
Abstract
Artemis is a hairpin-opening endonuclease involved in nonhomologous end-joining and V(D)J recombination. Deficiency of Artemis results in radiation-sensitive severe combined immunodeficiency (SCID) characterized by complete absence of T and B cells due to an arrest at the receptor recombination stage. We have generated several lentiviral vectors for transduction of the Artemis sequence, intending to complement the deficient phenotype. We found that transduction by a lentiviral vector in which Artemis is regulated by a strong EF-1α promoter resulted in a dose-dependent loss of cell viability due to perturbed cell cycle distribution, increased DNA damage, and increased apoptotic cell frequency. This toxic response was not observed in cultures exposed to identical amounts of control vector. Loss of cell viability was also observed in cells transfected with an Artemis expression construct, indicating that toxicity is independent of lentiviral transduction. Reduced toxicity was observed when cells were transduced with a moderate-strength phosphoglycerate kinase promoter to regulate Artemis expression. These results present a novel challenge in the establishment of conditions that support Artemis expression at levels that are nontoxic yet sufficient to correct the T−B− phenotype, crucial for preclinical studies and clinical application of Artemis gene transfer in the treatment of human SCID-A.
Introduction
NHEJ also plays a vital role in the rearrangement of immunoglobulin (Ig) genes and T cell receptor (TCR) genes (van Gent et al., 1996; Zhu et al., 1996). This site-specific rearrangement process begins when the RAG complex, composed of RAG-1 and RAG-2, is recruited to recombination signal sequences (RSS) flanking each V (variable), D (diversity), or J (joining) coding segment (Oettinger et al., 1990). The RAG complex introduces a nick adjacent to each RSS; the resulting 3′ hydroxyl group undergoes nucleophilic attack on the antiparallel DNA strand to form a hairpin structure at the coding ends (Roth et al., 1992; McBlane et al., 1995). The Artemis:DNA–PK complex then endonucleolytically cleaves the coding end hairpin and the DSB is processed and repaired through the NHEJ pathway (Ma et al., 2002).
Deficiency of Artemis interrupts Ig and TCR gene rearrangement, resulting in a radiosensitive B−T−NK+ severe combined immunodeficiency, designated SCID-A because of the founder mutation occurring in Athabascan-speaking Native Americans (Moshous et al., 2000, 2001; Li et al., 2002). Artemis deficiency can be treated by allogeneic hematopoietic cell transplantation (HCT); however, allotransplantation often results in incomplete reconstitution of B cell function and is also associated with complications such as graft failure and graft-versus-host disease (O'Marcaigh et al., 2001).
Genetic correction of autologous hematopoietic stem cells (HSCs) could provide an alternative therapeutic approach for SCID-A, thus avoiding the complications associated with allogeneic HCT. Two independent groups have reported correction of a SCID-A murine model, each using a lentiviral vector encoding human Artemis cDNA regulated by the phosphoglycerate kinase (PGK) promoter for transduction and transplantation of hematopoietic stem cells (Mostoslavsky et al., 2006; Benjelloun et al., 2008). SCID-A animals receiving HSCs transduced with PGK-regulated human Artemis displayed complete repopulation of both B and T cell compartments. However, Mostoslavsky and colleagues reported that RAG-1-deficient animals receiving SCID-A HSCs transduced with either cytomegalovirus (CMV)- or elongation factor (EF)-1α-regulated human Artemis lentiviral vectors were unable to repopulate B and T cells (Mostoslavsky et al., 2006; Benjelloun et al., 2008).
Our laboratory has also generated several lentiviral vectors expressing the human Artemis cDNA sequence, using various promoters, with the intention of complementing Artemis deficiency in vitro and in vivo. Upon characterization of these vectors, we found that transduction by a lentiviral vector in which Artemis is regulated by the strong EF-1α promoter resulted in a dose-dependent loss of cell viability that was not observed in cultures exposed to identical amounts of control vector. This toxic effect was reproduced in cultures transfected with the identical DNA construct, ruling out the possibility of toxicity associated with lentiviral transduction rather than Artemis expression. Mechanistically, Artemis overexpression was associated with an increase in DNA damage as determined by comet assay, G1 arrest of the cell cycle, and a relative increase in the proportion of apoptotic cells.
These results underscore the importance of regulating Artemis expression in transduced cell populations and present a novel challenge for effective correction of the B−T− SCID-A phenotype. Establishment of conditions that provide Artemis expression that is nontoxic and yet sufficient to correct the T−B− phenotype will be crucial for vector development in preclinical studies using Artemis-deficient mice and in clinical application to human SCID-A.
Materials and Methods
Lentiviral vectors
Lentiviral vector constructs were generated by standard molecular technique based on the pCSII and pCSIIEG lentiviral vectors, both of which have been previously described (Agarwal et al., 2006).
pCSIIEG/EPuro
The Streptomyces alboniger puromycin-N-acetyltransferase gene was amplified by polymerase chain reaction (PCR) from the pPUR plasmid (Clontech, Mountain View, CA), using the following oligonucleotides: forward, 5′-TCT
pCSII/EAIG
The murine Artemis cDNA sequence was excised from pGEMT/mArt (Li et al., 2005) with NotI and cloned into pCSII/EF1α-MCS (Agarwal et al., 2006) at the NotI site to generate pCSII/EF1α-mArtemis. The internal ribosome entry site (IRES)–green fluorescent protein (GFP) sequence was excised from pCSII/CMV-I2G (Gori et al., 2007) with NheI and XbaI and cloned into the XbaI site of pCSII/EF1α-mArtemis to generate pCSII/EAIG.
pLL-ABiG
The CLP (CpG-less promoter) regulatory element was excised from pCpG-free-mcs (InvivoGen, San Diego, CA;
pOK/EF1α-hArtemis
pOK/EF1α-MCS lentiviral vector was generated by restriction digest of pKT2/SE (Clark et al., 2007) with PvuII and SwaI to excise a minimal plasmid backbone containing the kanamycin resistance gene and the ColE1 origin of replication. pCSII/EF1α-MSC was digested with ScaI and PmeI, and then the long terminal repeat (LTR)-to-LTR fragment was ligated into the minimal plasmid backbone. The human Artemis cDNA sequence was excised from pCMV-SCIDA (Li et al., 2002), cloned into the BssHI restriction site of pSL301, excised with BamHI, and cloned into pOK/EF1α-MCS to generate pOK/EF1α-hArtemis.
pOK/PGK-hArtemis
The human PGK promoter sequence was amplified by PCR from pKT2/PGKi template (Clark et al., 2007) to include flanking XmaI restriction sites, using the following oligonucleotides: forward, 5′-AATATT
Mammalian cell culture
Murine embryonic fibroblasts (Artemis wild-type tMEFSCIDA+/+, heterozygous tMEFSCIDA+/−, or deficient tMEFSCIDA−/−; Li et al., 2005), HEK 293T, and murine NIH 3T3 tk− cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic–antimycotic at 37°C and 5% CO2.
Preparation and titering of lentiviral vectors
Vesicular stomatitis virus envelope glycoprotein (VSV-G)-pseudotyped vectors were generated by three-plasmid cotransfection of HEK 293T cells as previously described (Zufferey et al., 1997; Gori et al., 2007). Briefly, 24 hr pretransfection, 1.4 × 107 HEK 293T cells were seeded into poly-
Western blot analysis
tMEFSCIDA−/− cells were transduced with Artemis or control GFP vector at a multiplicity of infection (MOI) of 10. Forty-eight hours posttransduction, cells were sorted by FACSDiva (BD Biosciences, San Jose, CA) and GFP-positive cells were collected. Untransduced tMEFSCIDA+/+, tMEFSCIDA+/−, and tMEFSCIDA−/− cell cultures, and transduced tMEFSCIDA−− cell cultures, were subjected to nuclear extraction according to Schreiber and colleagues (1989). Briefly, cells were harvested by trypsinization, washed with phosphate-buffered saline (PBS), and then resuspended in ice-cold lysis buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol [DTT], 0.5 mM phenylmethylsulfonyl fluoride [PMSF]). After a 15-min incubation on ice, 10% Nonidet P-40 was added, and the lysate was vortexed vigorously and then cleared by centrifugation for 1 min at 16,000 × g. The supernatant (containing the cytoplasm) was discarded, and then the nuclei were resuspended in a second lysis buffer (20 mM HEPES [pH 7.9], 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF) and vigorously rocked at 4°C for 20 min. The nuclear extract was cleared for 5 min by centrifugation at 16,000 × g and the protein concentration was determined by Bradford analysis as formulated by Bio-Rad (Hercules, CA). Nuclear lysates (25 μg) were boiled in the presence of 5 × sodium dodecyl sulfate (SDS) loading buffer (300 mM Tris [pH 6.8], 25% glycerol, 20% 2-mercaptoethanol, 10% SDS, 0.02% bromophenol blue) for 5 min, electrophoresed through a 10% Tris-HCl polyacrylamide–SDS gel, and transferred onto polyvinylidene difluoride (PVDF) membrane, using a Bio-Rad Trans-Blot SD semidry system for 25 min at 12 V. The membrane was washed in 1 × Tris-buffered saline (1 M Tris [pH 7], 5 M sodium chloride) plus 0.05% Tween 20 (TBST), blocked for 1 hr with 5% milk (Bio-Rad) in 1 × TBST, washed again with 1 × TBST, and then incubated overnight at 4°C with a rabbit polyclonal anti-Artemis antibody (BioLegend, San Diego, CA) diluted 1:500 in 2.5% milk–1 × TBST. After washing with 1 × TBST, the membrane was incubated for 1 hr at 4°C with a secondary peroxidase-conjugated anti-rabbit IgG (whole molecule) (Santa Cruz Biotechnology, Santa Cruz, CA), diluted 1:3000 in 2.5% milk–1 × TBST. The signal was visualized with a SuperSignal West Pico chemiluminescent substrate detection kit (Thermo Fisher Scientific).
Hairpin-opening assay
Whole cell lysates were generated from MEF cells by freeze–thawing four times and cleared by centrifugation at 25,000 × g for 10 min in an Eppendorf microcentrifuge.
Assays were conducted in a 96-well format. Each reaction consisted of 25 μg of whole cell lysate and 300 nM hairpin substrate brought to a final volume of 100 μl in reaction buffer (25 mM Tris [pH 8], 50 mM KCl, 10 mM MgCl2, 1 mM DTT, bovine serum albumin [BSA, 50 ng/μl], and 5 mM ATP). The hairpin substrate was generated as a 19-base oligodeoxynucleotide (5′-TTTCGAGCTCATGAGCTCG-3′) modified at the 5′ terminus by the fluorophore 6-carboxyfluorescein (FAM) and at the 3′ terminus by the quencher 6-carboxytetramethylrhodamine (TAMRA). At room temperature a double-stranded, stem–loop configuration is predicted for this sequence, with a melting temperature (T m) of 72°C; once cleaved, the resulting product acquires a T m of 20°C and dissociates at room temperature, allowing for the release of FAM from TAMRA with ensuing fluorescence at 494 nm. FAM fluorescence was measured in real time at room temperature with an Eppendorf Realplex thermocycler. Realplex software was used to compile fluorescence readings once per minute for each reaction. Initial velocities were calculated by linear least squares and expressed as the change in fluorescence per milligram of protein per minute. The amount of substrate cleaved, expressed as moles per fluorescence unit, was determined as the fraction of fluorescence assessed after complete degradation of the hairpin substrate by DNase. Initial velocities were then expressed as nanomoles of substrate cleaved per milligram of protein per minute.
Comet assay
tMEFSCIDA−/− cells were transduced with lentiviral vectors and 5 days later sorted for the GFP+ population by FACSDiva separation. The comet assay was performed under alkaline conditions as described by Olive and Banath (2006). Briefly, a single-cell suspension of each population was prepared and submerged in 1% low melting point agarose at a concentration of 5 × 103 cells/ml. Each cell suspension was smeared onto a microscope slide precoated with a thin layer of 1% low melting point agarose and allowed to gel for 5 min. Slides were then fully submerged into A1 alkaline lysis solution (1.2 M NaCl, 100 mM Na2EDTA, 0.1% sodium lauryl sarcosinate, 0.26 M NaOH [pH >13]) overnight at 4°C in the dark. After lysis, slides were incubated with A2 solution (0.03 M NaOH, 2 mM Na2EDTA [pH ∼12.3]) at room temperature three times for 20 min each time. Slides were then placed in an electrophoresis chamber and fully submerged in fresh A2 solution. Electrophoresis was conducted for 25 min at 20 V. Slides were removed from the electrophoresis chamber, neutralized by washing with distilled water, and incubated in staining solution (propidium iodide, 2.5 μg/ml in distilled water) for 20 min. Fluorescent comet images were collected for each population with an Axioplan 2 imaging system (200-fold magnification; Carl Zeiss, Oberkochen, Germany). Images were analyzed with CometScore 1.5 software (
Cell cycle analysis
3T3 cells transduced with EF1α-hArtemis or control vector EF1α-Puromycin were harvested 48 hr posttransduction and stained overnight with propidium iodide as previously described (Nicoletti et al., 1991). Briefly, 0.5 × 106 transduced cells were harvested by trypsinization and pelleted at 377 × g. Cells were resuspended in hypotonic fluorochrome solution (propidium iodide [50 μg/ml], 0.1% sodium citrate, 0.1% Triton X-100 in distilled water) and incubated overnight at 4°C. Cell cycle distribution of transduced cell populations was determined by flow cytometry with a FACScalibur (BD Biosciences), with subsequent cell cycle analysis using FlowJo software (Tree Star, Ashland, OR).
Apoptosis
3T3 cells were transduced with CSIIEG and EAIG lentiviral vectors at increasing MOI. Twenty hours posttransduction, cells were harvested by trypsinization and subjected to Annexin V staining, in accordance with the Annexin V–biotin apoptosis detection kit protocol (BioVision, Mountain View, CA). Briefly, cells were resuspended in 1 × binding buffer and incubated with Annexin V–biotin at room temperature for 5 min. Cells were washed twice with binding buffer and then incubated with avidin–peridinin chlorophyll protein (PerCP) (BD Biosciences) at room temperature for 15 min. 7-Amino-actinomycin D (7-AAD) was added to samples immediately before flow cytometry. The GFP-positive transduced cell population was gated and analyzed for Annexin V (PerCP)- and 7-AAD-positive cells.
Statistical analysis
Data were statistically evaluated either by unpaired Student t test (hairpin-opening assays) or by analysis of variance (ANOVA) (all other analyses), using Prism 4 software (GraphPad Software, San Diego, CA), with p < 0.05 considered significant.
Results
Lentiviral transduction and expression of the Artemis gene
To establish effective conditions for restoring Artemis expression by gene transfer, lentiviral vectors containing the murine or human Artemis coding sequences were generated (Fig. 1). Initial vector constructs were also engineered for expression of green fluorescent protein (GFP) to facilitate tracking of transduced cells and quantitation of viral vector stocks by flow cytometry. Lentiviral vectors were packaged by transfection in human 293T cells as described in Materials and Methods. Transduction of mouse NIH 3T3 cells was verified 48 hr after exposure to vector by flow cytometry for GFP expression and by real-time quantitative PCR. Expression of Artemis protein was verified by Western blot analysis (Fig. 2).
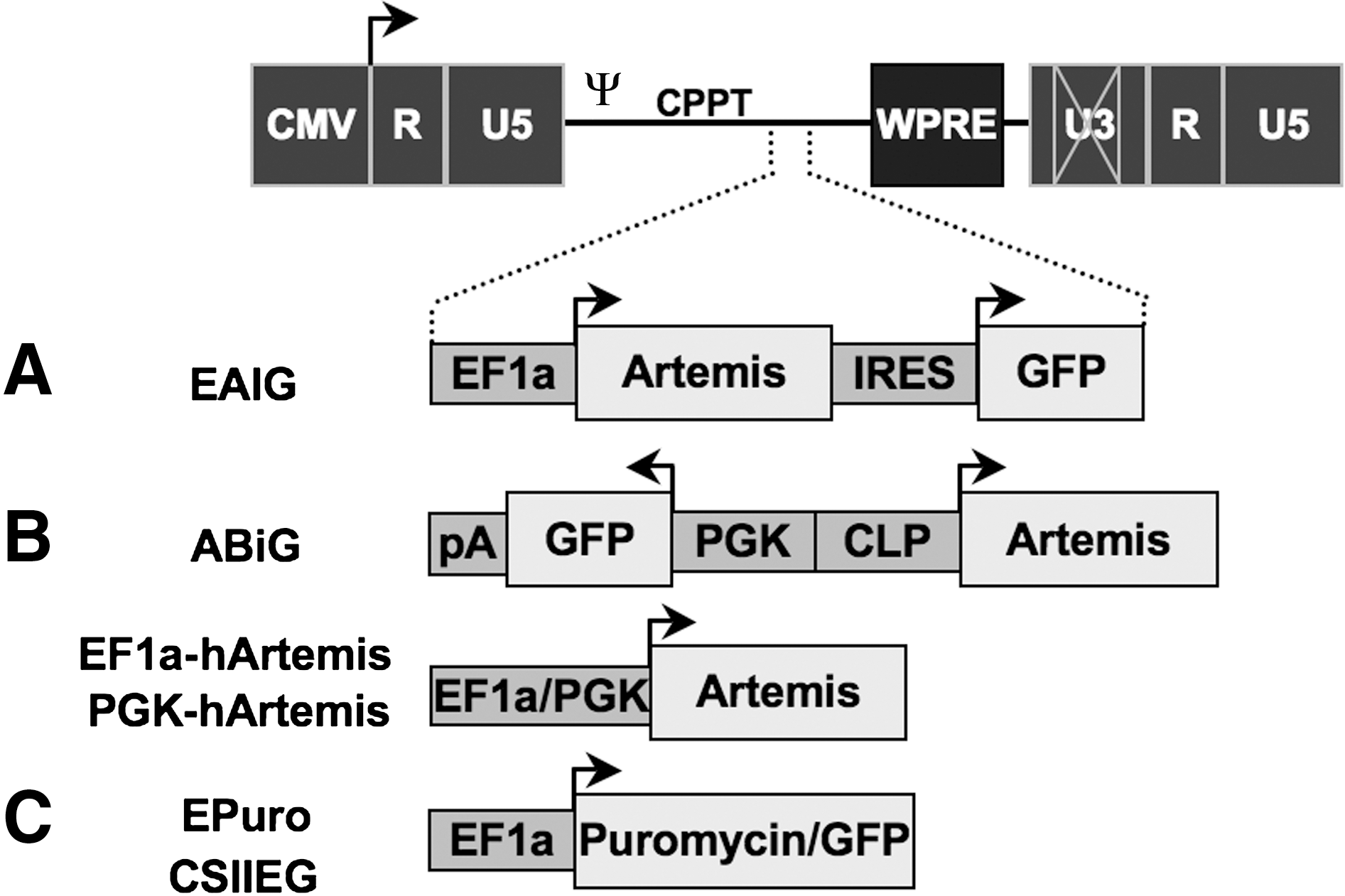
Lentiviral vector constructs. Several lentiviral vectors were constructed for analysis and complementation of Artemis deficiency. (
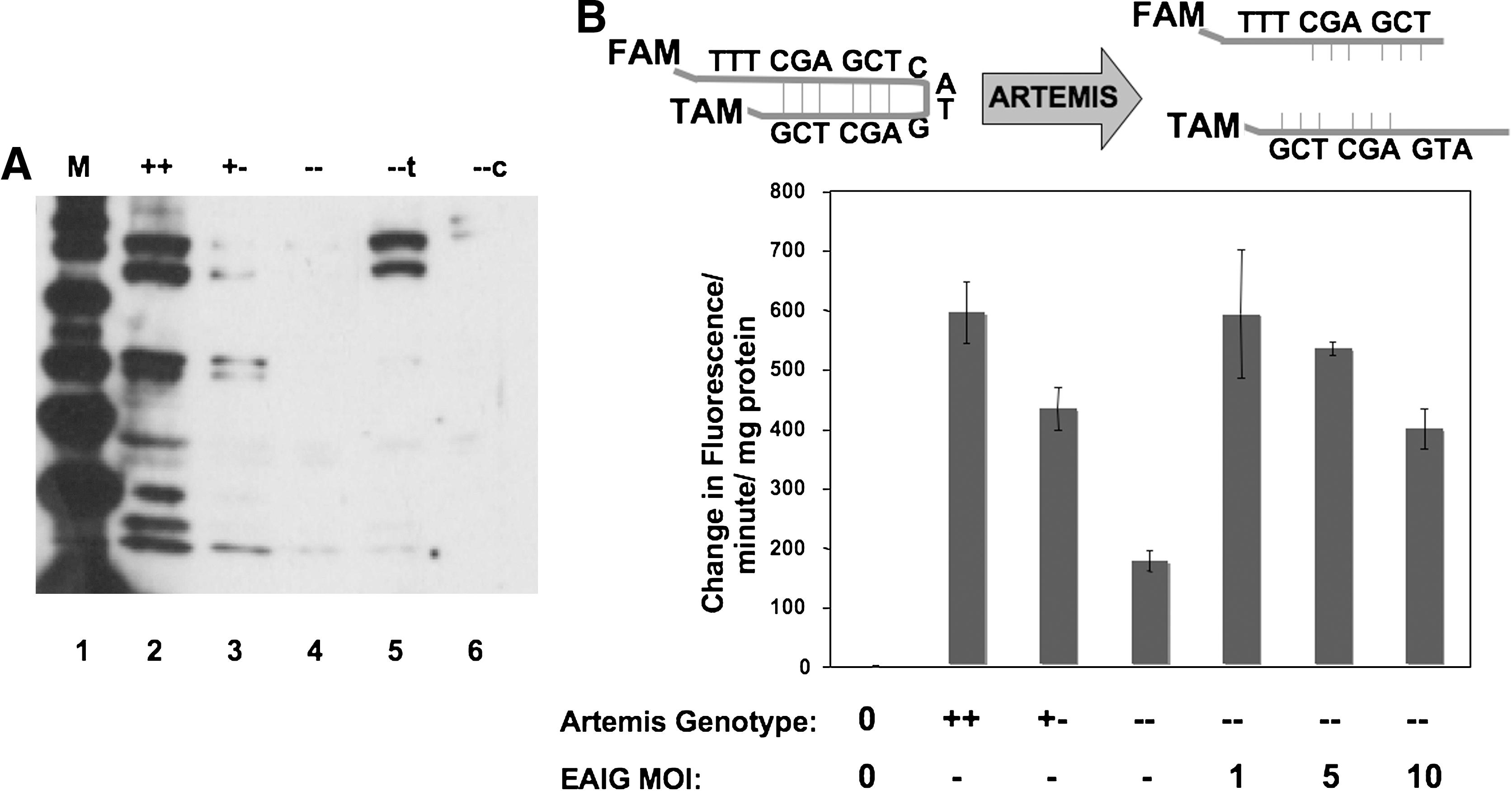
Analysis of Artemis expression in transduced cell populations. (
We developed a hairpin-opening assay to evaluate Artemis activity in cell extracts (Fig. 2). A 19-base oligodeoxynucleotide was designed to contain an internal stem–loop structure with a predicted melting temperature of 72°C. The hairpin oligodeoxynucleotide was synthesized with a FAM fluorophore at the 5′ terminus and a TAMRA quencher at the 3′ terminus. In hairpin configuration, FAM is quenched by TAMRA; however, upon hairpin cleavage the double-stranded molecule acquires a melting temperature of 20°C, with subsequent strand dissociation at room temperature, separation of FAM from TAMRA and fluorescence at 494 nm. Increasing FAM fluorescence was monitored in real time, using a Bio-Rad Realplex thermocycler as described in Materials and Methods.
Extracts were prepared from murine embryonic fibroblasts wild-type, heterozygous, and deficient for Artemis, and analyzed for hairpin-opening activity by increasing FAM fluorescence. As expected, Artemis wild-type cell lysates displayed greater hairpin-opening activity than did Artemis-deficient cell lysates (Fig. 2). Extracts from Artemis heterozygous cells exhibited an intermediate level of hairpin-opening activity (Fig. 2). A background of hairpin-opening activity was also observed in Artemis-deficient MEFs, most likely due to the presence of other nucleases in whole cell extracts.
Artemis-deficient MEFs were then transduced with various amounts of EF1α-mArtemis lentiviral vector (Fig. 2). Cell lysates were assayed for hairpin-opening activity, and, as expected, transduction with Artemis vector at an MOI of 1 restored hairpin-opening activity to wild-type levels (p < 0.005 vs. Artemis-deficient [−/−] MEFs). Surprisingly, exposure to greater amounts of Artemis vector resulted in a reduced level of hairpin-opening activity in cell extracts (p < 0.01 for an MOI of 10 vs. an MOI of 5) (Fig. 2). One potential explanation for this result is that the higher levels of Artemis expression brought about by transduction at higher multiplicity may have been toxic, resulting in loss of viability among those cells expressing the highest level of Artemis. We therefore tested the cytotoxic effect of Artemis overexpression, as described below.
Artemis-mediated growth inhibition
To test the possibility that overexpression of Artemis leads to a loss in cell viability, a dose–response experiment was carried out in which 3T3 cells were transduced with various amounts (MOI) of EAIG (Artemis) and CSIIEG (GFP control) lentiviral vectors. Integrant copy number was well controlled in this experiment because these vector preparations were also titered on 3T3 cells by quantitative PCR (see Materials and Methods). For five continuous days posttransduction, cell populations were assayed for viability by trypan blue exclusion, using a Vi-CELL (Beckman Coulter, Fullerton, CA). Cultures transduced with CSIIEG remained viable independent of transduction at increasing MOI; however, a dose-dependent decrease in cell survival was observed over time in cultures transduced with EAIG at increasing multiplicity (p < 0.005 for EAIG vs. CSIIEG at MOIs of both 3 and 10) (Fig. 3).
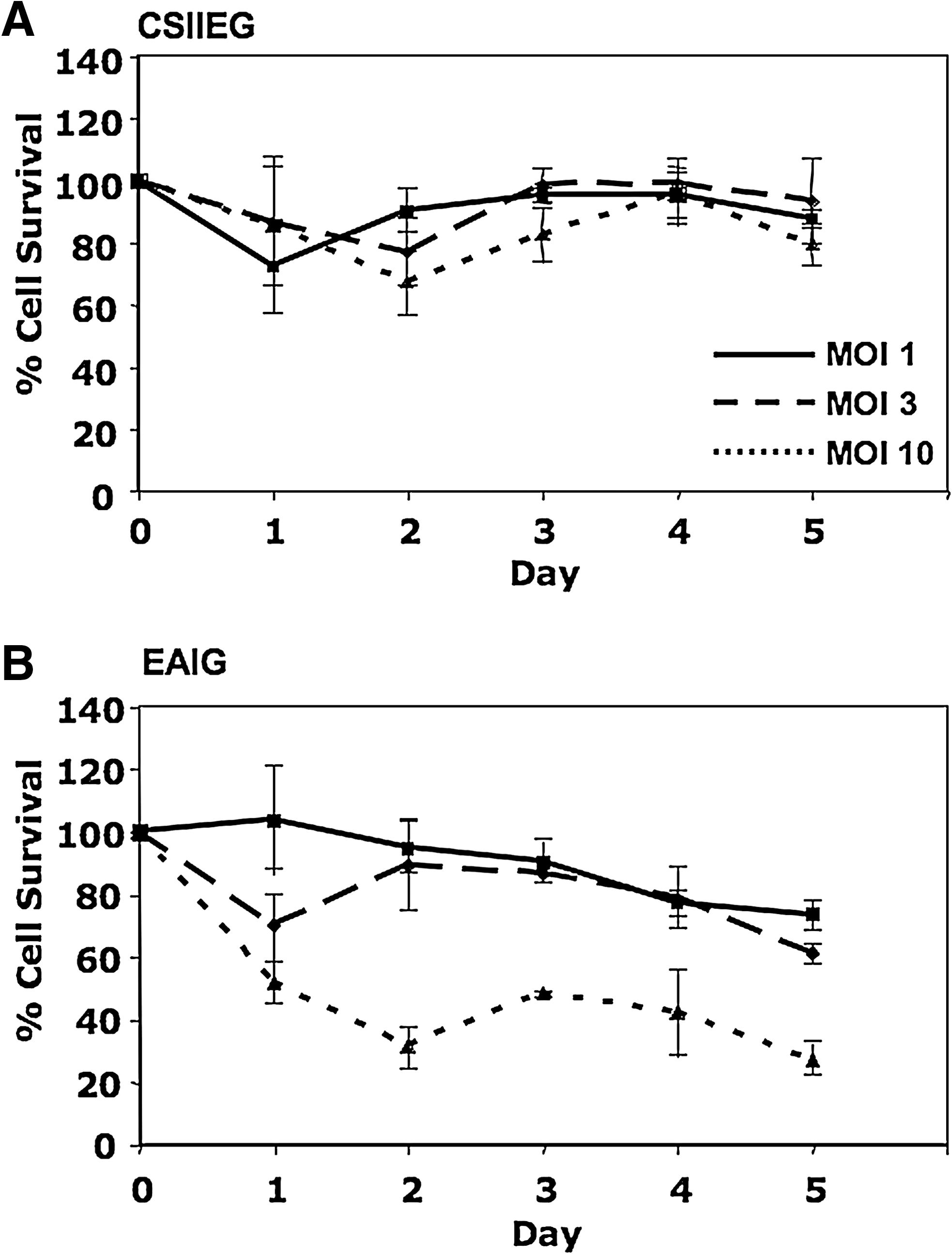
Cell survival response following Artemis transduction. Murine NIH 3T3 cells were transduced at increasing MOI using A) CSIIEG or B) EAIG lentiviral vectors as indicated. Cell survival was assessed over time, plotted as the percentage of cells surviving in control, untreated populations. Each value represents the mean of three replicates ± SD.
Decreased cell viability in the preceding experiment could have been the result of exposure to lentiviral vector at high multiplicity (MOI, 10) as well as to Artemis overexpression. To verify that the observed loss of cell viability was due to Artemis overexpression, 293T cells were transfected with increasing amounts of plasmid DNA EF1α-hArtemis or control EF1α-Puro (conferring resistance to puromycin), and cell viability was determined 4 days posttransfection by trypan blue exclusion (Fig. 4). 293T cells were used to assess growth response after Artemis transfection because of their high transfection efficiency. Cultures transfected with increasing amounts of Ef1α-hArtemis plasmid exhibited a significant decrease in cell survival in comparison with cells transfected with increasing amounts of EF1α-Puromycin plasmid (p < 0.001), which had no effect on cell viability (Fig. 4). These results confirm that the loss of cell viability observed after lentiviral transduction was due to overexpression of Artemis rather than to a toxic response to lentiviral transduction.
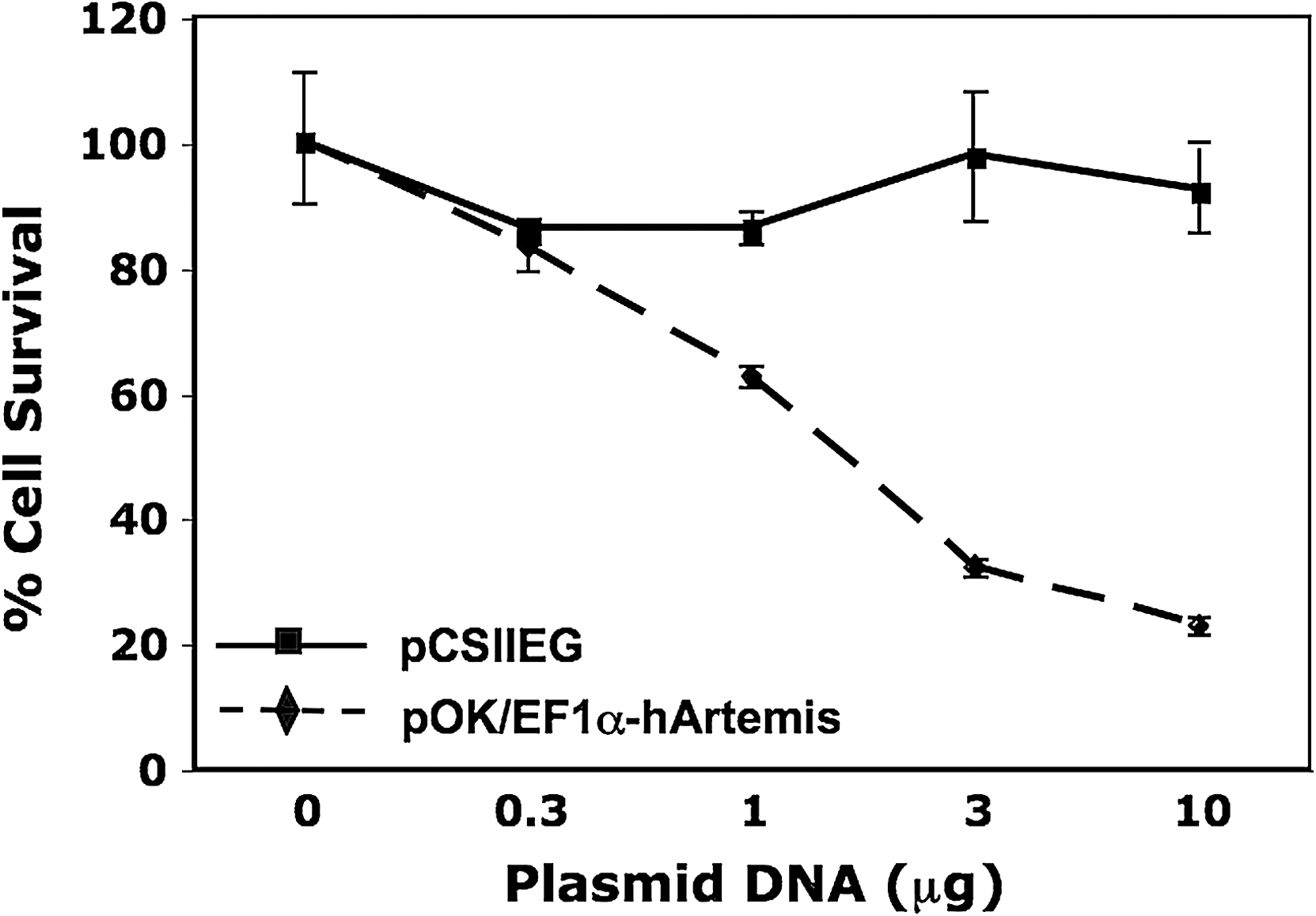
Growth response after Artemis transfection. 293T cells were transfected with increasing amounts of plasmid EF1α-hArtemis or EF1α-Puromycin, using the DNA-calcium phosphate coprecipitation technique. Cell survival was assessed 5 days later by trypan blue exclusion, expressed here as the percentage of an untreated control cell population. Each value represents the mean of three replicates ± SD.
If growth inhibition results from a high level of Artemis expression in cells transduced with an Artemis expression vector, this predicts that transduction with a vector in which Artemis is regulated by a more moderate-strength promoter will be less toxic. We therefore prepared a lentiviral vector in which the human Artemis gene is regulated by the phosphoglycerate kinase (PGK) promoter, and compared the effect of transduction with that of the EF1α-regulated human Artemis vector. The PGK promoter has been documented to mediate gene expression at more moderate levels in comparison with stronger promoter systems in the liver (Mikkelsen et al., 2003; Wilber et al., 2005), and in several cell lines including 293T and 3T3 as well as CD34+ cord blood progenitors (Ramezani et al., 2000; Schambach and Baum, 2007). Both EF1α- and PGK-regulated human Artemis vectors were used to transduce MEFs at various multiplicities of infection; moreover, MEFs wild-type (+/+), heterozygous (+/−), and deficient (−/−) for Artemis were used in this study to ensure that cytotoxicity associated with Artemis overexpression was not cell type or genotype specific (Fig. 5). Cultures transduced with increasing amounts of the control vector EF1α-Puromycin retained cell viability, whereas cultures exposed to increasing amounts of EF1α-hArtemis displayed significant growth inhibition (p < 0.001 for all three genotypes) (Fig. 5). Heterozygous MEFs transduced with increasing amounts of PGK-hArtemis displayed a less dramatic growth inhibition compared with heterozygous MEFs transduced with EF1α-Puromycin vector (p < 0.05). Moreover, wild-type and Artemis-deficient MEF cultures transduced with increasing amounts of PGK-hArtemis displayed growth curves statistically indistinguishable from those of cultures transduced with the EF1α-Puromycin vector. These data support the hypothesis that overexpression of Artemis leads to decreased cell growth. The endogenous level of Artemis expression (i.e., MEFs +/+, +/−, and −/− for Artemis) did not appear to contribute to the cytotoxicity associated with overexpression of Artemis after transduction.
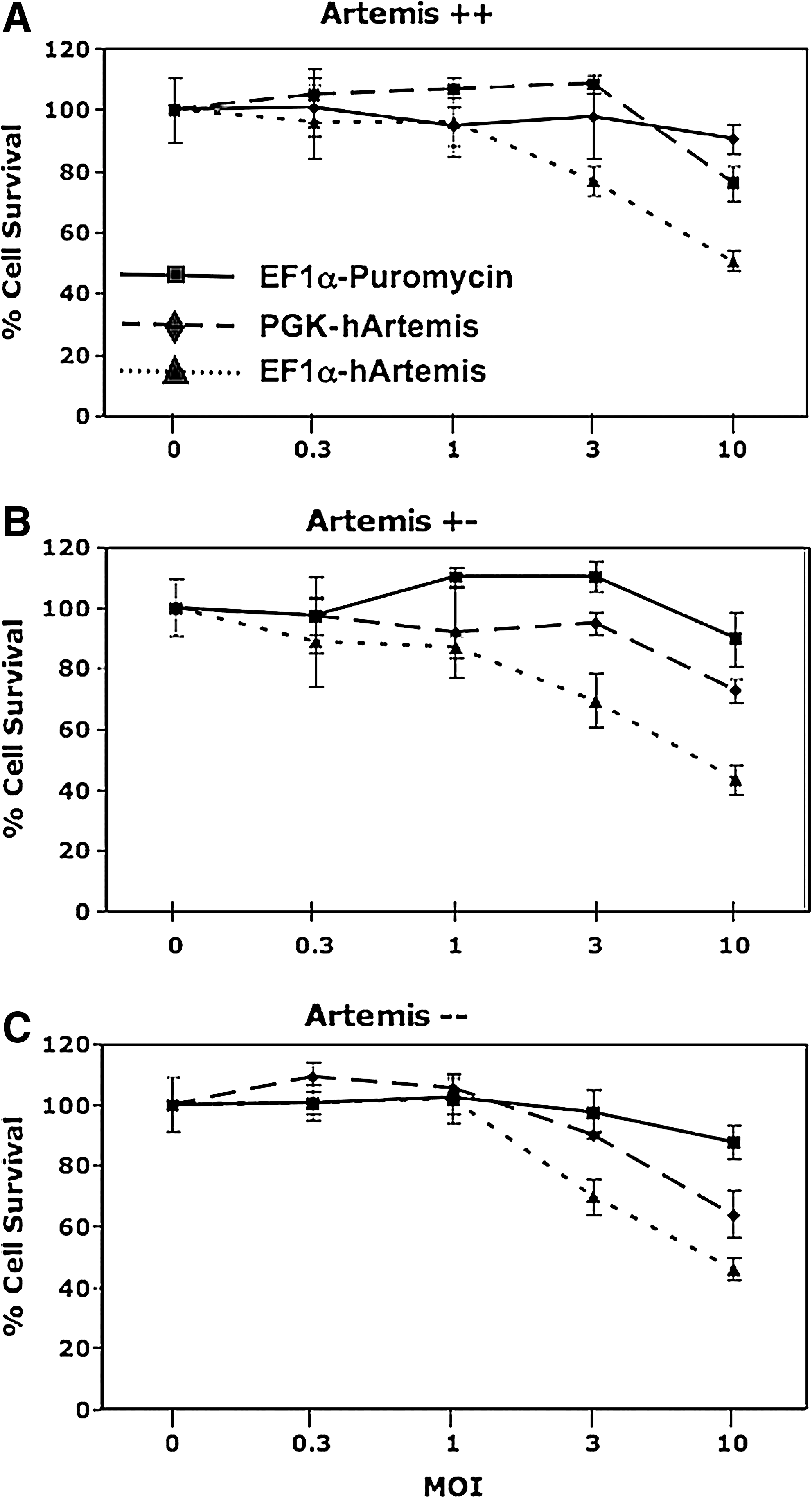
The effect of promoter strength on Artemis toxicity. The effect of promoter strength on Artemis toxicity was assayed by transducing (
Cellular responses to Artemis overexpression
To further characterize the cytotoxicity associated with Artemis overexpression, we investigated several possible cellular responses. Because Artemis is an endonucleolytic DNA hairpin-opening enzyme, we tested for global genomic instability by comet assay, conducted under alkaline conditions. The comet assay has been used extensively as an assessment of global DNA damage in cells subjected to nucleic acid-damaging agents. Briefly, cells are exposed to damaging conditions, suspended in low melting point agarose, and lysed. Upon electrophoresis, highly fragmented DNA migrates more rapidly through the agarose matrix than the cell body, thus generating a smear that mimics the tail of a comet. Exposure of cells to increasing concentrations of damaging agents causes increasingly fragmented DNA, which can be directly correlated with a longer comet tail.
Murine embryonic fibroblasts deficient for Artemis were transduced at increasing MOI with either the control CSIIEG vector or with EAIG, which expresses both murine Artemis and GFP. Four days posttransduction, GFP-positive cells from each transduced population were sorted by FACSDiva and subjected to comet assay. GFP-negative cells were also sorted from the EAIG-transduced (MOI, 1) population to be used as a negative control. In cultures transduced with EAIG, we observed a significant increase in comet tail length that was vector dose dependent up to an MOI of 3 (p < 0.001 for EAIG- vs. CSIIEG-transduced cells) (Fig. 6). An increase in tail length was not observed in cultures exposed to identical amounts of control vector. These data demonstrate that overexpression of Artemis resulted in an increase in total cellular DNA damage.

Comet assay of genomic DNA damage. The comet assay was performed on tMEFSCIDA−/− cells transduced with either CSIIEG or EAIG. Transduced cells were collected by GFP-positive cell sorting and subjected to alkaline comet assay as described in Materials and Methods. Mean tail length ± SD (n = 3) is plotted for Artemis-deficient MEFs transduced at various multiplicities with EAIG and CSIIEG lentiviral vectors.
Because Artemis has been reported to play a role in the cell cycle checkpoint response (Zhang et al., 2004), we conducted cell cycle analysis of 3T3 cells transduced with increasing amounts of EF1α-hArtemis versus EF1α-Puromycin. Cells were harvested 48 hr after transduction; nuclei were stained with propidium iodide and analyzed on a FACScalibur using FlowJo software. Interestingly, after transduction with a greater amount of Artemis vector, there was an accumulation of cells in the G1 stage of the cell cycle (Fig. 7). Cells overexpressing Artemis thus appear to arrest at the G1 phase checkpoint, preventing progression to DNA synthesis. When considered along with the results from the comet assay, these data indicate that Artemis overexpression may be inducing genomic DNA damage, resulting in cell cycle arrest at G1.
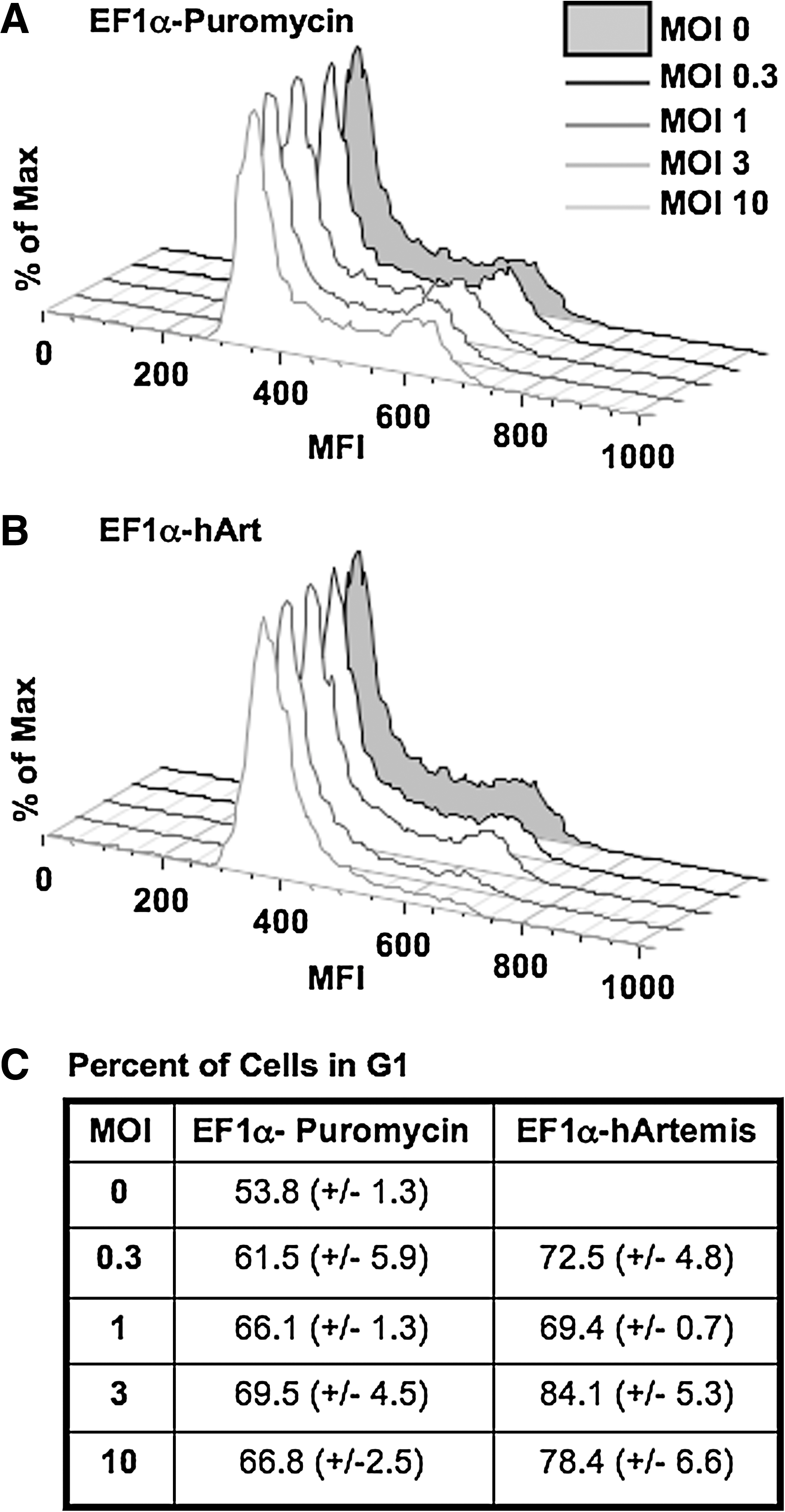
Effect of Artemis overexpression on cell cycle progression. NIH 3T3 cells were transduced with (
A potential outcome of Artemis-mediated DNA damage is the induction of an apoptotic response. To evaluate apoptosis in cells overexpressing Artemis, 3T3 cells were transduced with CSIIEG and EAIG lentiviral vectors at increasing MOI and after 20 hr stained with Annexin V and 7-AAD as described in Materials and Methods. The GFP-positive transduced cell population was gated and analyzed for Annexin V- and 7-AAD-positive cells. Both EAIG- and control CSIIEG-transduced cell populations exhibited similar levels of Annexin V single-positive staining (Fig. 8). However, the EIAG-transduced populations contained increased proportions of Annexin V/7-AAD double-positive cells (i.e., cells undergoing apoptosis) (p < 0.001 for EAIG- vs. CSIIEG-transduced cells). In addition, EAIG-transduced populations contained significantly increased percentages of 7-AAD-positive (Annexin-negative) cells (i.e., dead cells) (p < 0.001 for EAIG- vs. CSIIEG-transduced cells). Along with our data from the previously described comet assay and cell cycle analysis, these results indicate that Artemis overexpression induces genomic damage that ultimately induces apoptosis and loss of cell viability.

Induction of apoptosis in Artemis-transduced cells. NIH 3T3 cells were transduced at increasing MOI with (
Discussion
We found that Artemis overexpression by either transduction or transfection resulted in a dose-dependent cytotoxic response that was not observed in cultures exposed to identical amounts of control vector. On transduction with a lentiviral vector bearing an EF1α-regulated Artemis gene, we observed a direct correlation between overexpression and genomic DNA damage, arrest in cell cycle at G1, and apoptosis. Interestingly, the observed toxicity was diminished when the strong EF1α promoter was replaced with a moderate-strength PGK promoter, thus demonstrating the importance of restricting the level of Artemis expression.
Because of the inherent nucleolytic nature of Artemis, it is perhaps not surprising that increased expression levels may generate a greater extent of nonspecific breaks, thus causing genomic damage sufficient to trigger cell cycle arrest and apoptosis. It has been well documented that mammalian cells respond to DNA damage by activating cell cycle checkpoints and if the damage is not sufficiently repaired, arrest in cell cycle progression ultimately results in apoptosis to prevent replication of severely damaged DNA (Kuerbitz et al., 1992; Kastan and Kuerbitz, 1993). With these observations in mind, it becomes evident that achieving transgenic expression of Artemis may present a challenge.
Deficiency of Artemis results in SCID-A, characterized by complete loss of B cell and T cell function coupled with radiation sensitivity (Moshous et al., 2001; Li et al., 2002). At present, hematopoietic stem cell transplantation (HCT) is the most effective treatment of SCID-A; however, there are significant complications associated with HCT such as difficulty in identifying an HLA-matched donor, graft rejection, and graft-versus-host disease (O'Marcaigh et al., 2001). In addition, initial HCT studies reported that 11 of 12 SCID-A children treated displayed T cell reconstitution but no B cell reconstitution (O'Marcaigh et al., 2001). Because of the failure to reconstitute B cell immunity in patients with SCID-A after HCT, there is a considerable need for novel therapies that improve upon hematopoietic stem cell transplantation.
Gene transfer is currently emerging as a feasible and realistic alternative to HCT for effective long-term treatment of primary immunodeficiencies. Two independent studies have reported correction of X-linked SCID by ex vivo transduction of CD34+ hematopoietic stem cells, using a retroviral vector expressing the common cytokine receptor γ chain (common γ chain) (Cavazzana-Calvo et al., 2000; Hacein-Bey-Abina et al., 2002; Gaspar et al., 2004). Long-term engraftment of corrected stem cells was observed in the majority of patients, ultimately resulting in reconstitution of a functional lymphocyte compartment. In addition, successful long-term treatment of adenosine deaminase (ADA)-deficient SCID by gene transfer has been reported in which patients were infused with autologous CD34+ marrow cells transduced ex vivo with an ADA-expressing retroviral vector (Aiuti et al., 2009). After an average of 4 years posttreatment, these patients have shown evidence of stable engraftment, differentiation of transduced cells, and immune reconstitution of functional T lymphocytes (Aiuti et al., 2009). Clinical improvement of patients in these trials for X-linked SCID and for ADA deficiency demonstrates the potential effectiveness of gene transfer in the treatment of primary immunodeficiencies in general, including SCID-A.
Cytotoxicity associated with overexpression of Artemis after lentiviral transduction presents a challenge in the development of gene transfer as a therapeutic approach to correct SCID-A. Although sufficient Artemis expression will be necessary in order to restore immune function, our results suggest expression levels that trigger a toxic response will need to be avoided. This supposition has been confirmed in two independent studies that have reported correction of a murine model of SCID-A by ex vivo lentiviral transduction of hematopoietic stem cells (Mostoslavsky et al., 2006; Benjelloun et al., 2008). In both of these studies, the lentiviral vectors successfully used for treatment contained the human Artemis cDNA regulated by the moderate-strength PGK promoter. Optimally regulated Artemis expression would replicate endogenous conditions, so to this end we have isolated the human Artemis promoter region and are currently characterizing its expression in vitro and in vivo to be used as a potential regulator of Artemis in future preclinical studies (M. Multhaup, S. Gurram, K. Podetz-Pedersen, A. Karlen, D. Swanson, N. Somia, P. Hackett, M. Cowan, and R.S. McIvor, unpublished data).
The importance of therapeutic gene regulation has been exemplified in the case of gene transfer therapy for X-linked SCID. To date, 5 of 20 patients treated for X-linked SCID by gene transfer have developed clonal T cell outgrowth resulting in leukemia, from which one child has died (Hacein-Bey-Abina et al., 2008; Howe et al., 2008). Although insertional activation of the LMO2 oncogene was reported in three of the leukemic cases, overexpression of the common γ chain also likely contributed to these adverse events (Hacein-Bey-Abina et al., 2008; Howe et al., 2008). It has subsequently been demonstrated that overexpression of the common γ chain induces cellular proliferation that may have ultimately been responsible for the T lymphocyte clonal outgrowth (Amorosi et al., 2009).
Because allogeneic HCT in the correction of SCID-A has shown to be sufficient for T cell repopulation but has not been shown to support repopulation of the B cell compartment (Moshous et al., 2001), there clearly is a need for improved therapy. On the basis of its emerging success in the treatment of primary immunodeficiencies such as ADA and X-linked SCIDs, gene transfer may also be developed for the effective treatment of Artemis-deficient SCID. The results presented in this study, showing that overexpression of Artemis results in cytotoxicity, illustrate the need for regulated Artemis gene expression as a critical component to therapeutic vector design for gene therapy of SCID-A.
Footnotes
Acknowledgments
The authors acknowledge the assistance of the Flow Cytometry Core Facility of the Masonic Cancer Center, a comprehensive cancer center designated by the National Cancer Institute, supported in part by P30 CA77598. This work was supported by NIH grant R01 AI063340 to R.S.M. Work was done at the University of Minnesota- Minneapolis (Minneapolis, MN).
Author Disclosure Statement
No competing financial interests exist for the material described in this paper.