
Abstract
The expression of two or more genes from a single viral vector has been widely used to label or select for cells containing the transgenic element. Identification of the foot-and-mouth disease virus (FMDV) 2A cleavage peptide as a polycistronic linker capable of producing equivalent levels of transgene expression has greatly improved this approach in the field of gene therapy. However, as a consequence of 2A posttranslational cleavage the upstream protein is left with a residual 19 amino acids from the 2A sequence on its carboxy terminus, and the downstream protein is left with an additional 2 to 5 amino acids on its amino terminus. Here we have assessed the functional consequences of the FMDV 2A cleavage motif on two secreted proteins (interleukin [IL]-2 and transforming growth factor [TGF]-β) when expressed from a retroviral bicistronic vector. Whereas IL-2 expression and function were found to be unaffected by the 2A motif in either orientation, functional expression of secreted TGF-β was significantly abrogated when the transgene was expressed upstream of the 2A sequence. We believe this is a consequence of aberrant cleavage and intracellular trafficking of the TGF-β polyprotein. These results highlight that to achieve functional expression of secreted proteins consideration must be taken of the transgenic protein's posttranslational modification and trafficking when using 2A-based bicistronic cassettes.
Introduction
A consequence of 2A cleavage is the retention of 19 residual amino acids at the carboxy terminus of the upstream protein and 2–5 amino acids at the amino terminus of the downstream protein. To our knowledge analysis of cleavage or the functional consequences of the addition of these short peptides on the translated proteins has not been reported. The majority of published studies using 2A peptides have involved the expression of intracellular gene products such as transcription factors or cytoplasmic proteins, which, in general, do not require extensive posttranslational modifications. In this study we have investigated the effect of transgene position within the FMDV 2A cassette on the expression of two secreted cytokines, interleukin (IL)-2 and transforming growth factor (TGF)-β, that are subject to substantial posttranslational modification, in comparison with the reporter gene encoding green fluorescent protein (GFP) (Hardy et al., 2007; Ferguson et al., 2009; Molloy et al., 2009). Murine leukemia virus (MLV)-based retroviral vectors expressing these two therapeutically significant secreted proteins were generated and the expression and functionality of each gene product were determined when expressed both upstream and downstream of the 2A cassette.
Materials and Methods
Generation of vectors and general reagents
Full-length cDNAs of murine IL-2 and TGF-β were amplified from BALB/c RNA and cloned either upstream (rKat.I2G and rKat.T2G, respectively) or downstream (rKat.G2I and rKat.G2T, respectively) of an FMDV 2A cleavage site in conjunction with GFP (G) (Fig. 1A). Cell line 293T was grown in Dulbecco's modified Eagle's medium (DMEM) Glutamax (Invitrogen, Paisley, Scotland) and CTLL-2 and NIH-3T3 cells were grown in RPMI 1640 (Lonza, Braine-l'Alleud, Belgium), all supplemented with 10% fetal calf serum (FCS). Murine dermal fibroblasts (MDFs) were cultured as described previously (DiPersio et al., 1995). Recombinant human IL-2 (rhIL-2) was obtained from Chiron Pharmaceuticals (Amsterdam, The Netherlands) and used at a concentration of 100 U ml–1.
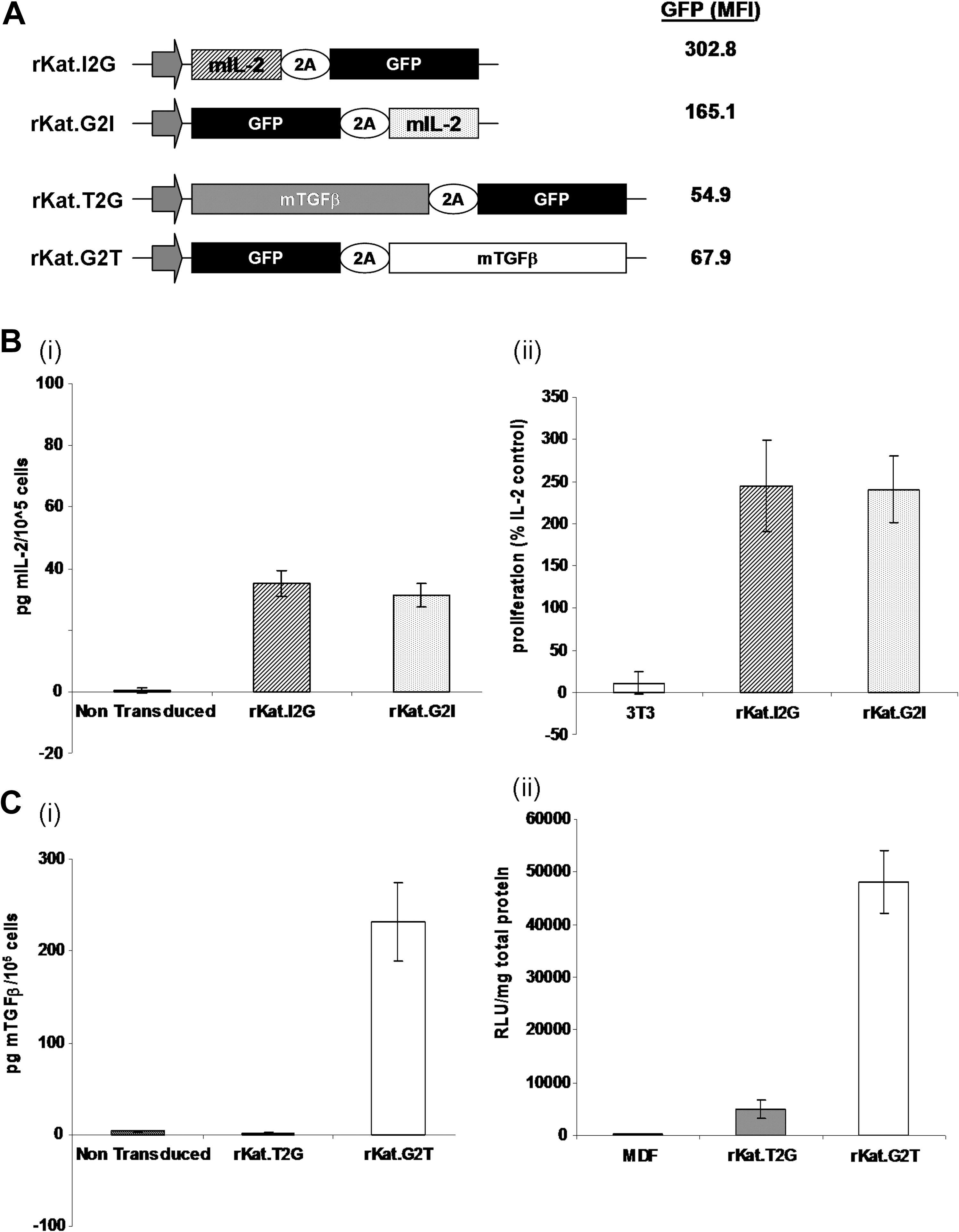
Expression and function of TGF-β, but not IL-2 and GFP, is position dependent in the FMDV 2A bicistronic cassette.
Transfecting 293T cells and transduction of target cells
Retroviral supernatants were generated from transiently transfected 293T cells for each vector as previously described (Finer et al., 1994). Target cells were transduced at a ratio of 105 cells/ml virus by centrifugation at 1200 × g for 180 min in the presence of Polybrene (6 μg ml–1). Cells were then grown for 72 hr before analysis. Transfected cells were sorted by fluorescence-activated cell sorting (FACS) for GFP-positive cells, with all samples being >95% positive postsorting (data not shown). When conditioned medium was analyzed for the expression of cytokine, medium was changed 18 hr before harvest to serum-free medium supplemented with 1 × B27 (Invitrogen).
Enzyme-linked immunosorbent assay for IL-2 and TGF-β
Supernatants were analyzed for IL-2 or TGF-β by enzyme-linked immunosorbent assay (ELISA), using matched antibody pairs. TGF-β (MAB-1835) and IL-2 (JES6-1A12/5H4) antibodies were obtained from R&D Systems (Abingdon, UK) and BD Pharmingen (Cowley, UK), respectively. TGF-β samples were prepared by acid (0.175 N) activation for 10 min at room temperature, followed by neutralization. Samples were added at a range of dilutions in RPMI medium to ensure that they lay within the limit of sensitivity of the assay (31.25 pg/ml).
CTLL-2 survival assay and conditioned media
CTLL-2 cells were grown for 48 hr in RPMI plus 10% FCS in the absence of IL-2 and then either conditioned medium from NIH-3T3 cells, transduced with rKat.I2G or rKat.G2I 72 hr earlier, or rhIL-2 was added. Proliferation was then measured 24 hr later by means of a WST-1 assay (Roche, Basel, Switzerland) as per the manufacturer's instructions.
Luciferase assay with TGF-β-responsive element in MDFs
A lentiviral vector was generated to express the firefly luciferase (Luc) gene under the control of the CAGA(12) SMAD3-responsive enhancer element, specific to TGF-β activation, and a murine minimal promoter (Dennler et al., 1998). This was used to transduce primary MDFs to generate CAGA(12)-Luc/MDF cells. MDFs transduced with the TGF-β-carrying retroviral vectors (multiplicity of infection [MOI], 50) were mixed 1:1 with CAGA(12)-Luc/MDF cells and Luc expression was assayed 72 hr later.
Western blotting
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer containing 1% Nonidet P-40 (NP-40). Protein concentrations were assessed using the Bio-Rad protein estimation assay (Bio-Rad, Herts, UK). Twenty micrograms of total protein was loaded per well mixed 1:1 with reducing or nonreducing loading buffer and boiled for 5 min or left untreated, respectively. All lysates were separated on 10% sodium dodecyl sulfate (SDS)–polyacrylamide gels, transferred to Hybond nitrocellulose membrane (Amersham Biosciences/GE Healthcare, Little Chalfont, UK), and probed for GFP (1:5000 dilution of goat anti-GFP and 1:5000 dilution of donkey anti-goat horseradish peroxidase [HRP]-conjugated IgG [Santa Cruz Biotechnology, Santa Cruz, CA]), mIL-2 (1:250 dilution of rat anti-mIL-2 [R&D Systems] and a 1:1000 dilution of rabbit anti-rat HRP-conjugated IgG [BD Pharmingen]), or TGF-β (1:1000 dilution of rat anti-mTGF [R&D Systems] and 1:1000 dilution of rabbit anti-rat HRP-conjugated IgG) diluted in Tris-buffered saline (TBS)–Tween/5% skimmed milk powder. Bands were resolved with an enhanced chemiluminescence (ECL) Western blotting detection kit (Amersham Biosciences/GE Healthcare) according to the manufacturer's instructions and the membranes were analyzed by exposure to Kodak X-ray film (Carestream Health, Rochester, NY).
Confocal fluorescence microscopy and intracellular staining
To further characterize intracellular localization, high-power confocal immunofluorescence colocalization was performed with GFP, 4′,6-diamidino-2-phenylindole (DAPI), and a range of intracellular markers on NIH-3T3 cells, using a TCS-SP2 confocal imaging spectrophotometer system (Leica, Wetzlar, Germany) and a × 63 oil immersion objective.
Results and Discussion
Position-dependent expression of transgenes
To determine the effect of expression from the FMDV 2A cassette on secreted proteins NIH-3T3 cells were transduced with MLV-based rKat retroviral vectors expressing two therapeutically significant secreted proteins: IL-2 (I) and TGF-β (T). Each transgene was positioned either upstream (rKat.I2G; rKat.T2G) or downstream (rKat.G2I; rKat.G2T) of the FMDV 2A cleavage site in conjunction with GFP (G). GFP expression was determined 72 hr posttransduction with similar expression levels seen in all four experimental groups (mean ± SEM: rKat.I2G, 51 ± 3%; rKat.G2I, 49 ± 5%; rKat.T2G, 45 ± 5%; rKat.G2T, 51 ± 4%; n = 4). Furthermore, GFP mean fluorescence intensity (MFI) values did not exhibit position variability in either construct set, although there was a 3- to 6-fold higher MFI in the IL-2 constructs compared with the equivalent TGF-β constructs (Fig. 1A).
In agreement with previous studies, expression of the GFP reporter gene was not position dependent, so transduced cells were sorted and selected on the basis of GFP expression and used for all further analyses. Conditioned medium from cells transduced with the four constructs was collected 72 hr after replating and 24 hr after culture in chemically defined medium to assay for secreted transgene expression. High levels of IL-2 protein were detected in conditioned medium from both rKat.I2G and rKat.G2I cells by ELISA, with no significant difference between the two expression constructs when normalized against GFP MFI to account for variation in relative integrations (rKat.I2G, 35.2 pg of mIL-2 per 105 cells; rKat.G2I, 31.3 pg of mIL-2 per 105 cells; n = 3) (Fig. 1B, panel i). Conditioned medium was used to stimulate the IL-2-dependent cell line CTLL-2 (Gillis and Smith, 1977). The proliferation rate of the CTLL-2 cell line increased equally in response to conditioned medium from both rKat.I2G- and rKat.G2I-transduced NIH-3T3 cells, showing that IL-2 produced from either orientation within the 2A vector was functional (Fig. 1B, panel ii).
In stark contrast, we detected no TGF-β in the conditioned medium from rKat.T2G-transduced cells whereas rKat.G2T-transduced cells produced >230 ± 41 pg of secreted TGF-β per 105 GFP-positive cells (±SEM; n = 6 from two independent experiments) (Fig. 1C, panel i). MDF-Smad2/3-Luc cells treated with rKat.G2T-conditioned medium showed a 2460-fold increase in luciferase expression over background, thereby confirming that the secreted TGF-β protein was also functional. However, as suggested by the ELISA analysis, conditioned medium from the rKat.T2G-transduced NIH-3T3 cells failed to induce any significant increase in luciferase expression above background in the MDF-Smad2/3-Luc cells (Fig. 1C, panel ii).
Processing of FMDV 2A proteolytic peptide
To investigate the efficiency of posttranslational 2A cleavage for all four vectors, transduced NIH-3T3 cell lysates were immunoblotted for each expressed protein. When blotting for IL-2 we observed bands corresponding to the predicted sized products (IL-2-2A, 19 kDa; 2A-IL-2, 17 kDa) in both the rKat.I2G and rKat.G2I constructs; however, the expression levels of IL-2 appeared higher in the rKat.I2G lysates (Fig. 2A). This was consistent with our GFP FACS data, which showed a higher GFP MFI in these cells and could be explained by higher insertion levels. Interestingly, some evidence of an uncleaved product (47-kDa band of a consistent size for uncleaved IL-2-2A-GFP proprotein detected by both the IL-2 and GFP antibodies) was observed in the rKat.I2G lysates (Fig. 2A). This did not appear to have a significant effect on relative expression or function of the IL-2 when the two constructs were compared (Fig. 1B, panels i and ii); this may be because the uncleaved IL-2-2A-GFP is still secreted and biologically active. Corresponding GFP blots corroborated the expression and cleavage data from the IL-2 blots.
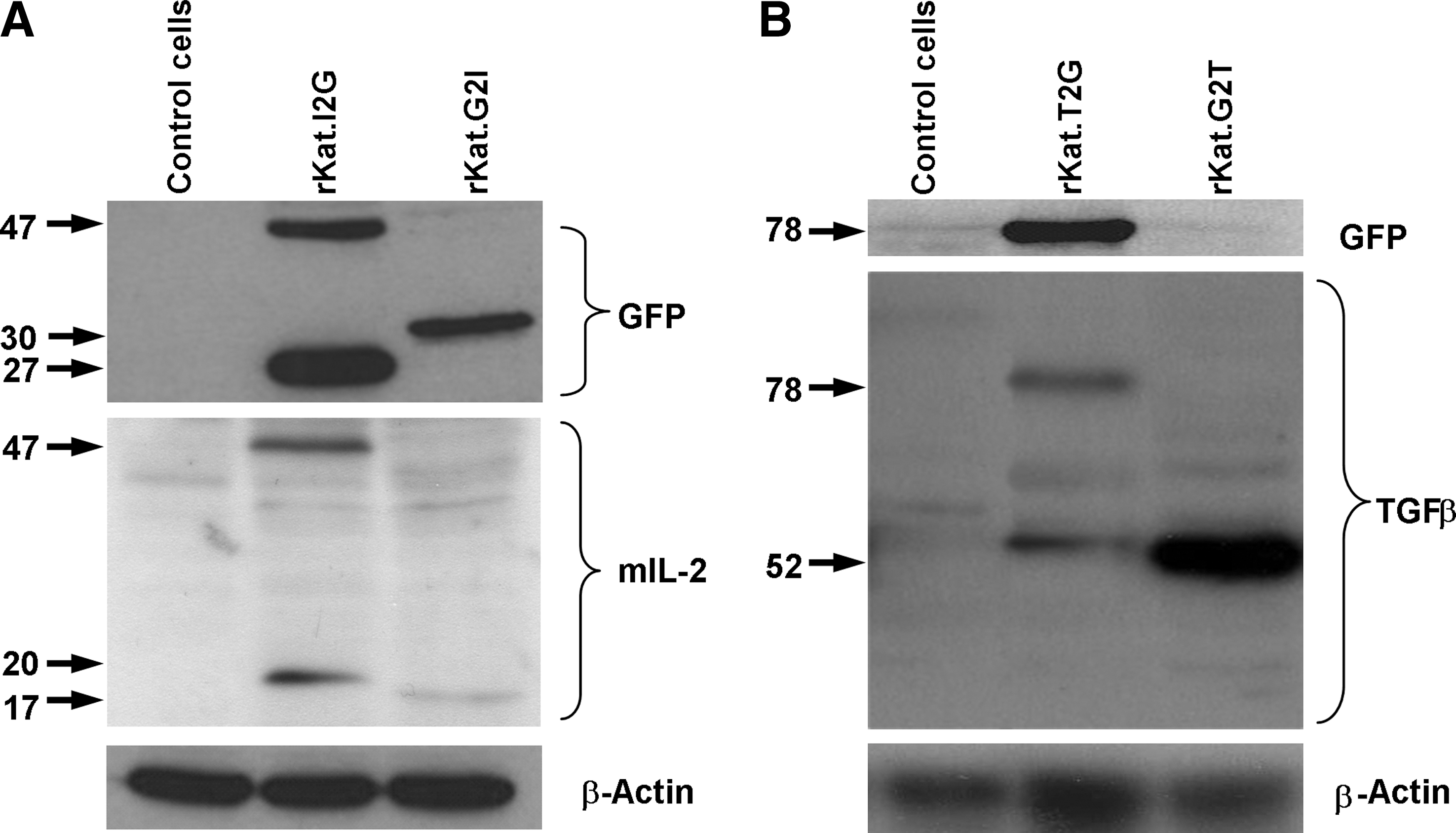
Cleavage of the bicistronic proprotein from a 2A cassette. NIH-3T3 cells were transduced with the rKat retroviral constructs and following FACS sorting lysed and analyzed by Western blot.
In lysates blotted for the presence of TGF-β there was a strong 50-kDa band present in G2T-transduced cells, consistent for the heterodimeric TGF-β protein compared with the nonfunctional rKat.T2G-transduced cells. In these cells there was also a strong 78-kDa immunoreactive band evident in lysates that was detected with both the TGF-β- and GFP-specific antibodies (Fig. 2B). This was of a size consistent for the complete TGF-β-2A-GFP proprotein, suggesting inefficient proteolytic cleavage. Interestingly, both TGF-β immunolabeled bands from the T2G lysate were weak as compared with that seen in the G2T lysate (loading normalized for total protein as evidenced with β-actin). Given the similar levels of GFP marker gene expression from both TGF-β constructs these observations suggest that the noncleaved protein may undergo intracellular degradation.
Cellular localization of expressed transgenes
To explore the intracellular fate of the uncleaved proprotein we transduced cells with all four constructs and analyzed them by high-power confocal fluorescence microscopy. NIH-3T3 cells transduced with the rKat.I2G and rKat.G2I constructs showed uniform cytoplasmic expression, typical for GFP and indicative of efficient cleavage (Fig. 3A and B). NIH-3T3 cells (Fig. 3D) and MDFs (Fig. 3F) transduced with rKat.G2T constructs again showed uniform cytoplasmic expression. In contrast, both NIH-3T3 cells (Fig. 3C) and MDFs (Fig. 3E) transduced with the inefficiently cleaved T2G construct showed punctate vesicular-like staining.
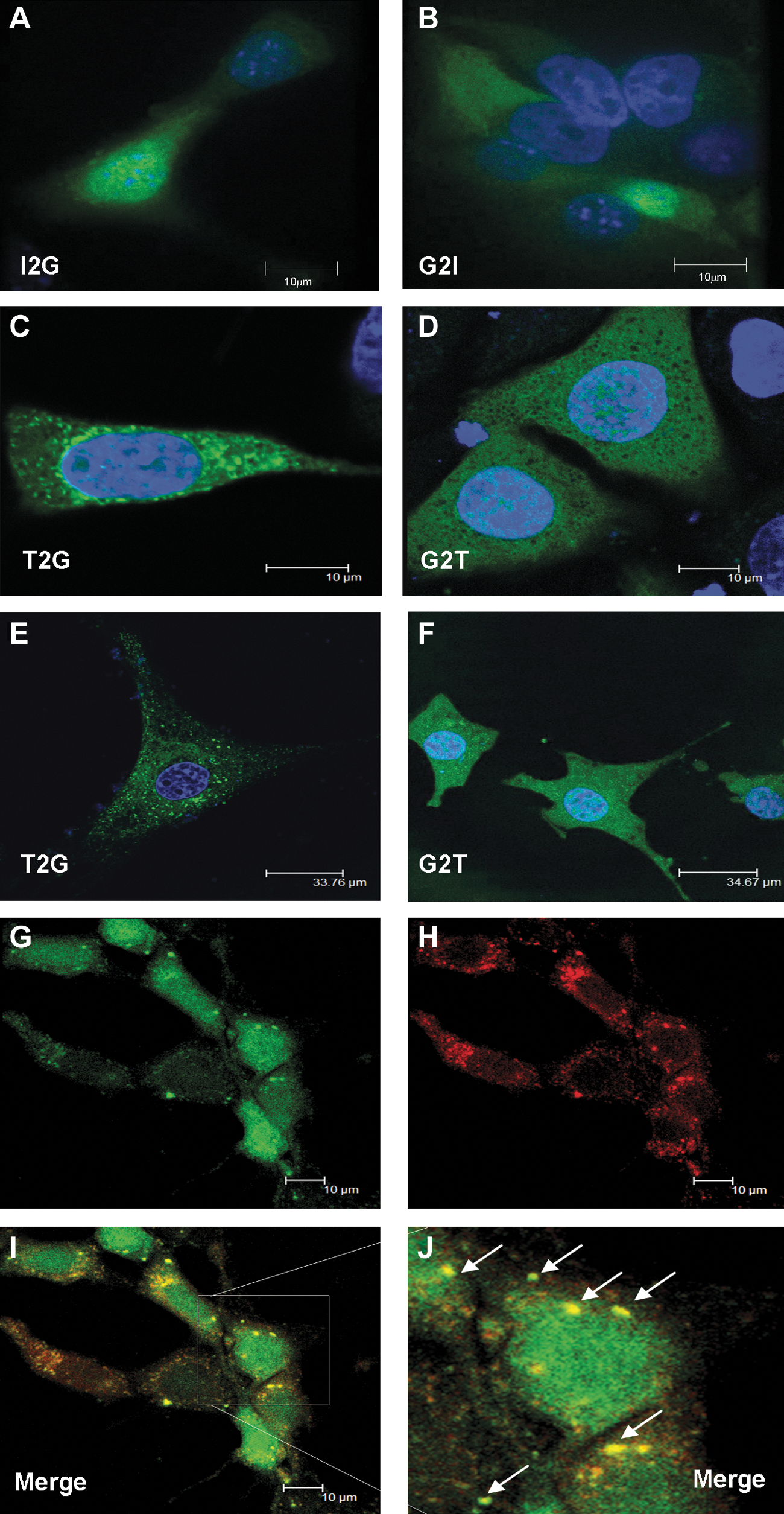
Intracellular distribution of uncleaved 2A linked proprotein. Cells transduced with the four constructs were analyzed by confocal fluorescence microscopy for the presence of GFP. NIH-3T3 cells were transduced with either I2G
In an effort to define these vesicles, cells were costained with a number of intracellular markers. Markers for lysosomes, mitochondria, endoplasmic reticulum (ER), and Golgi showed no evidence of colocalization (data not shown), although a pronounced and reproducible colocalization with Rab7, a late endosome marker, was observed (Fig. 3G, GFP; Fig. 3H, Rab7; Fig. 3I, merge; Fig. 3J, high resolution).
We show here that for some secreted proteins such as IL-2, there is little positional effect within a bicistronic FMDV 2A cassette in terms of function. However, there is significant evidence of uncleaved proprotein when IL-2 is upstream of 2A. The lack of consequence in terms of protein function due to this aberrant cleavage most likely suggests that the IL-2-GFP fusion is still bioactive. In contrast, another secreted cytokine, TGF-β, suffered a dramatic reduction in secretion and function evident when expressed upstream of the 2A motif but not downstream. Our data indicate that when TGF-β is upstream of 2A the complete bicistronic protein was not efficiently cleaved, with substantial levels of proprotein processed as a single unit. Consistent with previous reports, GFP was unaffected by relative position in any vector (Hu et al., 2009). One possible explanation for the complete lack of functional activity from the uncleaved TGF-β is that trafficking proteins bind the signal sequence as it exits the ribosome and this interaction causes a structural change that prevents ribosomal cleavage. Late endosomes are known to be upstream of lysosomal degradation, so it is also possible the proprotein is destined for degradation. No colocalization within the lysosome was observed but this could be because the protein had already been degraded. Early and late endosomes have been shown to be areas of intracellular signaling from receptor tyrosine kinases and receptor serine/threonine kinases such as the TGF-β receptor (Wang et al., 2004; Rajagopal et al., 2007). It is possible that the intracellular proteins responsible for vesicular transport may also inhibit cleavage of the T2G proprotein when binding at the ribosome. This would be an alternative explanation for the localization at the late endosome but warrants further investigation.
These studies show that the FMDV 2A bicistronic cassette can be a powerful and efficient cassette for equal expression of multiple genes. However, the sequence is derived from a virus where no protein product is secreted from the host cell and as such may create unpredictable results when fused to secreted protein products. Careful consideration of how the protein is processed and functions should be taken into account when designing such expression vectors. The orientation of the target protein with regard to the 2A cassette has potentially dramatic effects on its expression and analysis of the protein of interest should be undertaken before assumptions about expression and function are made.
Footnotes
Acknowledgments
This work was supported by a UK Centre for Tissue Engineering program grant, cofunded by the MRC, BBSRC, and EPSRC. D.G.R., D.E.G., and R.E.H. are also funded by Cancer Research UK and supported by the ATTACK FP6 programme. The authors thank Dr. Michael Milsom, Cincinnati Children's Hospital Medical Center (Cincinnati, OH) for the gift of the pSP72-2A plasmid and Dr. Louise Reynolds (CRUK, London, UK) for the gift of the luciferase plasmids containing Smad-responsive elements.
Author Disclosure Statement
No competing financial interests exist.