
Abstract
Adenoviral (Ad) vectors are widely used for gene therapy approaches. Because of the high abundance of the natural adenoviral receptors (coxsackievirus–adenovirus receptor and integrins) on a wide variety of cells, numerous methods have been developed to redirect the virions to specific receptors on target cell surfaces. Importantly, an increasing number of publications have shown that the success of targeting not only depends on receptor binding and cellular uptake, but also on intracellular trafficking processes. Therefore, improved knowledge about the intracellular fate of targeted Ad vector particles is mandatory for a rational design of targeted Ad vectors. However, the technologies currently available for fluorescent labeling of Ad vectors have significant limitations: (1) at present capsids are labeled all over the particle surface, and this imposes the risk of interference with particle infectivity; (2) capsomer-specific labeling requires extensive genetic modifications and has been demonstrated only at protein IX; and (3) two-color labeling approaches are not available. Here we present a novel, robust, and straightforward labeling procedure that overcomes these limitations. It allows for specific labeling of the capsomer's fiber, protein IX, or hexon and permits two-color labeling. We demonstrate the potential of this labeling technology by analyzing two different bioresponsive bonds that can be used for the attachment of shielding or targeting moieties to the capsids: disulfide and hydrazone bonds. We demonstrate that in contrast to disulfide bonds, hydrazone bonds are quickly hydrolyzed after uptake of the virions and are thus favorable for the generation of bioresponsive vectors.
Introduction
The most promising approaches to decrease specific and nonspecific interactions of Ad vector particles with cellular receptors and even noncellular body fluid components, including antibodies and blood coagulation factors, are based on chemical capsid modifications with synthetic polymers (Kreppel and Kochanek, 2008). Polymers such as polyethylene glycol (PEG) (O'Riordan et al., 1999; Croyle et al., 2000) and poly-N-hydroxypropylmethacrylamide (Fisher et al., 2001; Green et al., 2004) can be covalently linked to the virions and particles thus modified exhibit significantly decreased interactions with both their natural receptors and body fluid components such as antibodies (Weaver and Barry, 2008; Wortmann et al., 2008). Detailed molecular analysis has revealed that the most widely used amine-reactive synthetic polymers modify predominantly hexon, fiber, and penton base proteins (O'Riordan et al., 1999). Although little is known about the impact of such extensive capsid modifications on intracellular virion trafficking it seems highly probable that the dense steric shield generated by the polymers influences the interaction of the vector particles, for instance, with dynein, which is required for nuclear trafficking (Leopold et al., 2000).
As outlined previously, almost any capsid modification, including the generation of serotype chimeras, bears a significant risk of altering intracellular virion trafficking, with the potential consequence of decreased gene transfer efficiency despite efficient virion uptake into the target cells. Thus, it is obvious that reversible capsid modifications, in particular modifications that are reversed specifically after cell entry and thereby set back the virion capsid to its natural state, may be advantageous. Bioresponsive bonds, defined as dynamic covalent bonds that are able to undergo changes triggered by the physiological environment (i.e., pH or redox potential), have been consistently proposed for the attachment of ligands or shielding molecules (Wolff and Rozema, 2008).
Such bioresponsive bonds would allow for the separation of shielding polymers, ligands, or ligand–receptor complexes from the virions after uptake into the target cells and ensure efficient intracellular virion trafficking. In fact, a wide variety of potentially bioresponsive bonds may be used, including protease recognition sites and esters that could be a target for esterases. The development of bioresponsive nonviral gene transfer vectors has revealed a number of bioresponsive chemical bonds that respond to changes in the pH or redox environment (Wolff and Rozema, 2008). Two promising bioresponsive bonds are disulfide bonds and pH-responsive hydrazone bonds. Data were presented for a bioresponsive PEG-based polymer containing a pH-sensitive pyridyl-hydrazone bond (Walker et al., 2005; Fella et al., 2008). Release of PEG from targeted polyplexes at endosomal pH was verified by biophysical measurements and bioresponsive polyplexes showed greater in vitro and in vivo gene transfer efficiency than analogous, stably shielded polyplexes.
As outlined previously, the rational development of targeted Ad gene transfer vectors requires in-depth analysis of the intracellular trafficking of targeted vector particles. This can be achieved by confocal laser scanning microscopy in combination with fluorescently labeled vector particles. However, the most widely used technology to fluorescently label Ad vector particles is based on amine-reactive fluorescent dyes that are covalently linked to the vector particle surface. The use of amine-reactive dyes stems from the fact that the vector particle surface bears about 18,000 amino groups and thus amine-reactive dyes can be attached easily. However, this technology does not allow for the specific labeling of single capsomers or incorporated ligands and bears a significant risk of overlabeling, which in turn can interfere with particle infectivity. Furthermore, it cannot be used in conjunction with polymer modification of Ad vectors because amine-reactive shielding polymers attach to the same sites on the capsid as the fluorescent dyes.
One defined labeling approach has been published by Le and colleagues (2004). Le and colleagues were able to show that protein IX can be genetically tagged with green fluorescent protein (GFP), and this tagging allowed for the visualization of Ad particle trafficking in A549 cells. However, this labeling approach requires extensive genetic modification and is thus not suitable for the analysis of a wide variety of vectors, including vectors with targeting ligands attached to protein IX.
Here we present a simple and straightforward Ad particle-labeling technology that allows for specific chemical labeling of defined capsomers, is compatible with ligand-decorated vectors, and allows for the generation of vector particles labeled with two different fluorescent dyes. We demonstrate the proof-of-concept for this technology by analyzing two different bioresponsive bonds in the context of adenoviral vector particles: hydrazone bonds and disulfide bonds. Our results indicate that in contrast to redox-sensitive disulfide bonds, pH-dependent hydrazone bonds are favorable for the generation of chemically modified dynamic Ad vector particles. In summary, the data presented here stress the importance of analyzing the intracellular pathways of targeted Ad vector particles, provide robust tools for this analysis, and give insight into the use of bioresponsive bonds for Ad vector targeting and shielding.
Materials and Methods
Cell lines and cell culture
A549 cells (CCL-185; American Type Culture Collection [ATCC], Manassas, VA) were cultivated in minimal essential medium (MEM; GIBCO/Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (FCS) and 1% penicillin–streptomycin and subcultured twice weekly. CHO-K1 cells (CCL-61; ATCC) and Hepa 1-6 cells were cultivated in Dulbecco's modified Eagle's medium (DMEM; GIBCO/Invitrogen) supplemented with 10% FCS and 1% penicillin–streptomycin and subcultured twice weekly.
Adenoviral vectors
The adenoviral vectors used were Ad5-based E1-deleted first-generation vectors harboring a human cytomegalovirus (hCMV)-driven transgene expression cassette for the enhanced green fluorescent protein (EGFP).
AdControl is an Ad5 vector with wild-type capsid. The vector AdFiberCys contains the peptide motif LIGGGCGGGID inserted in the fiber HI loop and has been described by Kreppel and colleagues (2005). The vector AdpIX75Cys contains a cysteine-terminated 75-Å spacer fused to the C terminus of protein IX and has been described by Corjon and colleagues (2008). AdHexonCys carries an A → C point mutation in the hypervariable region 5 (HVR5) of hexon (amino acid 273; GenBank AY339865).
All vectors were generated by transfection of the corresponding infectious plasmid into the E1-trans-complementing N52E6 cell line (Schiedner et al., 2000) followed by subsequent vector amplification. Vectors were purified by subsequent discontinuous and continuous CsCl banding and desalted by gel filtration on PD-10 columns (GE Healthcare Biosciences, Piscataway, NJ) equilibrated with phosphate-buffered saline (PBS) (under reducing conditions; Kreppel et al., 2005). Vectors were stored in PBS supplemented with 10% glycerol. Vector titers were determined by a DNA-based slot-blot procedure (Kreppel et al., 2002).
Flow cytometric analysis of EGFP expression
Flow cytometric analysis of EGFP expression was performed with a FACSCalibur (BD Biosciences, San Jose, CA) without gating. Relative transduction efficiency was calculated from the mean fluorescence intensity.
Alexa dyes used for labeling
The following reactive fluorescent dyes were used in this study: Alexa Fluor 488 carboxylic acid; 2,3,5,6-tetrafluorophenyl ester (Alexa Fluor 488 5-TFP) *5-isomer* (A-30005; Molecular Probes/Invitrogen), called here Alexa488-TFP; Alexa Fluor 488 C5 maleimide, called here Alexa488-maleimide (A-10254; Molecular Probes/Invitrogen); and Alexa Fluor 633 C5 maleimide, called here Alexa633-maleimide (A-20342; Molecular Probes/Invitrogen).
Dyes were dissolved in dimethyl sulfoxide (DMSO) and kept in the dark at −80°C. For all modifications with genetically modified vector particles, manipulations were performed under an argon atmosphere with degassed and argon-saturated buffers. The reactions were performed at room temperature in the dark.
Random labeling of AdControl vector particles with Alexa488-TFP
AdControl vector particles (1 × 1012) were incubated with a 20-fold excess of chemically reactive Alexa488-TFP over the 18,000 amino groups present at the capsid surface for 2 hr at room temperature in an argon atmosphere, filled up to a final volume of 900 μl with HEPES buffer, pH 7.2. The reaction was stopped by incubation for 1 hr with a 5-fold excess of lysine over Alexa488-TFP. The labeled vector particles were then purified from excess dye by PD-10 column purification. The transduction efficiency of the modified vector was analyzed by transduction assay on A549 cells.
Specific labeling of genetically modified vector particles with Alexa488-maleimide
Vector particles (AdFiberCys or AdpIX75Cys; 1 × 1011) were incubated with a 20-fold excess of chemically reactive Alexa488-maleimide over fiber (AdFiberCys) or protein IX (AdpIX75Cys) for 2 hr at room temperature in an argon atmosphere, in a final volume of 200 μl (filled with HEPES buffer, pH 7.2). For AdHexonCys a 15-fold excess of Alexa488-maleimide over hexon was used. The reaction was stopped by incubation for 1 hr with a 10-fold excess of cysteine over Alexa488-maleimide. The labeled vector particles were then purified from unreacted dyes by PD-10 column purification. The transduction efficiency of the modified vector was analyzed by transduction assay on A549 cells and the intracellular trafficking of these particles was analyzed on A549 cells with a confocal laser scanning microscope.
Ligand attachment and labeling with fluorescent dye
Ligand attachment has been described elsewhere (Kreppel et al., 2005; Corjon et al., 2008). In brief, the protein molecules transferrin (Tf ) and bovine serum albumin (BSA) were rendered thiol-reactive with the heterobifunctional cross-linker N-hydroxysuccinimide–polyethylene glycol–maleimide (NHS-PEG-Mal; Nektar Therapeutics [San Carlos, CA], IRIS Biotech [Marktredwitz, Germany]) and purified by automated size-exclusion chromatography on a Superose 6 column (GE Healthcare Biosciences).
After activation with NHS-PEG-Mal and purification by fast protein liquid chromatography (FPLC) the ligands were reacted with Alexa488-TFP (a 5-fold molar excess over protein molecules).
Synthesis of Alexa488-SS-PEG-NHS and Alexa488-SS-PEG-HZN-NHS
For the synthesis of Alexa 488-labeled PEGylation reagents, 1 μmol of ω-2-pyridyldithio–polyethylene glycol–α- succinimidyl ester (OPSS-PEG-NHS; 10 kDa; IRIS Biotech) or ω-2-pyridyldithio–polyethylene glycol–α-carboxypyridylhydrazone–N-hydroxysuccinimide ester (OPSS-PEG-HZN-NHS, synthesized as described by Fella and colleagues, 2008) was mixed with 0.75 μmol of Alexa 488-(2-mercaptoethyl)-amide in dimethylformamide (DMF). The latter had been prepared by mixing Alexa 488–succinimidyl ester at a 1:1 molar ratio with cysteamine in DMF under argon. After 1 hr at 37°C the Alexa-labeled PEG reagents formed were stored at −80°C after snap freezing with liquid nitrogen.
Labeled PEG moieties
The PEG moieties of Alexa488-SS-PEG-NHS and Alexa488-SS-PEG-HZN-NHS (Fella et al., 2008) were investigated for their ability to act bioresponsively. Both molecules contain a disulfide (SS) bond; in addition, the second molecule contains an acid-degradable hydrazone (HZN) bond. The couplings were performed overnight at room temperature under an argon atmosphere.
Labeled PEG moieties were kept in the dark at −80°C. Because extensive PEGylation decreases receptor binding and uptake of the modified particles, titration experiments were performed, allowing the attachment of a sufficient number of PEG molecules while largely maintaining particle infectivity (data not shown). Modification of amino groups with the fluorescent PEG derivatives was performed at pH 8.5 to prevent premature hydrolysis of the hydrazone bonds.
Two-color labeling of adenoviral vectors
For two-color labeling the genetically introduced cysteines of AdHexonCys were first modified with Alexa633-maleimide (15-fold excess over cysteine residues) in argon-saturated HEPES, pH 7.2 (5 to 15 hr). The reaction was stopped by incubation for 1 hr at room temperature with a 5-fold excess of cysteine over Alexa633-maleimide. Subsequently, amino groups on the vector particle surface were labeled with Alexa488-TFP (20- to 60-fold excess over amino groups) at pH 7.2.
Preparation and transduction of cells for confocal laser microscopy
A549 or Hepa 1-6 cells (1 × 105) were seeded onto poly-
For the labeled bioresponsive PEG moieties the transduction protocol was adapted. Twenty-four hours after seeding the cells were transduced with 40,000 pMOI in 1 ml of PBS–10% FCS at 37°C. Fifteen minutes posttransduction the PBS–10% FCS was removed, the cells were washed twice with 2 ml of PBS, and 2 ml of PBS–10% FCS was added. The dishes were put on ice until they were placed in the 37°C incubation chamber of the confocal microscope.
LSM 510 META settings
Live cell imaging was performed with an LSM 510 META confocal microscope equipped with a × 63 water immersion objective (C-Apochromat × 63/1.20 W corr; Carl Zeiss) and with a humidified CO2-controlled incubation chamber.
The Alexa 488 dyes were excited with the argon laser (37% of the 488 nm wavelength laser, and with the laser set to 50% of its maximal power), and the emission was collected with an LP 505 filter (high-pass filter, wavelengths higher than 505 nm). The images were collected mainly on the z axis at the nuclear level, with a stack size of 512 × 512 pixels and 8-bit pixel depth, with two channels (Ch3-T1 fluorescence channel and ChD-T1 transmission light, with calibrated PH2 phase contrast) and as the sum of four images collected in 4–10 sec. The pinhole of the fluorescence channel was set to 1 Airy unit (which defines the optimal settings for the system, in this case 112 μm) and the signal intensity was adjusted from a gain of 1000 to a range of 900 to 950. The 10-μm scale and the merge of the two channels are shown for each image.
The Alexa 633 dye were excited with the helium/neon laser (at 100% of its maximal power) and the emission was collected with a filter for wavelengths between 654 and 718 nm. Multiple track images were taken (ChS1 [red] and Ch2 [green] fluorescence channel and ChD-T1 transmission light). The images were collected mainly on the z axis at the nuclear level with a stack size of 512 × 512 pixels and as the sum of four images collected.
The pinhole of the fluorescence channel was set to 1 Airy unit (in this case for channel ChS1: 180 μm and for channel Ch2: 181 μm) and the signal intensity was adjusted from a gain of 1000 to a range of 900 to 950. The 10-μm scale and the merge of the two or three channels, respectively, are shown for each image.
AxioVision software (Carl Zeiss) was used to take the images. Further analyses was done with LSM Image Browser (Carl Zeiss).
Results
Capsomer-specific fluorescent labeling of adenovirus
Chemical strategies to fluorescently label adenoviral vector particles have so far been based exclusively on the use of amine-reactive fluorescent dyes (Leopold et al., 1998, 2000; Martin-Fernandez et al., 2004). Such dyes are based mainly on N-hydroxysuccinimide ester (NHS) reactive groups and couple readily to capsid surface amino groups, of which about 18,000 are present on the surface of Ad5 virions (O'Riordan et al., 1999). Although it has been shown that such dyes can be used to generate fluorescently labeled Ad particles, the approach suffers from two important shortcomings. First, because of the presence of amino groups in all major capsid proteins, including those responsible for receptor binding and cell entry, there is a significant risk of particle overlabeling, with the consequence of reduced infectivity and labor-intense titration experiments after labeling. Second, the amine-directed labeling approach does not allow specific labeling of certain capsomers or ligands incorporated into the vector particle, making it difficult to analyze the intracellular trafficking of retargeted Ad vectors. Therefore, the development of a labeling strategy that uses a small number of dye molecules coupled specifically to selected positions in the capsomer would be a significant advance.
We have described a capsid modification technology that is based on a combination of genetics and chemistry (Kreppel et al., 2005). Cysteine residues are genetically introduced at defined capsid positions and subsequently used to specifically couple thiol-reactive moieties such as ligands or shielding polymers to the vector particle surface. Here we employed this technology to assess whether capsomer-specific labeling with fluorescent dyes can be performed and would allow us to monitor intracellular trafficking of the modified vector particles. To this end we used the technology to specifically label capsomer fiber, protein IX, and hexon with maleimide-activated (and thus cysteine-reactive) Alexa-based fluorescent dyes and compared the utility of these specifically labeled particles with that of conventionally labeled particles. We reacted the vectors AdFiberCys (cysteine introduced in fiber, 36 cysteines per particle), AdpIX75Cys (cysteine introduced in protein IX, 240 cysteines per particle), and AdHexonCys (cysteine introduced in hexon, 720 cysteines per particle) with a 15- to 20-fold molar excess of thiol-reactive dye over cysteines and purified the vector particles from excess dye by gel-filtration chromatography. In parallel, a vector with a wild-type capsid, AdControl, was labeled with a 20-fold molar excess of amine-reactive Alexa488-TFP over surface amino groups (18,000 groups per particle). After confirming that the labeling itself did not significantly interfere with particle infectivity (flow cytometric analysis and Western blot experiment: see Supplementary Figures 1 and 2 at
Images taken after amine-labeled Ad vector had been in contact with cells for 10 min showed association of the viral particles with the cell surface (Fig. 1A). Images taken after 25 min revealed that the vector particles were internalized, and after 50 min a significant portion of vector particles had accumulated around the nucleus. The kinetics of trafficking were in agreement with previous reports (Greber et al., 1993) and, therefore, the images allowed us to analyze the performance of capsomer-specific labeling (Fig. 1B–D).

Intracellular trafficking of randomly labeled AdControl and capsomer-specific labeled Ad Cys vectors. Trafficking of the various vectors was analyzed by transduction of A549 cells and subsequent confocal laser scanning microscopy. Left: Alexa Fluor 488 fluorescence. Middle: Transmitted light. Right: Merged fluorescence and transmitted light. (
Ad vector specifically labeled at the genetically introduced cysteine residues in the fiber HI loop (36 cysteines per particle) did not exhibit a robust fluorescent signal, either because the number of fluorescent dye molecules per vector particle was too small or because of very early shedding of fiber from the vector particle, which has been reported to partially occur even before cell entry (Nakano et al., 2000). In contrast, vector particles specifically labeled at the C terminus of protein IX generated fluorescent signals that were reliably detectable on the cell surface 10 min after vector addition and warming of the cells to 37°C, and showed regular vector trafficking routes and kinetics (Fig. 1C). A comparison of signal intensities for the amine-labeled AdControl and protein IX-labeled AdpIX75Cys vectors revealed a lower signal intensity for the latter, suggesting that an overall smaller number of dye molecules were coupled. In fact, we demonstrated in a previous report that coupling of small maleimide-activated molecules to cysteine residues genetically introduced at protein IX can be performed with an efficiency of >90% (Corjon et al., 2008). Therefore, at most 230 dye molecules were coupled to the AdpIX75Cys vector particles, whereas amine-directed PEGylation approaches are known to modify up to 10,000 amine groups per vector particle (O'Riordan et al., 1999), which in our case could be an underestimation because of the smaller size and hydrophilicity of the dye molecules. These results demonstrate that capsomer-specific labeling at protein IX results in sufficiently strong signals with a minimal amount of dye molecules per vector particle. Because, as far as is known to date, protein IX is neither involved in receptor binding nor directly involved in nuclear trafficking processes, the capsomer-specific labeling strategy using protein IX does not bear the risk of interference with vector infectivity. Nevertheless, because protein IX is known to traffic, together with the capsid remnants that escape from early endosomes, to the nucleus (Rosa-Calatrava et al., 2001), we could readily detect trafficking of the vector particles and their accumulation around the nucleus (30 min; Fig. 1C).
Specific labeling of the hexon capsomer with maleimide-activated Alexa 488 dye also revealed regular particle trafficking kinetics (Fig. 1D). As expected, the signal intensity of labeled AdHexonCys was stronger than that of labeled AdpIX75Cys. Analysis of the number of molecules coupled to hexon revealed that 80% of the hexon monomers could be modified by this strategy, which corresponds to 550–600 dye molecules per vector particle (data not shown). Importantly, the regular trafficking of vector particles labeled at the hypervariable region 5 (HVR5) of hexon was not affected: within 50 min the majority of vector particles reached the nuclear membrane.
These results indicate that using the genetically introduced cysteine residues at protein IX or hexon for specific coupling of maleimide-activated fluorescent dyes is sufficient to generate fluorescent signals that can readily be tracked by confocal laser scanning microscopy. Thus, capsomer-specific labeling without the risk of particle overlabeling is feasible and may allow for analysis of particles decorated with various ligands or shielding moieties without interfering with shielding efficiency or ligand performance. In addition, specific labeling at protein IX or hexon may allow for the generation of particles with two different fluorescent dyes, one attached specifically to protein IX or hexon and the other attached to amino groups on the vector particle surface. Because such a labeling strategy is crucial for the analysis of, for example, bioresponsive bonds used to attach ligands or shielding moieties to the vector particles, we performed two-color labeling experiments.
Direct two-color labeling of Ad vector particles
To combine the genetic–chemical labeling technology with conventional amine-directed particle labeling we used the AdHexonCys vector, because it exhibited stronger fluorescence after specific labeling when compared with AdpIX75Cys. AdHexonCys particles were first labeled with maleimide-activated red fluorescent Alexa 633 dye, specifically at hexon HVR5, and next amino groups were labeled all over the capsid with Alexa488-TFP. As a control we used vector particles labeled only at hexon with the red fluorescent dye (Fig. 2A). The images shown in Fig. 2A demonstrate that specific labeling of hexon with the red fluorescent dye (which exhibits weaker fluorescence compared with green fluorescent dyes) generates signals that can be detected readily. Overlays of red and green fluorescence for the images obtained with two-color-labeled particles (Fig. 2B) revealed that the intracellular trafficking was the same as for particles labeled with only one dye, suggesting that the biology of the vector particles was not altered because of labeling. These experiments demonstrated that a two-color-labeling strategy is feasible when combining capsomer-specific and capsomer-nonspecific labeling.
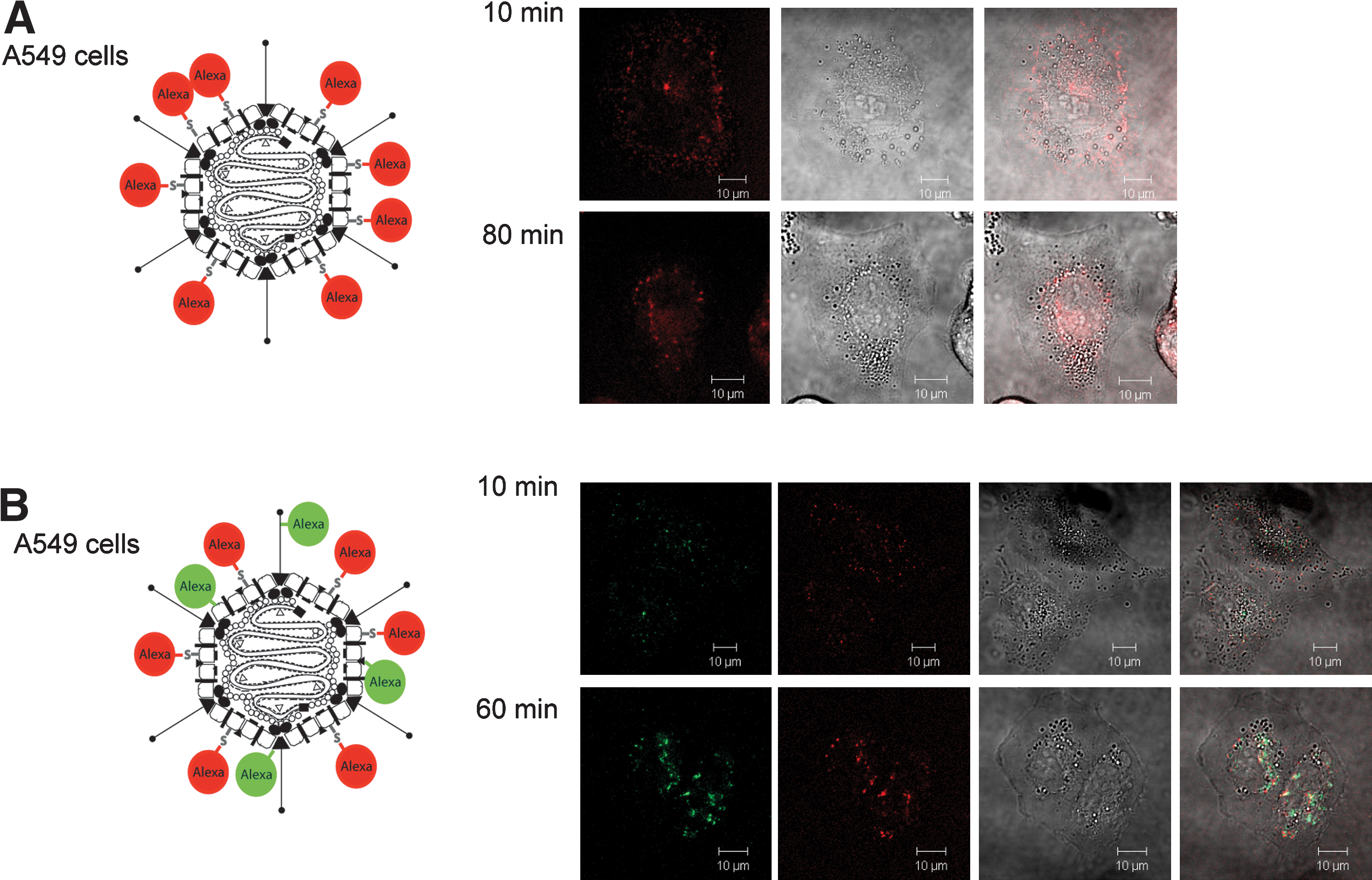
Two-color labeling of adenoviral vectors with red and green fluorescent dyes, and investigation of trafficking behavior. Trafficking of the various vectors was analyzed by transduction of A549 cells and subsequent confocal laser scanning microscopy. Panels ( from left to right): Alexa Fluor 488 fluorescence for two-color-labeled vectors; Alexa Fluor 633 fluorescence; transmitted light; merge (fluorescence and transmitted light). (
Indirect capsomer-specific labeling of Ad vector particles with fluorescently labeled ligands
As outlined previously, several groups have shown that the position of ligands on the capsid can determine the success of targeting. To analyze ligand position-dependent effects we attempted to use fluorescently labeled ligands specifically coupled to various capsomers for analysis of intracellular vector trafficking. Because maleimide-activated ligands can be covalently attached to the vector particle surface (Kreppel et al., 2005; Corjon et al., 2008), analogous to the fluorescent dyes, we attempted to couple fluorescently labeled ligands in order to generate targeted fluorescent vector particles. First, amino groups of the maleimide-activated model ligand transferrin (Tf ) were labeled with an amine-reactive Alexa 488 dye, purified from excess dye and coupled to cysteine residues of fiber, protein IX, and hexon. As a control molecule that has a similar size compared with transferrin we used maleimide-activated and amino-labeled bovine serum albumin (BSA) and coupled it to the various vector particles. We thus achieved capsomer-specific indirect capsid labeling that allowed analysis of the intracellular fate of ligand-decorated vector particles (Figs. 3 and 4). In addition to analysis by confocal fluorescence microscopy we performed flow cytometric analysis of ligand-decorated, unlabeled vectors 24 hr posttransduction to analyze transduction in terms of transgene expression (bar graphs; Figs. 3 and 4).

Correlation of flow cytometric data and LSM experiments of labeled ligand-decorated Ad particles: Transferrin. Flow cytometric data of unmodified and transferrin-decorated adenoviral vector (bar graphs) were compared with the results of LSM experiments with labeled transferrin-coupled vector (images). (

Correlation of flow cytometric data and LSM experiments of labeled BSA-decorated Ad particles. Flow cytometric data of unmodified and BSA-decorated adenoviral vector (bar graphs) were compared with the results of LSM experiments with labeled BSA-coupled vector (images). (
To analyze intracellular trafficking, adherent and comparatively large A549 cells were used. To confirm the results obtained on A549 cells an additional cell line was chosen: Hepa 1-6 cells, a mouse hepatoma cell line.
Whereas transferrin coupled to fiber led to a 10-fold increase in transgene expression in the model cell line K562, which is high in Tf receptor expression but low in CAR expression, the coupling of Tf to protein IX and to hexon significantly decreased transgene expression in these cells (bar graphs; Fig. 3A–C). These results corroborated the findings by Campos and colleagues, who suggested that only the fiber capsomer allows for robust vector targeting with various ligands (Campos and Barry, 2006).
Analysis by confocal microscopy revealed that, despite efficient uptake of all transferrin-decorated particles into the target cells, those particles that carried Tf at protein IX or hexon exhibited altered intracellular trafficking and the majority did not reach the nuclear envelope within 45–50 min (Fig. 3B and C).
The data obtained by laser scanning microscopy (LSM) and by flow cytometric analysis of transgene expression were in good agreement and suggest that vector particles modified at protein IX or hexon with Tf were not able to deliver their genomes to the nuclei of cells with the same efficiency as unmodified vector particles. To analyze whether the effect of Tf on protein IX or hexon is specific for Tf, we performed the same set of experiments with BSA as a control (Fig. 4). Interestingly, BSA-decorated particles exhibited the same altered trafficking pattern as Tf-decorated particles, suggesting that the decreased transduction efficiency observed with Tf coupled to protein IX or hexon is not a phenomenon specific to Tf.
The flow cytometry data shown in the bar graphs in Figs. 3 and 4 are based on quantification of EGFP fluorescence 24 hr posttransduction. The decreased transduction (compared with unmodified control vectors), as we observed it for vector particles modified at the capsomer pIX or hexon with transferrin and BSA, could be correlated with aberrant intracellular trafficking of these vector particles (Fig. 3B and C and Fig. 4A and B).
Direct fluorescent labeling to study the dynamic properties of bioresponsive bonds for Ad vector targeting
We attempted to evaluate the utility and to study the dynamics of two different bioresponsive bonds: disulfide bonds (SS) and hydrazone bonds (HZN) in the context of adenoviral vectors. Disulfide bonds may be cleaved in the endosomes or cytosol because of the reducing environment in these cell compartments. Hydrazone bonds respond to changes in pH and are quickly hydrolyzed on acidification in the endosomal uptake compartment. Our strategy to analyze these bonds was based on the two-color labeling strategy shown in Fig. 2. We first specifically labeled the hexon capsomer with Alexa633-maleimide in order to be able to monitor particle trafficking to the nucleus. After carefully analyzing the detargeting capacity of the PEG molecules by well-established methods (Wortmann et al., 2008) two different fluorescent PEGs were coupled to amino groups on the vector particle surface (Fig. 5) in a way that retains particle infectivity to a significant degree and thus enables analysis of intracellular trafficking: Alexa488-SS-PEG-NHS (Fig. 5A) contains a potentially redox-degradable disulfide (SS) bond. Alexa488-SS-PEG-HZN-NHS contains, in addition to the disulfide bond, a pH-sensitive hydrazone (HZN) bond (Walker et al., 2005; Fella et al., 2008). If the PEG moieties cannot be cleaved from the adenoviral vector particles, a sustained colocalization of red and green dyes would be expected over the whole time of observation. In the case of cleavage the dyes would be colocalized in the beginning, but with ongoing trafficking a different behavior of cleaved (green) dye and still vector particle-coupled (red) dye should be observed.
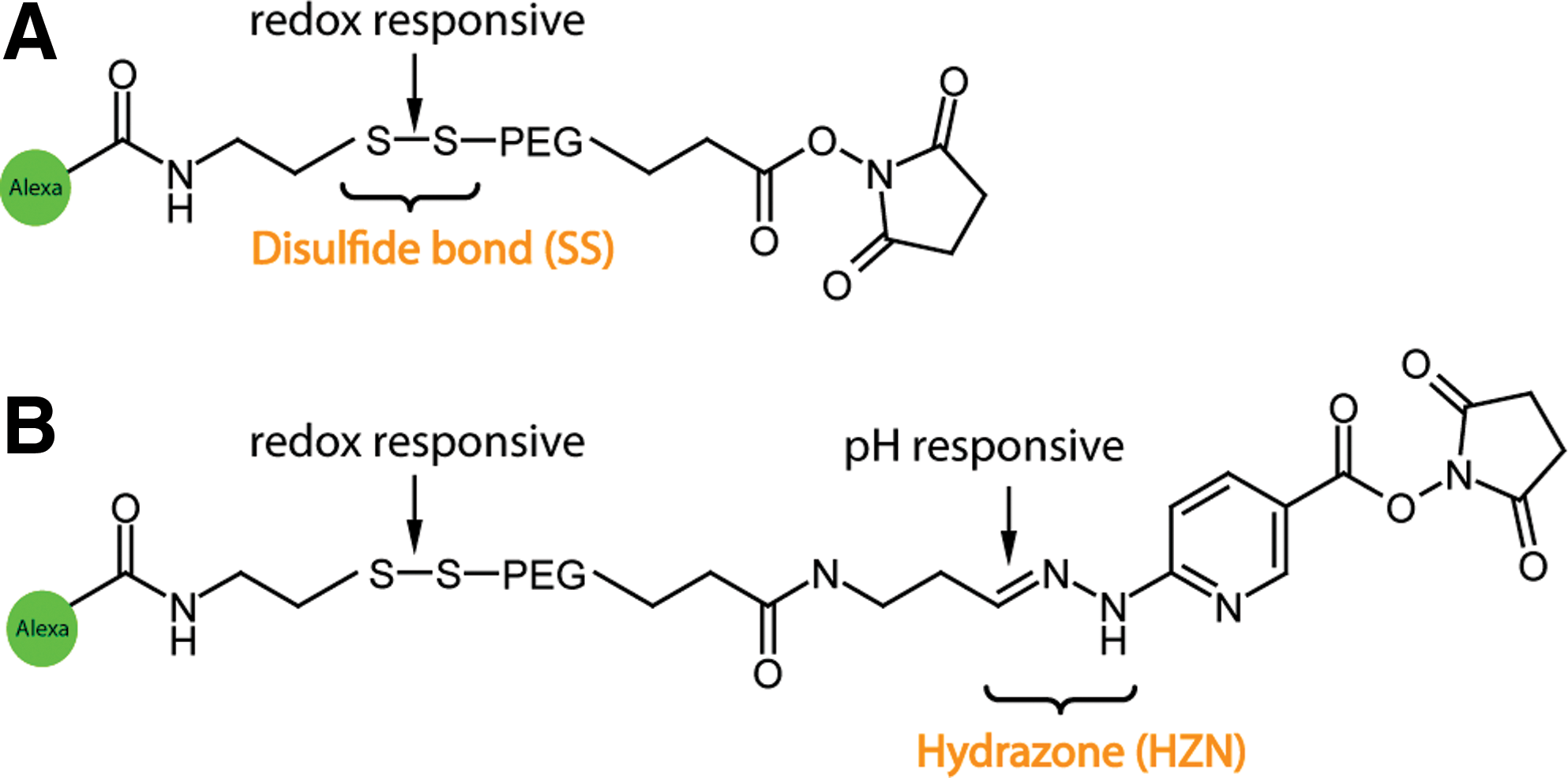
Structure of bioresponsive ligands. (
AdHexonCys labeled with Alexa488-SS-PEG-NHS (Fella et al., 2008) revealed a relatively low signal intensity and impaired trafficking was observed in the LSM experiment (Fig. 6A). The low signal intensity likely reflects slightly decreased infectivity due to vector PEGylation.
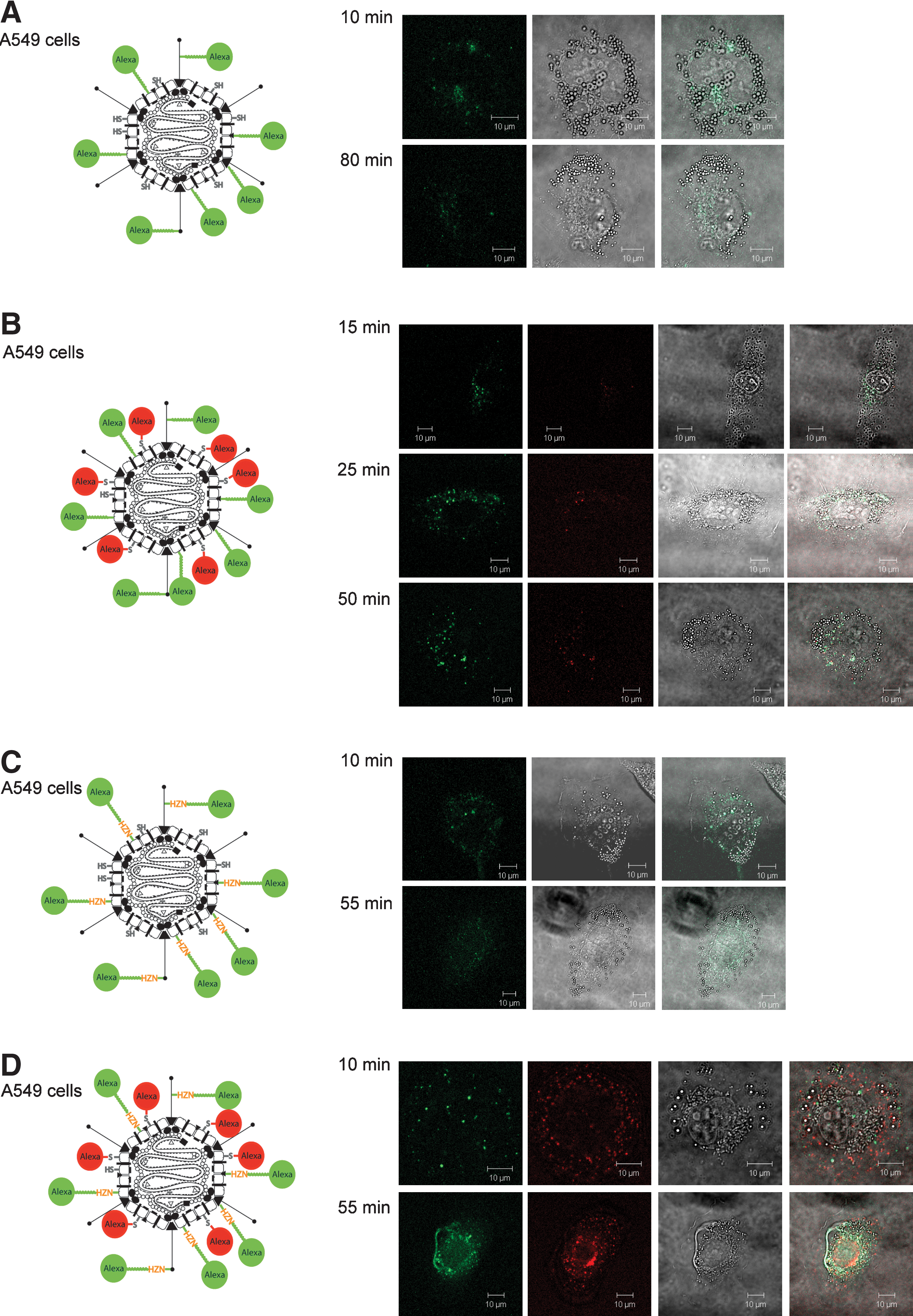
Dynamic behavior of AdHexonCys vectors with modified bioresponsive bonds on A549 cells. Trafficking of the various modified vectors was analyzed by transduction of A549 cells and subsequent confocal laser scanning microscopy. Panels ( from left to right): Alexa Fluor 488 fluorescence for two-color-labeled vectors; Alexa Fluor 633 fluorescence; transmitted light; merge (fluorescence and transmitted light). (
The two-color (Alexa633-maleimide and Alexa488-SS-PEG-NHS)-labeled vector also showed in late observation images the colocalization of red and green fluorescent dyes throughout the cell (Fig. 6B), indicating that particle trafficking to the nuclear membrane was inefficient within the observation time. These results suggested that the disulfide bond of the Alexa488-SS-PEG-NHS molecule was not cleaved completely during the observation time (within 80 min) and was relatively stable in the cells tested.
In contrast, with the Alexa488-SS-PEG-HZN-NHS-labeled AdHexonCys vector we observed after a certain time span the green signal becoming more and more diffuse (Fig. 6C). This suggested the hydrolysis of the hydrazone bond present in the molecule. To be able to detect trafficking of the particles two-color labeling was performed. Two-color (Alexa633-maleimide and Alexa488-SS-PEG-HZN-NHS)-labeled vector particles exhibited the same diffuse signal of the green dye after about 30 min and at the same time showed regular particle trafficking to the nuclear membrane as evidenced by the red color dye on the hexon capsomer (Fig. 6D). Interestingly, trafficking apparently occurred with a slight delay compared with two-color (Alexa633-maleimide and Alexa488-TFP)-labeled AdHexonCys vector.
The observation made with two-color-labeled, potentially bioresponsive adenoviral particles in A549 cells was validated in Hepa 1-6 cells (see Supplementary Fig. 3 at
Although residual colocalization of green and red dyes in Fig. 6D suggests that not all HZN bonds became cleaved within the observation time, HZN bonds appear to be favorable over SS bonds for the generation of bioresponsive bonds between Ad vector particles and coupling moieties. However, our findings may not be generalized because of differences in the intracellular redox status of various cell lines. Our findings suggest, rather, that HZN bonds can be a suitable tool for bioreversibility—when the target cells support their hydrolysis.
Discussion
The data presented here provide an improved labeling strategy for adenoviral gene transfer vectors. Although Ad vector particles randomly labeled at amino groups of the vector particle surface have been used successfully in several studies (Leopold et al., 1998, 2000; Martin-Fernandez et al., 2004), they show several limitations for precise analysis of the intracellular fate of Ad particles. First, because of the high abundance of amino groups on the Ad capsid there is a significant risk of overlabeling, with the consequence of reduced infectivity of the vector particles. Labor-intense titration experiments must then be performed in order to determine the amount of dye that can be coupled to amino groups without affecting particle infectivity. Second, because amino groups are present in all major outer capsid proteins of Ad, capsomer-specific effects are difficult to analyze by this random labeling strategy. Last, ligand motifs that are inserted genetically into the Ad capsid for targeting purposes are usually based on relatively small peptides that are likely to be modified by the amine-based labeling strategy. This labeling of the peptide ligands may well lead to reduced receptor affinities. The labeling strategy we present here is based on the introduction of specific amino acid residues at defined capsid positions that can be specifically labeled with maleimide-based fluorescent dyes. This strategy dramatically reduces the number of dye molecules needed for labeling: a 20-fold excess of Alexa488-TFP over 18,000 amino groups on the vector surface requires in fact more than 30-fold more dye molecules than for the 15-fold excess of Alexa488-maleimide over 720 cysteine residues. Furthermore, the maleimide-based dyes have a significant longer half-life compared with NHS (or TFP) in aqueous solution and this leads therefore to a higher reproducibility.
We could show that the capsomer protein IX and hexon can be specifically labeled with such dyes without affecting particle infectivity and that the labeled particles yield fluorescent signals that can be detected readily by confocal laser scanning microscopy. Furthermore, we could demonstrate that indirect labeling of the vector particles with large fluorescently tagged ligands is feasible and that this technology aids in deciphering the intracellular trafficking of targeted Ad vector particles. In fact, the data we obtained by LSM were in agreement with the data obtained by analysis of transgene expression.
In a previous study we showed that the ligand RAP (receptor-associated protein) coupled to AdpIX75Cys vector led to an increase in transduction efficiency in flow cytometric analysis of CHO-K1 cells, which are known to carry the LRP (low-density lipoprotein receptor-related protein) receptor on the cell surface. In the LSM experiment we observed rapid delivery to the nuclear membrane (further results described elsewhere; Corjon et al., 2008). RAP uses a histidine switch to regulate its interaction with LRP (Lee et al., 2006). After escorting the receptor to the Golgi, RAP dissociates from the receptor. The histidines were protonated as a consequence of pH changes and this leads to a modulation in binding/release of RAP from LRP receptor.
Taking this pH-dependent switch as a paradigm we used a bioresponsive labeled PEG moiety to investigate the influence of reversibly coupled ligands/shielding moieties on the trafficking behavior of vector particles.
In addition, the labeling strategy as presented here allows expansion of the random amine labeling of vector particles to generate particles tagged with two different fluorescent dyes. We have demonstrated that such particles can be used to analyze the performance of bioresponsive bonds used to attach ligands or shielding moieties to the vector particle surface. We believe that this is of high significance for rational targeting approaches because it has now been shown by several groups that targeting efficiency may not only be ligand dependent but may also depend on the position of the ligand on the capsid.
Coupling of bioresponsive PEG moieties in combination with a specific coupling of the red dye to hexon allowed us to investigate the trafficking of both the PEG moiety and the Ad vector in an independent manner.
Our finding that disulfide bonds do not become reduced within the observation time is in agreement with observations by Saito and colleagues (2003). It is believed that reduction of disulfides in the endosome may be limited to certain cell types and proteins (Wolff and Rozema, 2008). The fact that we could not observe significant reduction after endosomal release in the reducing environment of the cytosol may be explained by the limited observation time. If the reduction of disulfide bonds is slow compared with hydrolysis of HZN bonds only some of the disulfide bonds may have become reduced within 80 min. These results clearly indicate that the HZN bonds are favorable for the generation of bioresponsive shields or for the generation of reversible linkages between vector particles and ligands and may be used to overcome the limitations of intracellular trafficking processes as shown for the ligand Tf.
In summary, we present a simple and straightforward technology for specific chemical labeling of certain capsomers of adenoviral vectors. This technology is compatible with ligand-decorated vectors and with polymer-shielded vectors and allows for the generation of vector particles labeled with two different fluorescent dyes. Further, this technology was employed to analyze the use of bioresponsive bonds (hydrazone bonds and disulfide bonds) in the context of adenoviral vector particles. The results indicate that pH-responsive hydrazone bonds are favorable for the generation of chemically modified dynamic Ad vector particles to overcome the hurdles in the trafficking process. This finding may guide further rational developments of targeted and shielded Ad vectors for in vivo gene delivery. Last, we believe that the technology presented here may be used to generate vector particles labeled with, for example, quantum dots that allow trafficking analysis in vivo.
Footnotes
Acknowledgments
The authors thank Frank Dolp for technical help with the confocal laser scanning microscope (Carl Zeiss LSM 510 META). This work was supported by the German Research Foundation (SPP1230) and by the European Union (FP6-2003-GIANT-Project 512087).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.