. A 0–4 severity of scoring system was used: 0, minimal damage; 1+, mild damage; 2+, moderate damage; 3+, severe damage; 4+, maximal damage. A single pathologist conducted all histological assessments in a blinded manner.
Bronchoalveolar lavage cell counts
Bronchoalveolar lavage was performed twice at the end of the experiment, using 3 ml of normal saline infused into the left mainstem bronchus while clamping the right hilum. The recovered fluid was then centrifuged at 1000 × g for 10 min at 4°C. The cells were harvested and stained with various fluorescent cell-specific (macrophage, granulocyte, CD4, CD8, and CD25) antibodies and percent cell fluorescence and mean channel fluorescence were recorded by flow cytometry.
Cellular apoptosis in the lung
Cellular apoptosis in the lung tissue was analyzed by detecting activated caspase-3 by immunohistochemistry (IHC), using a SignalStain cleaved caspase-3 (Asp175) IHC detection kit (Cell Signaling Technology, Danvers, MA) and the procedures were carried out as per the manufacturer's directions. Briefly, thin (4-μm) formalin-fixed paraffin-embedded sections were mounted on slides. The slides were deparaffinized and rehydrated, using a standard combination of xylene followed by decreasing concentrations of ethanol baths as recommended by the manufacturer. Antigen unmasking was achieved by incubating the samples in subboiling 0.01 M sodium citrate buffer (pH 6.0) for 10 min. After cooling, the samples were quenched with peroxidase, washed with water, and blocked for 1 hr at room temperature. Tissue sections were then incubated with primary antibody or negative control solution at 4°C overnight. After washing, the biotinylated secondary antibody was applied for 30 min. The slides were washed, reagent A-B was applied for 30 min, and they were washed again. The substrate chromogen (NovaRED; Vector Laboratories, Burlingame, CA) was applied and washed away once the samples began to turn reddish-brown (∼5 min). Hematoxylin counterstain was applied, the slides were then dehydrated, and a permanent mounting medium and coverslip were applied before performing light microscopy examinations.
Apoptosis in the lung tissue was also analyzed with an ApopTag peroxidase in situ oligonucleotide (oligo) ligation (ISOL) apoptosis detection kit (S7200; Chemicon/Millipore, Billerica, MA) as per the manufacturer's directions. The ApopTag ISOL kit detects DNA fragmentation by end-labeling the DNA, taking advantage of the prevalence of blunt ends and 3′ single-base overhangs characteristic only of apoptotic cells. This allows clear differentiation of cells undergoing apoptosis from cells undergoing necrosis. Briefly, formalin-fixed paraffin-embedded tissue sections (4 μm) mounted on slides in triplicate were deparaffinized and rehydrated, using a standard combination of xylene followed by decreasing concentrations of ethanol baths. Antigen unmasking was achieved by quenching in 3% hydrogen peroxide for 5 min followed by a rinse and then incubation with proteinase K for 15 min at room temperature. After three rinses the samples were placed in 1× equilibration buffer for 10 sec and the excess liquid was removed followed by humidified incubation with biotin-labeled oligo A (3′ overhang) or oligo B (blunt end), which will bind to an exact complementary, double-stranded DNA end in a reaction catalyzed by DNA ligase at 22°C for 16 hr. The samples were washed with PBS and deionized water. Streptavidin–peroxidase conjugate (which binds tightly to the biotin on the oligo) was added to each sample and allowed to incubate in a humidified chamber for 30 min at room temperature. After rinsing, diaminobenzidine (DAB), a chromogenic peroxidase substrate (which produces a brown precipitate), was added to each slide for 7 min at room temperature. The slides were rinsed in deionized water, counterstained with methyl green, and decolorized in an acetic acid bath. Last, the slides were dehydrated and permanent mounting medium and coverslip was applied. The apoptotic cells (brownish) were identified and counted by high-power (×400) bright-field microscopy, using Image-Pro Plus software (Media Cybernetics, Bethesda, MD).
Statistical analysis
All values are described as means ± standard deviation (SD). Two-group comparison was performed by Student t test for independent samples. Multiple group statistical analysis was performed by one-way analysis of variance (ANOVA) followed by the Bonferroni test for multiple comparisons. Statistics were calculated with GraphPad Prism 5 (GraphPad Software, La Jolla, CA). p < 0.05 was considered statistically significant.
Results
Ex vivo vIL-10 production
Ex vivo-expanded MSCs were efficiently transduced with 75% rvIL-10 retrovirus supernatants and incubated for 6 hr (transduction was repeated at 24 hr). The transduced cells (MSC-vIL10) produced ∼4 ng of vIL-10 per 106 cells in 24-hr culture supernatants, as measured by ELISA. The engineered MSC survival in G418 medium, and expression of vIL-10, demonstrated successful gene transduction.
In vivo vIL-10 production
After MSC-vIL-10 administration, intralung vIL-10 mRNA message was detectable by RT-PCR analysis in the vIL-10-preconditioned animals (Fig. 1). As expected, no vIL-10 mRNA expression was detected in the control animals administered empty vector-MSCs or in reverse transcriptase-negative PCR controls (Fig. 1).

Viral interleukin-10 (vIL-10) mRNA message was detected by reverse transcription-polymerase chain reaction in the lung homogenates of warm lung ischemia–reperfusion injury-induced animals with MSC-vIL10 treatment 7 days postreperfusion. (
Lung function was significantly improved in MSC-vIL10-treated animals
As early as 4 hr after IR injury mean arterial blood oxygenation (PaO2/FiO2 [P:F] ratio, mmHg) was significantly (p < 0.05) higher in the MSC-vIL-10 group (319 ± 95; n = 7) compared with the untreated saline group (63 ± 19; n = 6) or MSC-empty vector control group (Fig. 2 and Table 1). At 24 hr post-IR injury the mean P:F ratio was higher (p < 0.05) in the MSC-vIL10 group (353 ± 105; n = 10) compared with the MSC-empty vector group (138 ± 85; n = 9) and saline group (88 ± 39; n = 10). By days 3 and 7 the P:F ratios approached normal in the MSC-vIL-10 group (475 ± 55; n = 10 and 434 ± 33; n = 9), but not in the MSC-empty vector (238 ± 165; n = 10 and 217 ± 158; n = 11) or saline (198 ± 141; n = 9; and 187 ± 149) control groups (Fig. 2 and Table 1). In sham controls, the base P:F ratio was 482 ± 24 mmHg (Fig. 2).
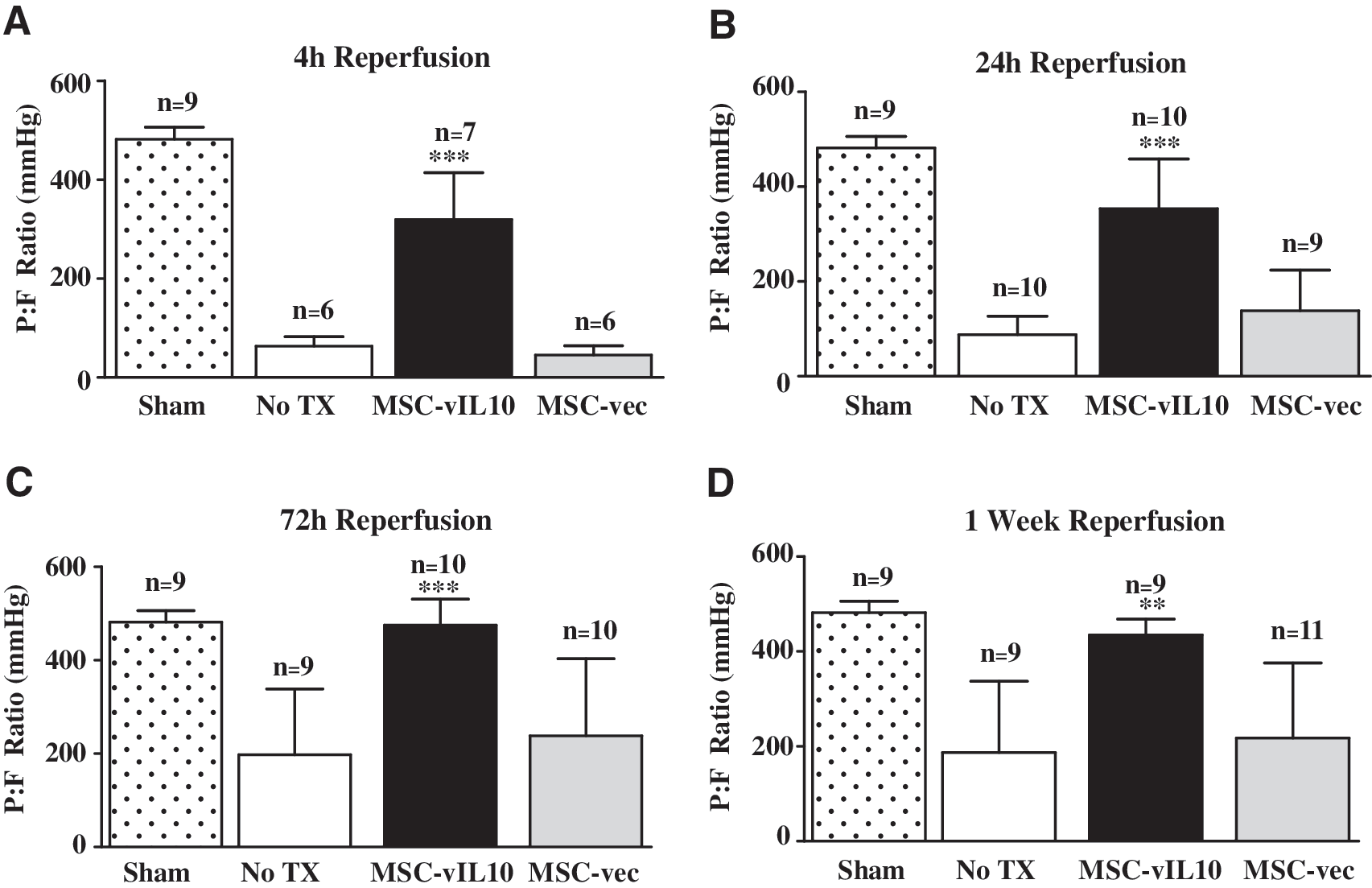
Lung function was monitored by blood gas analysis of samples obtained from the left pulmonary vein, and data are represented as the ratio of arterial oxygen pressure (PaO2) to the inspired fraction of oxygen (FiO2) (P:F, mmHg). (
Abbreviations: FiO2, fraction of inspired oxygen; MSC-vIL-10, viral interleukin 10-engineered mesenchymal stem cells; PaO2, arterial oxygen pressure.
Means ± SD; ratio of PaO2 to FiO2 (mmHg).
Means with common superscript (in rows) did not vary significantly (p < 0.05) between groups; n ≥ 6/group.
Lung wet-to-dry weight ratio was reduced in MSC-vIL10-treated animals
At 4 hr after reperfusion the wet-to-dry ratio of the lung was markedly lower in MSC-vIL10-treated animals (6.9 ± 0.3; n = 3) compared with untreated IR injury-induced controls (10.3 ± 1.2; n = 3). In sham (no IR injury) controls the wet-to-dry ratio was 5.7 ± 0.4 (n = 3) (Fig. 3A). However, at 24 and 72 hr there was no significant (p < 0.05) difference between the MSC-vIL10-treated and untreated groups.
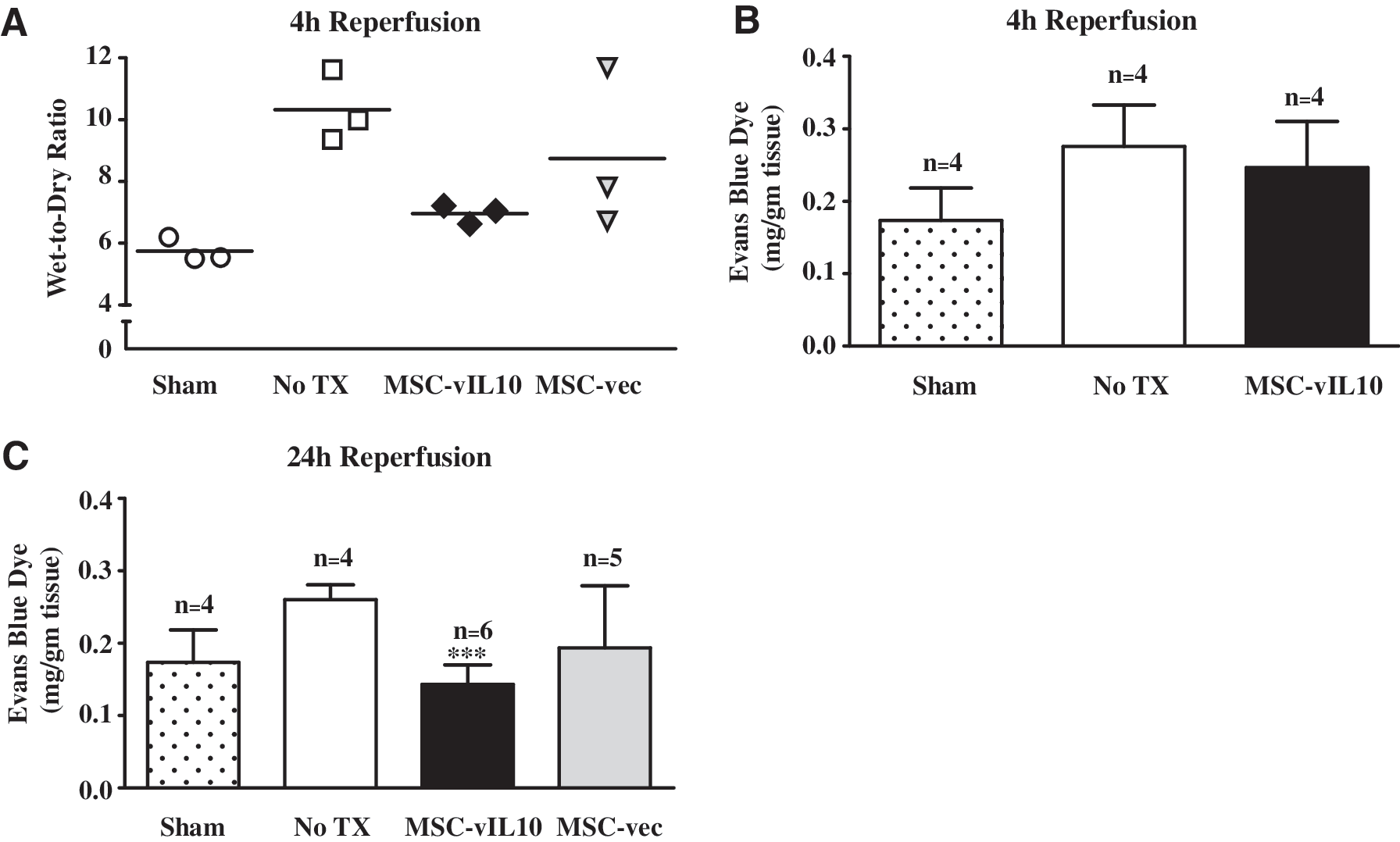
Lung edema was measured by lung wet-to-dry ratio at 4 hr reperfusion (
Lung microvascular permeability was lower in MSC-vIL10-treated animals
At 4 hr after reperfusion the lung microvascular permeability as determined by Evans blue dye extravasation was not sensitive enough to detect difference between the groups (Fig. 3B). Therefore, we chose to analyze this parameter at 24 hr after reperfusion. At 24 hr postischemia reperfusion the lung microvascular permeability was significantly (p < 0.01) lower in MSC-vIL10-treated animals (0.14 ± 0.04 mg of Evans blue/g of tissue; n = 6) compared with untreated controls (0.26 ± 0.02 mg/g of tissue; n = 4; Fig. 3C). In sham controls (no IR injury) it was 0.17 ± 0.04 mg/g of tissue (n = 4). However, at later time points (72 hr and 7 days) there was no significant (p < 0.05) difference between the groups.
Lung histopathology demonstrated reduced IR injury score in MSC-vIL10-treated animals
After ischemia–reperfusion, lungs were harvested at 4 hr, 24 hr, 72 hr and 7 days and analyzed for histopathology by examining hematoxylin and eosin (H&E)-stained sections to determine injury scores. The baseline mean injury score was significantly (p < 0.05) higher at 4 hr (2.0 ± 1.4; n = 5), compared with IR injury scores of lungs harvested at later time points, that is, 24 hr (1.1 ± 0.7; n = 5), 72 hr (0.5 ± 0.5; n = 5), and 7 days (0.8 ± 0.3; n = 4) (Fig. 4). In sham control animals with no IR injury the score was 0.2 ± 0 (n = 5). At 4 hr after IR injury the MSC-vIL10 group had a significantly (p < 0.05) lower injury score (0.9 ± 0.4; n = 7) compared with the no-treatment control group (2.5 ± 1.4; n = 6; Fig. 4). However, injury scores at 24 hr, 72 hr, and 7 days were not significantly (p < 0.05) different between the MSC-vIL10-treated and untreated groups.
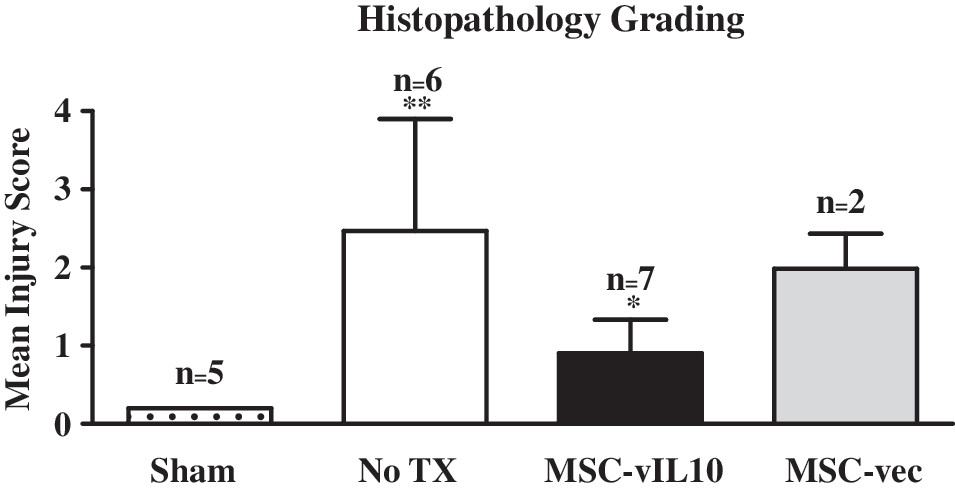
The ischemia–reperfusion (IR) injury score of lungs was measured on the basis of histopathology (cellular infiltration, edema, hemorrhage, and alveolar congestion) and graded on a scale of 0 to 4 at 4 hr reperfusion. The injury score was significantly (p < 0.01) higher in the MSC-vIL10-untreated group (No TX, no treatment but had IR injury) compared with normal (sham group with no IR injury) animals. In the MSC-vIL-10-treated group the lung injury score was significantly lower (p < 0.05) compared with the MSC-empty vector (MSC-vec)-treated and untreated (No TX) groups.
Bronchoalveolar lavage cell counts and characterization
At 4 hr reperfusion the total number of cells (Fig. 5a, panel A) and number of macrophages and granulocytes (Fig. 5a, panel B) in bronchoalveolar lavage showed no significant (p < 0.05) difference between the MSC-vIL10-treated and untreated groups. Therefore, we chose to analyze this parameter at 24 hr reperfusion. At 24 hr reperfusion the total bronchoalveolar lavage cell count was significantly (p < 0.05) lower in the MSC-vIL10-treated group (7.9 ± 4.9 × 106; n = 6) compared with untreated (18.0 ± 4.9 × 106; n = 6) controls; in the sham control group (no IR injury) it was the least, 2.0 ± 1.0 × 106 (n = 4) (Fig. 5b, panel A). At 24 hr after reperfusion the total number of granulocytes was slightly but not significantly lower in the MSC-vIL10-treated group (2.7 ± 1.5 × 106; n = 4) compared with untreated (3.4 ± 3.1 × 106; n = 4) or MSC-empty vector-treated controls (5.5 ± 2.3 × 106; n = 4) (Fig. 5b, panel B). Total lung macrophages (ED1/CD68 staining) did not differ between MSC-vIL-10-treated and untreated control groups. Bronchoalveolar lavage at 24 hr reperfusion demonstrated a significantly (p < 0.05) lower number (at least 2-fold) of CD4+ and CD8+ T cells in the MSC-vIL10-treated group compared with untreated controls (Fig. 5b, panel C). There was no significant (p < 0.05) difference in CD4+CD25+ or CD8+CD25+ cells between the MSC-vIL-10-treated and untreated groups.
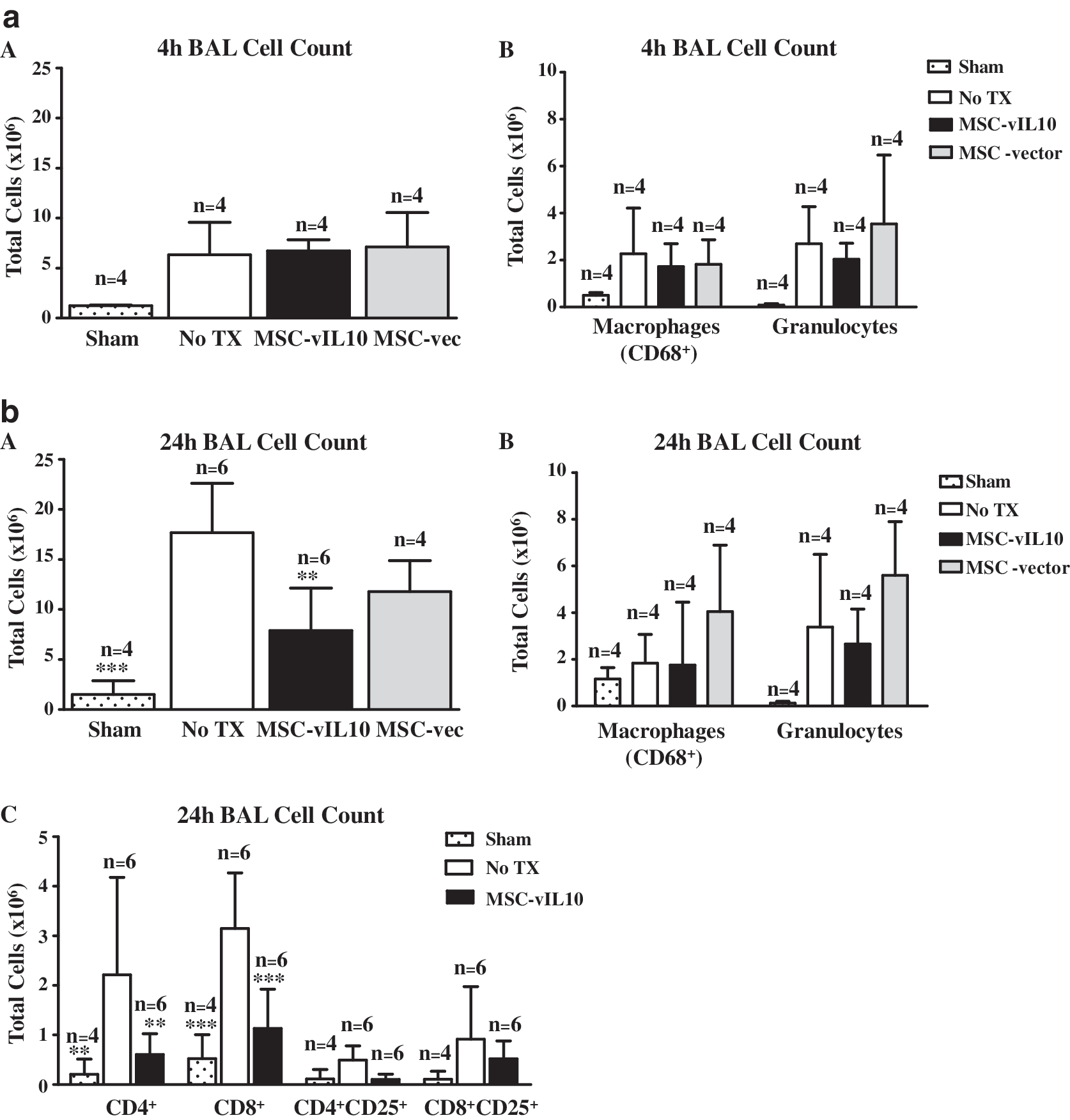
Bronchoalveolar lavage (BAL) was collected from lungs and analyzed for total cell number and cell types by flow cytometry. Data on cellular components of BAL in various groups of animals (MSC-vIL-10 treated [MSC-vIL10], MSC-empty vector treated [MSC-vec], no treatment [No TX], or sham group [no ischemia–reperfusion injury]) at 4 hr (Fig. 5a) and 24 hr (Fig. 5b) reperfusion are presented. (
Myeloperoxidase levels in the lung
Myeloperoxidase and chlorination assays were performed as a measure of infiltrating neutrophils in the lung. However, there was no significant (p < 0.05) difference in myeloperoxidase activity in the vIL-10-MSC-treated group compared with the untreated group at 4 or 24 hr reperfusion.
Cellular apoptosis in the lung
Apoptosis analysis by ISOL (in situ oligo-mediated ligation for DNA fragmentation detection) and activated caspase-3 staining showed a significant (p < 0.05) reduction in the number of apoptotic cells at 4 hr in MSC-vIL10-treated animals compared with untreated controls and MSC-empty vector-treated control animals (Table 2 and Fig. 6a). Quantitative measurements of caspase-3 and ISOL-positive cells are shown in Fig. 6b. Caspase-3-positive cells were significantly (p < 0.01) lower in MSC-vIL10-treated animals (16 ± 14/ × 400 field; n = 10 fields) compared with the untreated group (183 ± 83/ × 400 field; n = 10 fields) and the MSC-empty vector-treated group (70 ± 41/ × 400 field; n = 10 fields). A similar relationship was observed with ISOL staining, as well (Table 2). However, we did not perform the apoptotic assay on normal lungs (uninjured), as it has been demonstrated by several groups that apoptosis is negligible (<1%) in normal rat lungs (Fischer et al., 2000).
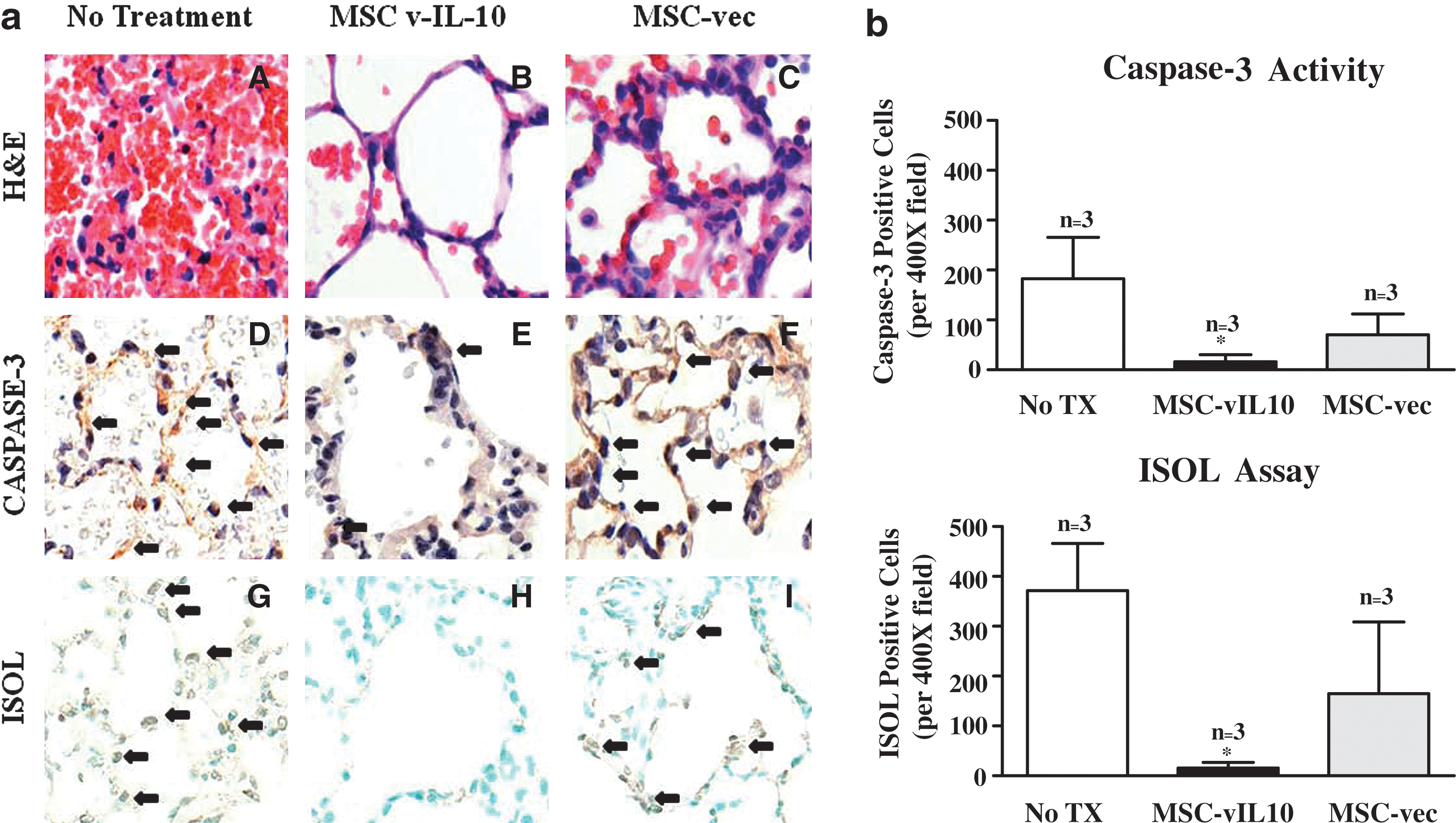
Lung tissue harvested from various groups of animals (MSC-vIL-10 treated [MSC-vIL10], MSC-empty vector treated [MSC-vec], or no treatment [No TX]) at 4 hr reperfusion was analyzed for ischemia–reperfusion injury and cellular apoptosis on the basis of histopathology (
Abbreviation: ISOL, in situ oligonucleotide ligation (for DNA fragmentation detection).
Means ± SD.
Means with at least one common superscript (in rows) did not vary significantly (p < 0.05) between the groups; n = 3/group.
Discussion
The present study demonstrates a novel approach to prevent IR injury by a combination of MSCs and gene therapy. We demonstrated that vIL-10 delivery via autologous MSCs significantly (p < 0.01) improved blood oxygenation (P/F ratio) in the rat lung IR injury model. The improvement in lung function was demonstrable as early as 4 hr after reperfusion (319 ± 19 mmHg), with further improvement that approached normal values by 72 hr (475 ± 55 mmHg) with MSC-vIL-10 treatment. In comparison, untreated (only saline administered) and MSC-empty vector-administered animals demonstrated significantly (p < 0.05) reduced blood oxygenation. Fischer and colleagues (2001, 2003) have demonstrated superior lung function after transfection with AdhIL-10 versus empty vector or vector diluent administration, with PaO2:FiO2 values of 240 ± 31 versus 98 ± 17 versus 120 ± 11 mmHg, respectively. AdhIL-10 gene transfection improved the function of transplanted lungs in rats in their study.
The literature presents several methods to evaluate the degree of pulmonary edema (Iglesias et al., 1998; Koike et al., 2000; Koksoy et al., 2001; Dowdall et al., 2002). In the current study we used the lung wet-to-dry ratio and Evans blue dye extravasation to assess changes in pulmonary microvascular permeability. Evans blue dye extravasation was significantly (p < 0.05) lower in MSC-vIL10-treated animals compared with untreated (saline) and empty vector-treated controls at 24 hr after reperfusion. However, at 4 hr after reperfusion the Evans blue dye extravasation method was not sensitive enough to detect differences between the groups studied. In MSC-vIL10-treated animals the wet-to-dry ratio 4 hr after reperfusion was markedly lower compared with untreated (saline) and empty vector-treated controls. This suggested that the reduced lung edema was possibly due to MSC-vIL-10 treatment.
These observations are consistent with previous reports in which human IL-10 (hIL-10) gene therapy attenuated lung injury caused by intestinal ischemia–reperfusion injury (Koksoy et al., 2001; Kabay et al., 2005). Kabay and colleagues (2005) injected plasmid DNA encoding hIL-10 or empty vector into mice intraperitoneally 24 hr before inducing mesenteric ischemia–reperfusion, and demonstrated that administration of hIL-10 (liposome-mediated gene delivery) attenuated dye extravasation and leukocyte sequestration, and reduced pulmonary tissue injury compared with the empty vector-injected controls. In the present study the detailed mechanisms by which vIL-10 attenuates the development of IR-related lung edema were not completely studied. The mechanism(s) by which vIL-10 attenuates microvascular permeability could be related to neutrophil–endothelial interaction and plugging, or could be due to inhibition of endothelial cell apoptosis and damage. Previous studies have shown that the increased appearance of activated neutrophils in the circulation exacerbated lung edema (Kuzu et al., 2002; Kabay et al., 2005). Moreover, the synchronous decrease in number of activated neutrophils and degree of pulmonary microvascular permeability in the hIL-10-treated (via gene therapy) group in an intestinal ischemia–reperfusion model (Kabay et al., 2005) also supports the idea that neutrophils have a major role in the development of pulmonary microvascular leakage. Koksoy and colleagues (2000) have demonstrated endothelial and smooth muscle dysfunction, pulmonary vasoconstriction, and a damaged microvascular barrier contributing to the pulmonary edema after intestinal IR injury. However, the effects of MSC-vIL-10 in the context of lung injury and edema warrant further studies.
Lung IR injury is promoted by a rapid accumulation of activated inflammatory cells in the lung interstitium and airways, leading to graft tissue destruction. In the present study, the total number of cells in the bronchoalveolar lavage of the vIL-10-treated group was markedly lower than in untreated controls. However, in the bronchoalveolar lavage samples the number of granulocytes in MSC-vIL-10-treated animals was lower, but not significantly different, when compared with untreated or empty vector-treated controls. Total ED1+ (CD68+) macrophages in the BAL of IR-injured lung, irrespective of vIL-10 treatment, was significantly (p < 0.01) higher compared with sham-operated controls. However, there was no difference in total macrophages between the MSC-vIL-10-treated animals and the controls, suggesting that vIL-10 treatment did not alter macrophage infiltration in our model system.
In a mouse model of lung injury caused by intestinal IR injury, Kabay and colleagues demonstrated that hIL-10 overexpression via gene therapy reduced leukocyte sequestration in the lung tissue. Other treatments such as sphingosine 1-phosphate (Okazaki et al., 2007) and adenosine A2A activation (Ellman et al., 2008) have been shown to reduce inflammatory cell (neutrophil) infiltration in the lung tissue and improve function after IR injury. In the present study the granulocyte number in BAL or myeloperoxidase activity in IR-injured lung tissue was not significantly altered between MSC-vIL10-treated and untreated controls, suggesting improved lung function with vIL-10 could be due to reduction in the number of other inflammatory cell types or mechanisms.
In the present study there was a significant reduction in the number of CD4+ and CD8+ cells in MSC-vIL-10-treated animals compared with untreated controls. Numerous studies involving a variety of organ systems have identified T cell trafficking into organs after IR injury. T cells have been demonstrated to have a direct role in mediating IR injury, and have been identified in the postischemic brain within 24 hr of reperfusion and localize to the stroke boundary zone near blood vessels (Schroeter et al., 1994; Jander et al., 1995; Linfert et al., 2009). Yilmaz and colleagues (2006) demonstrated that when Rag–/– mice, which are deficient in both T and B cells, were subjected to middle cerebral artery occlusion, they developed significantly smaller cerebral infarct size and less neurological deficit compared with wild-type control mice. Mice deficient for CD8+ T cells, CD4+ cells, and interferon-γ had a reduced volume of cerebral infarct compared with wild-type mice, as well. These findings demonstrated that T cells, and possibly B cells, play a vital role in brain IR injury. In a syngeneic rat lung transplant model, De Perrot and colleagues (2003b) showed that recipient CD4+ T cells infiltrated lung grafts within 1 hr of reperfusion and upregulated the expression of CD25 over the ensuing 12 hr. However, in the present study CD25 was not upregulated significantly on CD4+ or CD8+ T cells harvested from BAL, and this may be partially due to the fact that in the present study we used a warm IR injury model (Fig. 5b-C). Geudens and colleagues (2007) demonstrated the pathogenic role of T cells in a lung IR injury model in severe combined immune deficiency (SCID) mice. T lymphocytes have also been demonstrated to be pathogenic during myocardial IR injury by comparing RAG1–/– mice versus wild type (Yang et al., 2006). In the present study, there was a significant (p < 0.001) decrease in CD4+ and CD8+ T cells in the MSC-vIL-10-treated group compared with untreated groups. This was possibly one of the factors in reducing IR injury and improving the lung function with MSC-vIL-10 therapy.
vIL-10 delivery via MSC treatment successfully reduced the lung injury score as determined by histopathological grading of lungs when compared with untreated or empty-vector-treated control groups (Fig. 4) at 4 hr. This clearly suggested the protective role of MSC-vIL-10 treatment in IR injury. The finding observed in the present study is in agreement with hIL-10 treatment via gene therapy in the pulmonary damage induced by intestinal IR injury in rats (Kabay et al., 2005).
In this study we have demonstrated that there was vIL-10 production in the lung of MSC-vIL-10-treated animals. In addition, we could not detect the presence of vIL-10 in the peripheral blood. These data along with other reports (Devine et al., 2001; Jorgensen et al., 2003; Deng et al., 2004) showed that MSCs, when injected intravenously, home preferentially to lungs, especially injured lung. This suggests that the MSC-vIL-10 effect observed in the current study appears to be a local effect, and not a systemic one.
Ischemia has been shown to be a potential inducer of apoptosis (Majno and Joris, 1995). Because apoptotic cells in the lung are usually phagocytosed by macrophages before their membranes break down and intracellular enzymes are released, this mode of cell death does not lead to significant inflammation, which is characteristically prominent with necrosis (Majno and Joris, 1995). In human lungs it has been shown that apoptosis does not occur in grafts during cold or warm ischemia. However, in the early phase after graft reperfusion, the number of apoptotic cells increased to 34% (Fischer et al., 2000; Fischer and Keshavjee, 2001). As a consequence of this observation in lung transplantation, it would be essential to focus on regulatory mechanisms that are involved in this dramatic loss of cells in lung grafts and develop strategies to prevent cell death in the transplant setting. In the present study the most significant observation comes from the analysis of apoptotic cells in the lungs of both MSC-vIL-10-treated and untreated controls. There was a significant (p < 0.01) decrease in apoptotic cells in the vIL-10-MSC-treated animals compared with untreated controls. The majority of apoptotic cells were found in the alveolar septal region. The increased apoptotic cells in the untreated controls were possibly pneumocytes, as the blood oxygenation potential was significantly (p < 0.01) reduced compared with MSC-vIL10-treated (which prevented apoptosis) rats.
In the present study MSC-vec administration did not significantly improve lung function after IR injury when compared with MSC-vIL-10 administration. However, there was a distinct effect at the cellular/molecular level as observed by reduced histopathological injury score and reduced apoptotic cell numbers when compared with untreated control animals. Several groups have demonstrated immunosuppressive and antiinflammatory properties of bone marrow-derived mesenchymal stem cells (Tang et al., 2006; Stagg, 2007). The already known paracrine capabilities of mesenchymal stem cells, such as vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF) secretion (Neuss et al., 2004), and their ability to transdifferentiate into endothelial phenotype (Oswald et al., 2004) and lung epithelial cells (Kotton et al., 2001), hold particular promise in the treatment of ischemia–reperfusion injury of the lung. Mesenchymal stem cells have a self-renewable capacity and can easily engraft even in the allogeneic host (Allay et al., 1997; Harrington et al., 2002; Jorgensen et al., 2003); they are poorly immunogenic and suppress allogeneic or autologous T cell responses (Djouad et al., 2003; Tse et al., 2003; Plumas et al., 2005; Stagg, 2007); and they escape the immune system (recognition by allogeneic T cells) because of the absence/low expression of MHC class I or II, or T cell-costimulatory molecules B7-1, B7-2, CD40, and CD40L (Deans and Moseley, 2000; Jiang et al., 2002; Saito et al., 2002; Deng et al., 2004). MSCs have been shown to ameliorate tissue damages triggered by renal ischemia–reperfusion injury (Tang et al., 2006; Semedo et al., 2007). MSCs home preferentially to bone marrow, thymus, lung, cartilage, and injured sites (Devine et al., 2001; Jorgensen et al., 2003; Deng et al., 2004), and more so to the injured organs after intravenous injection. It is highly likely that exogenous MSC (unengineered) administration would exert some of these properties in the lung in vivo and minimize IR injury, and thus would be an excellent candidate for cell therapy in addition to IL-10-engineered MSCs to reduce lung IR injury. The paracrine, immunosuppressive, nonimmunogenic, transdifferentiation, gene transduceable, self-renewable, and homing properties of MSCs could be exploited to provide protection to the lung grafts from IR injury. Our future studies are focused on using unengineered MSCs for cell therapy to prevent IR injury and transplant rejection.
In summary, in this study we successfully delivered vIL-10 via MSCs to the ischemic lung. vIL-10-engineered MSCs had a significant (p < 0.05) impact in reducing IR injury as demonstrated by (1) reduced lung injury score (histopathology), (2) reduced pulmonary microvascular permeability, (3) reduced T lymphocytes in the bronchoalveolar lavage, (4) reduced apoptotic cells, and (5) increased lung function as measured by blood oxygenation. In conclusion, vIL-10 delivery via MSCs significantly reduced lung IR injury and improved lung function. vIL-10 and MSC therapy appears to be a promising strategy to reduce IR injury in lung transplantation to improve graft survival and prevent acute and chronic graft loss.
Our future studies are to use short-term and long-term cultured MSCs, either alone, or as a gene delivery vehicle, to protect against organ IR injury. Our preliminary data strongly support the protective effects of MSCs in lung IR injury. Further studies are warranted to delineate the effects of passage number, specific MSC phenotype involved, and dose and route of administration in reducing lung IR injury. In conclusion, vIL-10-engineered MSC therapy seems to be promising and attractive cell therapy approach to prevent lung transplant IR injury and graft dysfunction. We have begun to evaluate the effects of MSC and MSC-vIL-10 therapy in a lung transplant model and are hoping to move the field forward in order to mitigate IR injury in transplanted organs.
Footnotes
Acknowledgments
The authors thank Ms. Mireya Hernandez, Ms. Melissa St. Pierre, and Ms. Dania Mateu for technical assistance and Ms. Felisa Flores for administrative support in this study. Supported in part by the Department of Surgery and by the Transplant Foundation (Miami, FL).
Author Disclosure Statement
For all authors of this report, no competing financial interests exist.