
Abstract
The broad application of retroviral vectors for gene delivery is still hampered by the difficulty to reproducibly establish high vector producer cell lines generating sufficient amounts of highly concentrated virus vector preparations of high quality. To enhance the process for producing clinically relevant retroviral vector preparations for therapeutic applications, we have integrated novel and state-of-the-art technologies in a process that allows rapid access to high-efficiency vector-producing cells and consistent production, purification, and storage of retroviral vectors. The process has been designed for various types of retroviral vectors for clinical application and to support a high-throughput process. New modular helper cell lines that permit rapid insertion of DNA encoding the therapeutic vector of interest at predetermined, optimal chromosomal loci were developed to facilitate stable and high vector production levels. Packaging cell lines, cultivation methods, and improved medium composition were coupled with vector purification and storage process strategies that yield maximal vector infectivity and stability. To facilitate GMP-grade vector production, standard of operation protocols were established. These processes were validated by production of retroviral vector lots that drive the expression of type VII collagen (Col7) for the treatment of a skin genetic disease, dystrophic epidermolysis bullosa. The potential efficacy of the Col7-expressing vectors was finally proven with newly developed systems, in particular in target primary keratinocyte cultures and three-dimensional skin tissues in organ culture.
Introduction
To produce retroviral vectors packaging cell lines were developed, based mainly on mouse or human cell lines that stably express the retroviral helper genes (gag and pol) as well as the transfer vector (Miller and Buttimore, 1986; Miller et al., 1991; Danos and Mulligan, 1988; Markowitz et al., 1988; Cosset et al., 1995; Miller and Chen, 1996; Davis et al., 1997). The incorporation of envelope proteins (Env) from different viruses (pseudotyping), eventually combined with specific genetic modification of Envs, allowed researchers to alter and broaden the host spectrum (Miller et al., 1991; Cosset et al., 1995; Lavillette et al., 2001; Ward et al., 2003), to label viral particles (Spitzer et al., 2003), and to generate viral particles with improved stability during preparation (Burns et al., 1993; Ory et al., 1996) and also stability toward human serum during treatment (Pensiero et al., 1996; Rigg et al., 1996; Spitzer et al., 1999). Although relatively high-titer virus production could be established with these packaging cell lines, the procedure for producing a new therapeutic virus is still time and labor intensive (Sinn et al., 2005), mainly because the generation of producer cells is highly random and requires extensive screening. Further, using this classical strategy, production frequently relies on integration of multiple transfer vector DNA copies. This is critical in terms of safety because the chromosomal sites of each integration event would have to be mapped to monitor and eventually exclude transduction of readthrough transcripts comprising unwanted neighboring cellular sequence elements. Also, these multiple vector copies may be subjected to recombination leading to loss of titer and/or rearrangement of vector DNA.
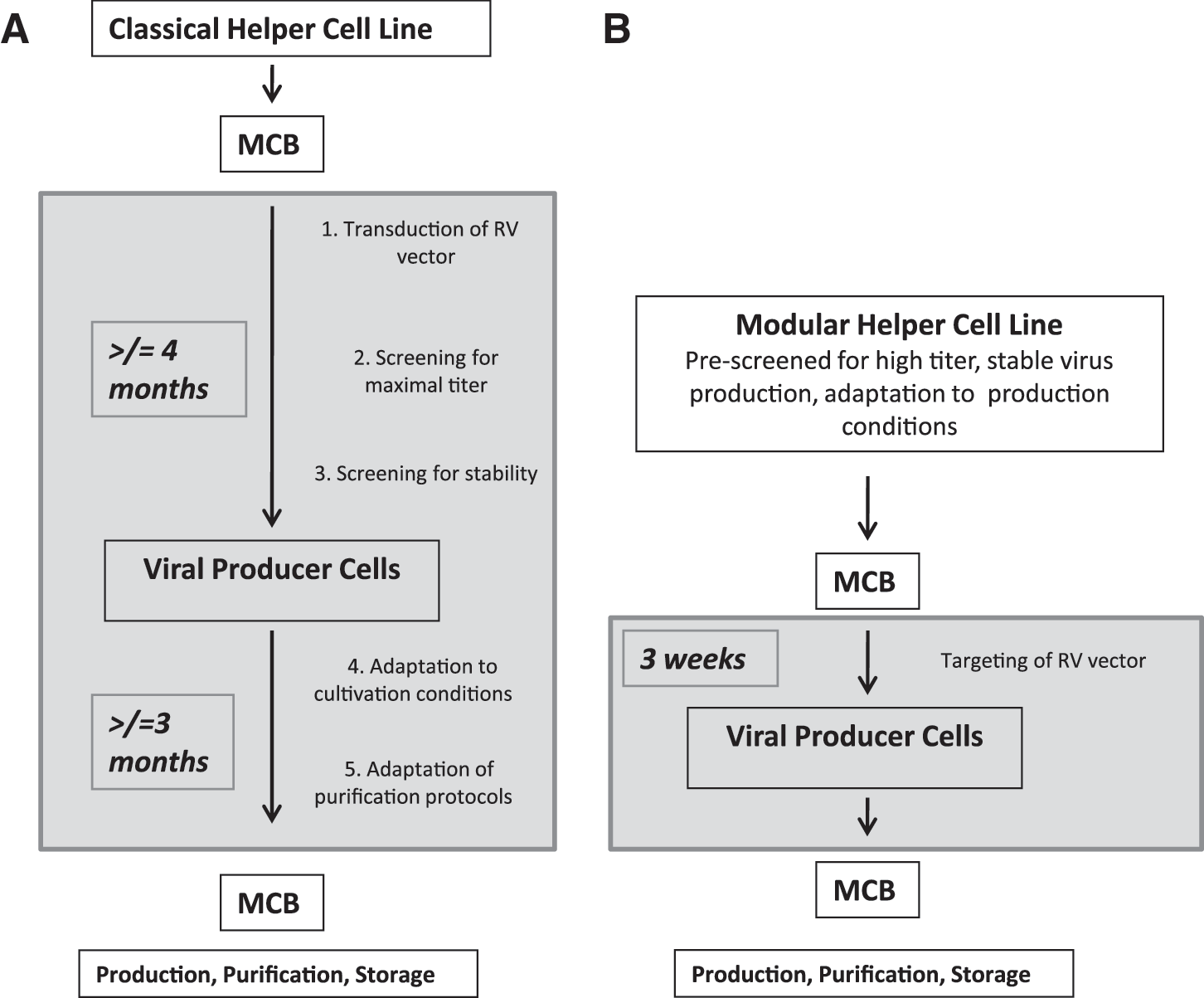
Once having developed a retroviral producer cell line with a stable vector titer, it must be optimized with respect to culture conditions, including the adaptation to production media (Merten, 2004). This is again tedious work that must be conducted for each producer cell clone independently. For clinical applications, a Good Manufacturing Practice (GMP)-compliant production process must be established. This requires a detailed safety study, including a precise characterization of the producer cell and the stability of virus production. In total, the process starting from the establishment of a packaging cell line to a suitable clone usually takes at least 6 months (Sinn et al., 2005) (Fig. 1A) and even then the performance in terms of virus production is not guaranteed.
Comparison of the conventional strategy to obtain retroviral producer cells (
Experimental Section
We have designed a new concept that comprises all steps required to produce therapeutic retroviral vectors including the rapid generation of high-titer retroviral vector-packaging cells, their cell culture process, and a robust purification scheme. The emphasis was placed on the development of an integrated process that can be carried out in compliance with standard GMP requirements and at the same time provides high flexibility.
In the following sections, the outline and characteristics of the processes of the individual steps are described in detail.
Modular helper cell lines
A major limitation in the generation of retroviral vector production systems is the randomness of integration of the therapeutic gene transfer vector into the genome of the packaging cell line. As a consequence, the viral titer and stability of production of individual producer clones, generated from pools of transfected cells, cannot be foreseen. Therefore, for each new therapeutic vector a large number of producer cell clones must be screened and thoroughly characterized. The strategy reported here is based on two previous findings: (1) Retroviral vector transcription is the major limiting factor and must be as high as possible to allow for maximal infectious virus production; and (2) to enable high-level retroviral vector production, the translation of Gag-Pol and Env proteins must be high and balanced. Accordingly, our first priority focused on an extensive screening for a highly expressed and stable chromosomal locus for retroviral DNA integration that would support efficient vector RNA transcription from a single-copy integration event. In a second step the helper functions were transfected into one of these cell lines with high vector RNA transcription and we screened for balanced expression of gag-pol and env products needed for optimal virus packaging. To render this strategy generic, we employed a novel method for the reuse of a validated retroviral vector integration site for a therapeutic vector of choice and thus exclude random integration of the retroviral vector into undesired loci in the packaging cell chromosome. This was facilitated by a recombinase-based approach, called recombinase-mediated cassette exchange (RMCE) (Wirth et al., 2007), that allows the exchange of a retroviral vector DNA integrated at an optimal validated locus with any vector expressing a therapeutic gene of choice (Verhoeyen et al., 2001) (Fig. 2A). The outlines of the “cassette exchange” process are depicted in Fig. 1B. In contrast to the classical technology (Fig. 1A), the efforts and time needed to establish retroviral producer cells and the production of clinical-grade viral particles are significantly reduced in the new approach (Fig. 1B). Use of the optimal well-characterized retroviral integration site is performed by Flp recombinase-mediated cassette exchange according to Fig. 2. On systematic screening for suitable safe chromosomal integration sites of a certified HEK293 cell clone, a series of high retroviral vector-expressing cell clones were identified. These cell clones served for the stable transfection of the helper viral genes gag-pol and env, thereby generating fully competent producer cell lines. These were subjected to long-term culture and finally resulted in the selection of two stably expressing cell clones. To produce different envelope pseudotypes, in the case of one cell line, Flp293A, an amphotropic MLV envelope (4070A strain) was used (Schucht et al., 2006); for the 293Flex cell line, the gibbon ape leukemia virus (GaLV) envelope was employed (Coroadinha et al., 2006).
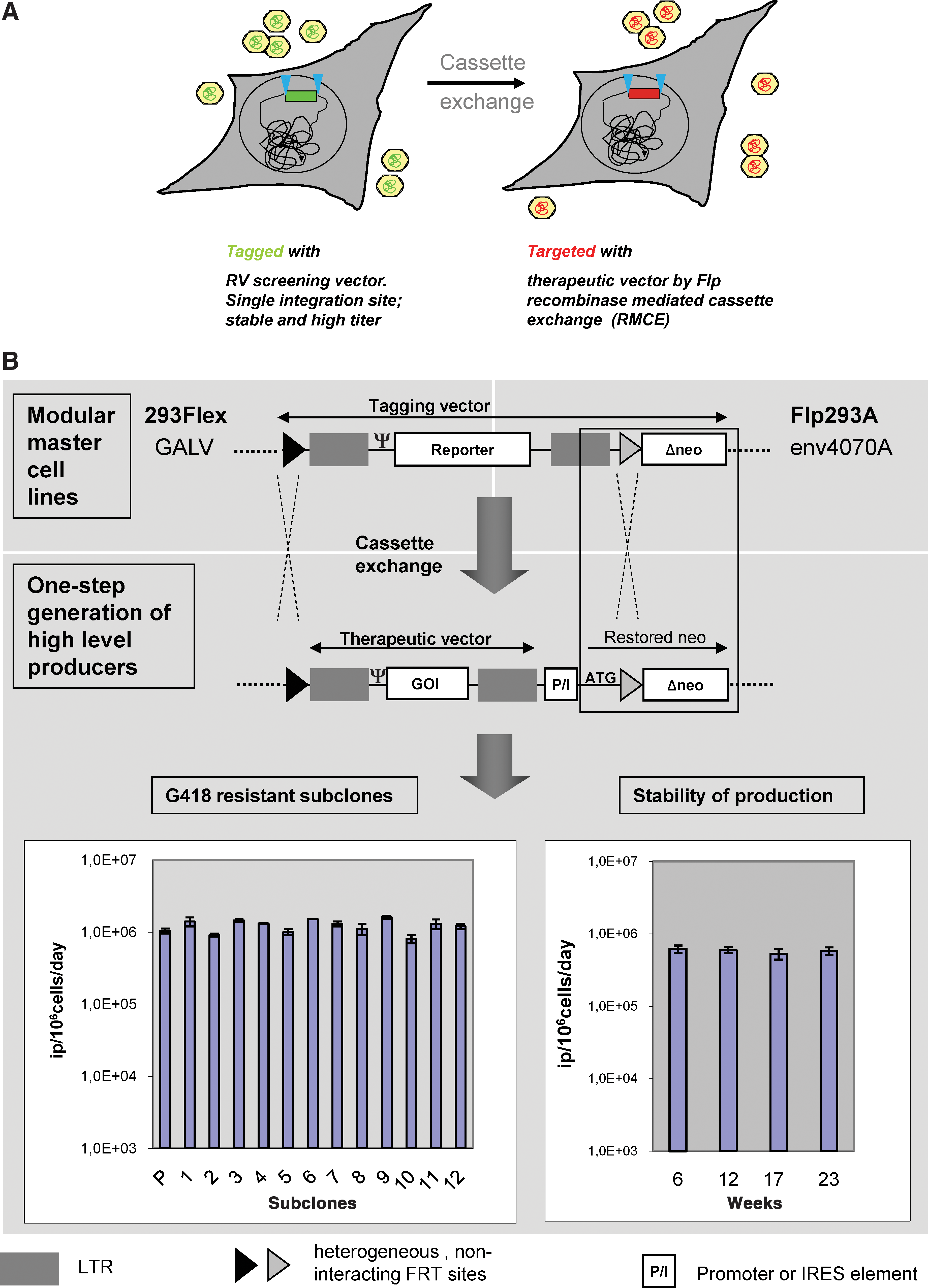
Application of recombinase-mediated cassette exchange (RMCE) for retroviral vector production. (
In the establishment of producer cell lines giving high retroviral titers, the need for balanced expression of the three viral components, Gag-Pol, Env, and the retroviral vector, was taken into account (Yap et al., 2000; Lei and Andreadis, 2005). A thorough analysis in 293Flex cells showed that a single copy of the retroviral vector, as long as it is highly expressed, is sufficient to cover high levels of Gag-Pol helper functions. Indeed, for the tagged chromosomal loci in 293Flex selected for high retroviral vector expression we observed that increasing amounts of Gag-Pol did not result in the generation of empty particles. This indicates that for the tagged cell lines the expression level of the retroviral vector is not limiting even on overexpression of Gag-Pol. However, the titer could be improved on increasing Env expression, indicating that expression of the envelope helper functions limits the production of infective retroviral particles (Carrondo et al., 2008). The study further showed that noninfectious particles in the preparation decrease significantly the transduction efficiency of the retroviral particles (Carrondo et al., 2008). Considering these findings we could establish the modular helper cell lines Flp293A and 293Flex with maximized infectious particle production and minimized production of noninfective particles on screening for optimal stoichiometry between Gag-Pol and envelope helper functions (Coroadinha et al., 2006; Schucht et al., 2006).
Both producer cell lines, 293Flex and Flp293A, provide high titers of retroviral particles even under nonoptimized production conditions (between 1 × 106 and 5 × 106 infectious particles [IP]/ml in 24 hr with an average cell density of 1 × 106 cells/ml). It is important to note that high viral titer is achieved from a single copy retroviral vector integrated into the tagged chromosomal site. Furthermore, retroviral vector production was proven stable for more than 12 months (Fig. 2 and Schucht et al., 2006).
Both Flp293A and 293Flex packaging cell lines serve as a platform for the production of retroviral vectors and are thus designated as modular helper cell lines. New retroviral producer clones are generated from these modular helper cell lines by exchange of the integrated retroviral cassette with a new retroviral cassette of choice according to Fig. 2. Importantly, the resulting vectors are selection marker free because the inherent neomycin resistance gene (neo) in the vector is not transduced. The resultant newly integrated retroviral vector copy provides a highly consistent production level (Fig. 2B), which makes further screenings superfluous. This is attributed to the fact that the integration site is predetermined and, accordingly, the cells generated on targeting are isogenic. In the protocols used, Flp recombinase is provided on cotransfection of the targeting vector together with a Flp-encoding plasmid. Usually, this does not result in integration of the Flp vector. It must be noted, however, that the targeted vector is stably integrated and cannot be excised even if the Flp-encoding plasmid is continuously expressed. This is due to the heterologous, noninteracting Flp recognition target (FRT) sites flanking the cassette.
An extensive exploration of these modular helper cell lines was undertaken by integration of a series of different retroviral vectors into the tagged site. More than 50 different retroviral vectors have been integrated into these modular helper cell lines, including both classical retroviral vectors and self-inactivating (SIN) vectors and the highly consistent vector production was thus confirmed (Coroadinha et al., 2006; Schucht et al., 2006; Gama-Norton et al., 2010; and data not shown). In addition, it could be shown that optimization of the retroviral vector construct design can increase the titer to 2.5 × 107 IP/106 cells/24 hr.
It was also found that the respective retroviral integration sites in Flp293A and 293Flex lines support the production of a given retroviral vector to different extents. This again indicated that the unique integration sites influence the expression of retroviral vectors significantly. A systematic study was carried out to reveal individual vector performance in the given integration sites in Flp293A and 293Flex packaging cells. The results indicate that interactions of internal retroviral vector elements (promoter, enhancer, etc.) with flanking chromosomal elements govern the performance of the vectors in the individual integration sites. Indeed, the modular helper cell lines have enabled, for the first time, rational approaches to address the question of cell genome–retroviral vector interactions and have already allowed the definition of optimal combinations of viral vectors and the requirements for a given integration site (Gama-Norton et al., 2010).
One critical safety concern arising in the production of retroviral vectors in the packaging cell lines is related to transcriptional readthrough due to the weak polyadenylation/termination signal present in the 3′ long terminal repeat (LTR) of the viral DNA. As much as 10% of viral transcripts are estimated to be a consequence of this illegitimate readthrough (Zaiss et al., 2002). Because these transcripts are readily packaged, the readthrough is suggested to be most relevant for the adventitious transduction of cellular genes derived from the retroviral helper cell lines. The modular helper cell lines provide a highly defined situation with a unique and well-characterized integration site in which the neighboring elements are known; therefore accidental transduction of harmful genes by readthrough is excluded. Even unwanted transduction of the neighboring neo gene sequences can be overcome, for example, on reverse integration of the retroviral vectors (Gama-Norton et al., 2010; Loew et al., 2010).
Conditions for cultivation of modular helper cell lines and retrovirus production
The production technology and culture media for retrovirus production are tightly linked to the specific producer cell line (Merten, 2004). Thus, the use of modular helper cell lines derived from 293 cells can facilitate the establishment of generic processes.
The retroviral producer cells were adapted to a standard medium that supported high cell densities. It could be shown that both the master modular helper cell lines and the daughter cell lines as established by RMCE can be cultivated under these conditions. This is attributed to the short time needed to create the new producer cell clones (3 weeks).
Most of the packaging cell lines used for the production of retroviral vectors are anchorage dependent and thus traditionally adherent culture systems have been used, that is, monolayer cultures (e.g., Nunclon Δ Cell Factories [Nunc/Thermo Fisher Scientific, Waltham, MA] or CellCube [Corning Life Sciences, Lowell, MA]) or microcarriers in stirred bioreactors (Warnock et al., 2006). HEK293-derived cells are able to grow in suspension as single cells or as small aggregates, particularly when serum-free media are used, facilitating scaleup.
Several commercial serum-free media suitable for 293 cells were tested with our master modular helper cell lines, namely, EX-CELL 293 (SAFC Biosciences/Sigma-Aldrich, St. Louis, MO), EX-CELL GTM-3 (SAFC Biosciences/Sigma-Aldrich), HyQ (HyClone, Logan, UT), CD 293 (Invitrogen, Carlsbad, CA), and 293 SFM II (Invitrogen). All serum-free media supported cell growth to similar or even higher densities than Dulbecco's modified Eagle's medium (DMEM) with 10% (v/v) fetal bovine serum (FBS) in monolayer cultures. In suspension cultures the producer modular cells grow as single cells; EX-CELL 293 and GTM-3 supported maximal cell densities greater than 3.5 × 106 cells/ml without optimization. Further long-term adaptation of the cells and optimization of the cell culture parameters could result in enhanced cell growth.
However, the viral titers obtained in most serum-free media were comparably low. The influence of serum on retroviral titers has been observed previously and its effect has been shown to be cell line dependent (Merten, 2004). The relationship between serum and viral production in the master modular helper cell lines was further analyzed for 293Flex and found to be correlated with lipid nutritional needs (Rodrigues et al., 2009). The infectious viral titer obtained was observed to be dependent on the serum concentration used in the culture media. Interestingly, we were able to reduce serum concentration in the culture media down to 1% (v/v) of FBS without significantly affecting cellular growth; however, the titers of infectious particles decreased by about 10-fold. The use of delipidated serum confirmed lipids as the serum component required in retrovirus production. Indeed, results show that on supplementation of fatty acids and cholesterol viral production titers in monolayer cultures can be recovered and even improved in the absence of serum when compared with the standard 10% (v/v) FBS supplementation (Rodrigues et al., 2009).
Last, after screening more than 13 serum-free media, we identified OptiPRO SFM (GIBCO; Invitrogen) as allowing high-titer virus production from the modular helper cell lines. Titers on the order of 1.5 × 106 IP/ml were obtained in OptiPRO SFM, and these could be further improved by the addition of lipid supplements.
Further, various cultivation conditions for growing the modular master cell lines in suspension with serum were evaluated. It was possible to cultivate cells in suspension either in a medium with reduced Ca2+ concentration or in a normal serum-containing medium under elevated shear stress induced by a pitched blade agitator. However, because suspension culture was associated with a considerably (20-fold) reduced specific vector production rate in comparison with a culture in serum-containing medium in a fixed-bed reactor culture, serum-containing conditions with elevated shear stress were retained.
Microcarrier culture
The use of microcarriers was considered and Cytodex 1 and Cytopore 1 carriers (GE Healthcare Life Sciences, Piscataway, NJ) were evaluated for cell growth and vector production. Cytodex 1 carriers were chosen as the reference carrier because they are frequently used for reactor cultures of many different adherent cells (Merten et al., 1996). Cytopore 1 carriers were chosen because they provide a much larger culture surface than solid carriers such as Cytodex 1. For the modular master cell lines on Cytodex 1 carriers (used at a concentration of 1 g/liter), 40 to 50% of the cell inoculum remained in suspension and the cells attached to the microcarriers were not able to spread. On further cultivation, the cells present in the microcarriers attached to each other, forming macroaggregates including several microcarriers. In contrast, on Cytopore 1 carriers Flp293A and 293Flex cells were completely attached a few hours after inoculation, and the carriers were found to be filled up with cell biomass at later stages of the culture, with, however, significant clumping in the late phase of cultivation. Thus, Cytopore 1 microcarriers turned out to be the method of choice for small-scale as well as large-scale cultures in stirred tank reactors. Importantly, under these conditions, the high titers obtained in standard culture dish cultures were confirmed. Thus, Cytopore 1 microcarriers can support cell growth and vector production, both at small and bioreactor scales.
Cell Factories
Large-scale production was optimized for both modular helper cell lines Flp293A and 293Flex, using Cell Factories. The general procedure was as follows: Cells were seeded in Cell Factories (area, 6300 cm2) at a concentration of 5 × 104 cells/cm2 for both modular helper cell lines. After 48 hr, the supernatant was replaced with 900 ml of DMEM, supplemented with Benzonase (10 U/ml; Merck, Darmstadt, Germany). Twenty-four hours after medium exchange the supernatant was collected. Titers obtained depended on the transgene and levels of 1–5 × 106 IP/ml were obtained for retroviral vectors encoding Col7 and green fluorescent protein (GFP) (Fig. 3).
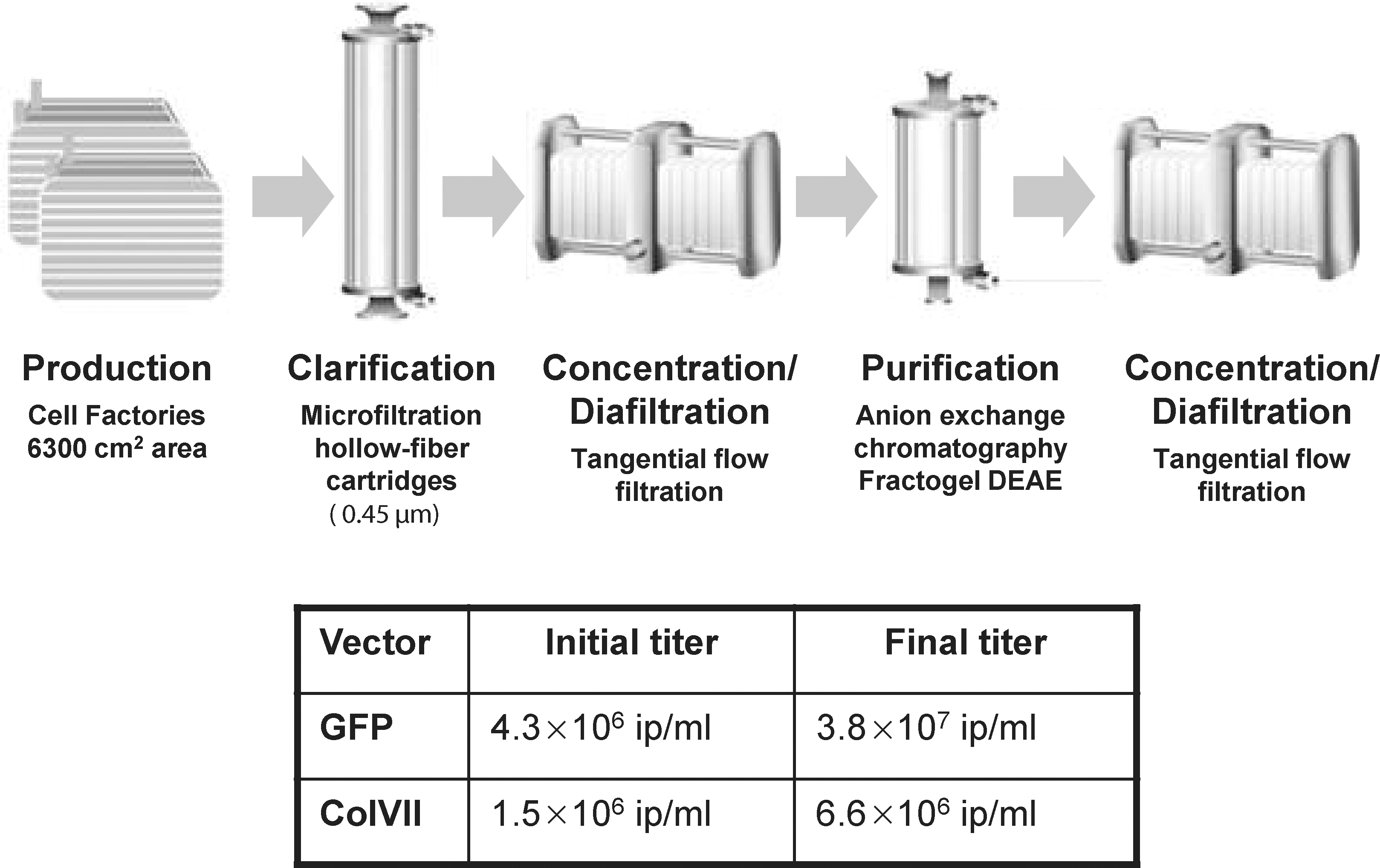
Process for production and purification of retroviral vectors from modular helper cells. The supernatant from Cell Factories is clarified and concentrated, using membrane processes. Further purification is achieved by anion-exchange chromatography and final polishing and concentration is carried out by ultrafiltration. (Equipment images were kindly provided by Sartorius). Further, the initial titers obtained from a representative Cell Factory production process as well as the final titers on purification are given. GFP, green fluorescent protein; ColVII, collagen type VII.
Virus purification and storage
The establishment of an efficient downstream processing strategy for retroviral vector preparations largely depends on the viral properties and cell culture characteristics. Starting with the modular cell lines, rather than the standard packaging lines, a large part of this variability can be eliminated and an optimal combination of established methods applied in the purification of retroviral vectors can be defined for these preparations (see Rodrigues et al., 2007a, for a review). However, further optimization and development of new specific operation steps may have to be done, depending on the particular characteristics of the retroviral vector and its final clinical application.
In the particular case of a retroviral vector expressing Col7, we have developed a scalable purification strategy and optimized it for Flp293A packaging cells expressing the amphotropic envelope. This purification strategy was shown to be applicable to other retroviral vectors with similar yield and purity. The procedure is based on membrane separation and anion-exchange chromatography (AEXc) techniques (Rodrigues et al., 2007b). A combination of micro- and ultrafiltration membranes with AEXc processes was applied (Fig. 3) (Rodrigues et al., 2007b). Retroviral vectors produced by modular helper cell lines and encoding GFP, and targeted vectors encoding Col7, were successfully purified from the supernatant of two Cell Factories (area, approximately 1800 ml), resulting in 5- or 20-ml purified vector preparations. The purification yield of the retroviral vectors produced was similar for the GFP and Col7 vectors, reaching final titers of 3.8 × 107 and 6.6 × 106 IU/ml, respectively.
Storage of retroviral vectors has been previously studied, with the most successful methods being ultralow-temperature freezing (–80 and −196°C) and lyophilization followed by low-temperature freezing (–20°C) (Cruz et al., 2006). These methods allow the retention of more than 70% of the infectious retroviral vector titer over a period of 4 months, thus enabling widespread application of these vectors.
GMP cell bank
In contrast to standard helper cell systems, in modular helper cell lines the rapid and highly defined integration of the therapeutic vector does not significantly change the properties and safety features of these cells, reducing the need of extensive adaptation and extensive characterization to achieve qualification for GMP production. For the modular helper cell line Flp293A the entire set of quality control (QC) tests required to fully qualify a “master cell bank” (MCB) as the final producer cell line for a clinical application were carried out, in particular identity tests; potency test (viral titer); absence of replication-competent retroviruses (RCRs); in vitro and in vivo detection of adventitious agents; detection of specific human, bovine, and porcine viruses; and confirmation of the absence of mycoplasma, bacteria, and fungi. The results suggest that no further problems are expected from these cells. An MCB in full compliance with current Good Manufacturing Practice (cGMP) conditions from the modular helper cell line Flp293A was manufactured. Cryotubes (250, each with 2.3 × 106 cells showing excellent recovery features for up-scaling) have been stored under appropriate conditions. Further, on cell expansion, retroviral vector cassette exchange as well as production and purification were confirmed with an MCB sample.
Production of efficient vectors for treatment of epidermolysis bullosa
The methods describe previously for the construction of modular helper cells and the established production/purification conditions for retroviral vectors were implemented in the preparation of retroviral vectors expressing human Col7 for the treatment of dystrophic epidermolysis bullosa (DEB). DEB is an inherited skin disease characterized by local detachment of the epidermal layer from the dermal tissue, resulting in skin blistering (Tamai et al., 2009). The genetic defect is characterized by mutations in the Col7 molecule, which is a major constituent of anchoring fibrils, present in the basal membranes between the epidermal and dermal layers. Ectopic expression of functional Col7 molecule, in either the epidermal or dermal cells adjacent to the basal membrane, may restore functional anchoring fibrils, thus preventing the detachment of the two skin layers and subsequent blistering. Correction of this genetic defect by gene therapy approaches would require stable long-term expression of a functional Col7-encoding gene in either epidermal stem cells or in stable mesenchymal cells in the dermal layer.
We selected this therapeutic gene for the present study because production of high-titer retroviral vector for the treatment of DEB, using standard production strategies, has been compromised by the fact that the mRNA encoding the Col7 protein exceeds 8.9 kb, the generally communicated limit of transgene RNA packaging into the retroviral vector (Pagès and Bru, 2004). The retroviral vector genomic RNA packaging limitation excludes the construction of a vector encoding both the Col7 gene and a reporter/selection gene in order to screen for a high-titer producer cell.
Col7 vector-producing cells were prepared by cassette exchange according to Fig. 2, based on the Flp293A and 293Flex packaging cells. Five different vector designs were evaluated including classical vectors with LTR promoter-derived transcription as well as SIN-based retroviral vectors. The mouse stem cell virus (MSCV)-derived vector pMSIRCOL7 (described by Schucht et al., 2006), giving rise to 1.2 × 105 IP/106 cells in cell culture flasks (here called RV Col7), was selected and for production for an envisaged phase I clinical trial and for detailed toxicity and efficacy analysis.
A production process was established for RV Col7-targeted producer cell lines. Because only relatively small quantities of vector are required for the phase I clinical trials, classical adherent culture conditions were employed using Cell Factories as described previously. In parallel, the parental modular helper cell line 293FlpA was handled under the same conditions. These cells produce a retroviral vector that encodes a reporter gene (GFP). The GFP vector was produced at approximately 3-fold higher infectious titers than the Col7 vector (4.3 × 106 vs. 1.5 × 106 IP/ml, respectively). The protocols for purification and storage were employed accordingly, resulting in the final titers given in Fig. 3.
Toxicity and efficacy of the retroviral Col7 vectors
Within our comprehensive strategy, we subjected the therapeutic Col7 transducing vector to a preclinical efficacy evaluation. Two relevant biological systems were applied in the study: skin tissue in organ culture (Kunicher et al., 2008) and cultured primary keratinocytes (Green, 2008). Primary keratinocytes in culture represent a convenient system with which to study turnover of the epidermal cell layer and to produce autologous epidermal sheets for transplantation in burn patients. This cell culture system is amenable for ex vivo gene transfer using RV Col7 and subsequent transplantation of the engineered epidermal sheets to patients with DEB. The results presented in Fig. 4A (top row) indicate efficient transduction of human keratinocytes in culture at a low multiplicity of infection (MOI, 0.1). As the cultured human keratinocytes are derived from a normal individual, some Col7 expression is expected in the control cells transduced with a comparable control RV GFP (Fig. 4A, panel I, top). Notwithstanding, immunostaining of human primary epidermal cell cultures transduced with RV Col7 indicates high cytoplasmic expression of Col7. In parallel, a transformed human keratinocyte cell line (HaCat) was transduced with RV Col7 and extensive cytoplasmic expression of Col7 is clearly evident (Fig. 4A, bottom row).

Collagen type VII (Col7) expression in skin cells and tissue. (
Next, we analyzed the tropism of RV Col7, pseudotyped with the amphotropic Env, to the various cell types constituting skin tissue by applying a novel organ culture system composed of the epidermal and dermal layers in their native three-dimensional organization (Kunicher et al., 2008). Mouse skin tissue in organ culture was transduced ex vivo with RV Col7 and either immunostained as “whole-mount tissue” preparations with antibody specific to human Col7, or subjected to immunostaining after preparation of histological slices (thickness, 5 μm). The images depicted in Fig. 4B indicate infection of cells mostly in the dermal layer. Further immunostaining with a mesenchymal cell marker (vimentin) confirmed that transduced cells within the three-dimensional structure of the skin are mesenchymal, possibly of fibroblast origin.
Last, possible toxicity of RV Col7 to the skin was investigated, using organ cultures derived from human and mouse skin tissues. Several independent cell viability assays were applied in this analysis, that is, the MTT assay (Mosmann, 1983), the glucose uptake assay (Brill-Almon et al., 2005), activation of caspase-3 (Yaacov et al., 2008), and histological observation of the transduced tissues (Kunicher et al., 2008). Using all of these assays, we could not observe any adverse toxic changes to the skin after transduction with RV Col7 (data not shown).
Taken together, the results indicate the potential and safety of the RV Col7 described in this paper for the treatment of patients with DEB.
Summary and Outlook
Our study gives evidence of an integrated process for fast and reliable production of therapeutic retroviral vectors based on master modular helper cell lines. This procedure comprises several important features. The benefits of the integrated process described herein are summarized in Table 1.
In the long run, one could imagine that, if the generic modular helper cell lines are fully certified and if the vector targeting can be performed in a controlled environment (Good Laboratory Practice [GLP] or GMP), the QC testing plan could be reduced to a certain point, as is the case, for instance, for master versus working cell banks. This could have a significant impact on the manufacturing of clinical preparations of RVs as it would result in a reduction of the time needed to release the MCB of the final producing cell line and on manufacturing costs by avoiding costly analytical tests.
Footnotes
Acknowledgments
This work was supported by grants from the EU (InsertAGene, QLK3-CT-2002-01949; Clinigene, LSHB-CT-2006-018933; Therpeuskin, LSH-2003-1.2.4-8), the Deutsche Forschungsgemeinschaft (Wi 2624 and REBIRTH Cluster of Excellence), the Bundesministerium für Bildung und Forschung (FKZ0313940), the Fundação para a Ciência e a Tecnologia–Portugal (PTDC/BIO/69451/2006), and the Israeli Academy of Sciences.