
Abstract
Despite the advantages of using adenoviral vectors for specific antigenic gene delivery in the development of antigen-presenting cell (APC)-based vaccines, the lack of the coxsackievirus–adenovirus receptor (CAR) on APCs limits the use of adenoviral vectors for in vitro gene delivery. In this study, we used a recombinant adapter protein, CFm40L, which consists of the ectodomain of CAR genetically fused to the ectodomain of CD40 ligand (CD40L) via a trimerization motif, to target Her-2/neu- or human papillomavirus 16 (HPV16) E6/E7-encoding adenoviruses to CD40 on dendritic cells (DCs) and B cells. Targeting CD40 enabled the enhancement of tumor antigen delivery and simultaneous activation of APCs via the CD40–CD40L interaction. We found that these transduced DCs and B cells substantially enhanced the CTL response against human Her-2/neu- and HPV16 E6/E7-expressing tumors, resulting in significant inhibition of tumor growth in a murine tumor model. In addition, the use of the CFm40L adapter protein in combination with gemcitabine treatment allowed for a successful immune response against a self-tumor antigen, murine Her-2/neu. Our results suggest that targeting adenovirus to APCs via CD40, using CFm40L, represents a great improvement in anticancer cellular vaccines.
Introduction
Antigens can be loaded onto APCs by incubation with peptides, proteins, or tumor lysates and by genetic manipulations using DNA, RNA, or viral vectors encoding protein antigens. Genetic manipulations have the advantage of persistent antigen presentation over time and generate more potent antitumor immunity than the former incubation methods (Kirk and Mule, 2000; Jenne et al., 2001; Nakamura et al., 2005). Much attention has been paid to the recombinant adenoviral vectors because of their gene transfer efficiency, ability to encode relatively large genes of interest, and ability to transduce both proliferating and resting cells (Xia et al., 2006). Moreover, adenoviral transduction can induce DC differentiation and maturation (Lyakh et al., 2002; Mehrotra et al., 2004), and adenoviral vectors can serve as an adjuvant to simultaneously stimulate both innate and adaptive immune responses (Appledorn et al., 2008).
Unfortunately, DCs and B cells are relatively resistant to adenoviral transduction because of their lack of coxsackievirus–adenovirus receptors (CAR) surface expression, which mediates adenoviral entry. It has been reported that targeting adenovirus to an alternative receptor, particularly CD40, using bispecific antibody conjugates or recombinant adaptor proteins that bind to both the adenovirus and CD40, can overcome the CAR deficiency of DCs and enhance gene transfer in addition to promoting DC maturation (Tillman et al., 1999, 2000; Pereboev et al., 2002). Pereboev and colleagues reported a recombinant adaptor protein, CFm40L, consisting of CAR fused to mouse CD40L via a trimerization motif. The trimerization motif increases the binding avidity of the adenovirus and stabilizes a trimeric conformation of CD40L for efficient CD40 binding and function. Thus, CFm40L efficiently targets adenovirus to DCs, enhances adenovirus-mediated gene transfer, and also activates DCs. After administration in vivo, CFm40L-complexed adenovirus induced enhanced antigen-specific T cell responses compared with noncomplexed adenovirus (Pereboev et al., 2004; Huang et al., 2008).
In this study, we extended previous studies on the use of CFm40L to demonstrate an antitumor effect when transducing tumor antigens. We applied this system to treat tumors, using Her-2/neu and human papillomavirus 16 (HPV16) E6/E7 as antigens. A DC-based vaccine showed a greater antitumor effect when transduced with CFm40L-complexed adenovirus. Furthermore, we applied this system to a B cell-based vaccine and improved its antitumor effect. Our results suggest that targeting adenovirus to APCs via CD40, using CFm40L, represents a significant improvement in anticancer cellular vaccines.
Materials and Methods
Mice
BALB/c and C57BL/6 mice were purchased from Charles River Laboratories (Seoul, Korea) at the age of 6 weeks and were kept under pathogen-free conditions in the Animal Center for Pharmaceutical Research at Seoul National University (Seoul, Korea). All experiments were approved by the Institutional Animal Care and Use Committee of Seoul National University.
Antibodies
Antibodies from hybridomas GK1.5 (anti-CD4) and 2.43 (anti-CD8) were obtained from ascites. These antibodies were injected intraperitoneally (200 μl/mouse) to deplete CD4+ T cells or CD8+ T cells. Phycoerythrin (PE)-labeled anti-Her-2/neu antibodies (BD Biosciences, San Jose, CA), fluorescein isothiocyanate (FITC)-labeled anti-CD19 antibodies, and PE-labeled anti-CD80, anti-CD86, and anti-I-A/I-E antibodies (all from Biolegend, San Diego, CA) were used for flow cytometry analysis.
Preparation of CFm40L
CFm40L recombinant protein was prepared as previously described (Pereboev et al., 2004). Briefly, stable CFm40L-expressing cells were generated from 293 cells transfected with the pcDNA3/CAR/F/m40L plasmid vector. CFm40L protein was subsequently collected and purified from the cell culture medium. The binding specificity of the CFm40L fusion protein to both the adenovirus serotype 5 (Ad5) recombinant fiber knob and the ectodomain of mouse CD40 produced by bacterial expression was tested by enzyme-linked immunosorbent assay (ELISA).
Construction of recombinant adenoviruses
A recombinant adenovirus encoding the extracellular and transmembrane domains of Her-2/neu (AdHM) was constructed by Viromed (Seoul, Korea) as previously described (Kim et al., 2008). Briefly, a cDNA encoding the HM gene was cloned into the pAxCAwtit cosmid, resulting in the pAxCA-HM cosmid. AdHM was generated by cotransfection of 293 cells with the pAxCA-HM cosmid together with Ad5 DNA–terminal protein complex (TPC). A recombinant adenovirus encoding HPV16 E6/E7 (AdE6E7) was constructed by Genexine (Pohang, Korea), using the AdEasy vector system (Qbiogene/MP Biomedicals, Irvine, CA) as previously described (Jin et al., 2005). To reduce the risk of oncogenicity, mutations in the E6/E7 gene were simultaneously introduced at positions Cys-63 and Cys-106 of E6 and at Cys-24 and Glu-26 of E7, all resulting in translation as glycine. A cDNA encoding E6/E7 was subcloned into the KpnI and XbaI sites of an adenoviral shuttle vector (pShuttleCMV). The pShuttleCMV/E6E7 construct was cotransformed with the adenoviral backbone vector pAdEasy into Escherichia coli BJ5183 by electroporation to achieve homologous recombination. The recombinant plasmid was transfected into 293 cells, and recombinant adenovirus was isolated. E3 was absent from the viral vectors.
Generation of DC- and B cell-based vaccines
Dendritic cells were generated by culturing murine bone marrow cells in RPMI medium containing 10% fetal bovine serum (FBS) and granulocyte-macrophage colony-stimulating factor (GM-CSF, 20 ng/ml) (R&D Systems, Minneapolis, MN) for 6 days, and B cells were magnetically isolated from the spleen, using anti-B220 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). To prepare DC- and B cell-based vaccines DCs and B cells were transduced at a multiplicity of infection (MOI) of 100 with CFm40L-complexed adenovirus (Ad-CFm40L) or noncomplexed adenovirus (Ad). The adenovirus was complexed with CFm40L by preincubating adenovirus with CFm40L for 20 min at room temperature. CFm40L was used at 250 ng for 1 × 109 plaque-forming units (PFU) of adenovirus. After culturing overnight, unbound virus was washed out, and 2 × 106 DCs or B cells were injected intravenously into syngeneic mice.
Real-time polymerase chain reaction
Total RNA was isolated from 5 × 106 DCs or B cells, using an RNeasy mini kit (Qiagen, Hilden, Germany). Reverse transcription was performed with Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen, Carlsbad, CA), and quantitative real-time polymerase chain reaction (PCR) was carried out with SYBR Premix Ex Taq (Takara Bio, Otsu, Shiga, Japan). The following primers were used: E7, F5′-ACGAGTACATGCTGGATCTG-3′, R5′-GATGTCTACGTGGGTGCTCT-3′; interleukin (IL)-4, F5′-TCGGCATTTTGAACGAGGTC-3′, R5′-CAGTGATGTGGACTTGGACTCA-3′; tumor necrosis factor (TNF)-α, F5′-AGCAAGGGACTAGCCAGGAG-3′, R5′-CCCATCTTTTGGGGGAGTGC-3′; IL-6, F5′-CCACTTCACAAGTCGGAGGC-3′, R5′-TGGTACTCCAGAAGACCAGAGG-3′; IL-12 p35, F5′-ACCCAGTTGGCCAGGGTC3′, R5′-CAAGGCACAGGGTCATCATC-3′; IL-12 p40, F5′-ATGGCCATGTGGGAGCTGGA-3′, R5′-TTTGGTGCTTCACACTTCAG-3′. The gene expression was normalized to GAPDH.
In vivo cytotoxicity assay
Syngeneic lymphocytes were either primed with a 1-μg/ml concentration of cytotoxic T lymphocyte (CTL) epitope peptide (human Her-2 p63 [TYLPTNASL] peptide for hHer-2/neu, a mixture of E641–50 [EVYDFAFRDL] and E749–57 [RAHYNIVTF] peptides for HPV16 E6/E7, and murine Her-2 p63 [TYLPANASL] peptide for mHer-2/neu) or left unprimed. Lymphocytes were then labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen); 20 μM was used for primed cells, whereas 2 μM was used for unprimed cells. Equal numbers of CFSEhigh and CFSElow cells were mixed and injected intravenously into immunized mice. After 24 hr, splenocytes from the injected mice were analyzed to assess peptide-specific target lysis. Specific lysis was calculated as follows: r (ratio) = (% CFSElow/% CFSEhigh), % lysis = [1 – (r unprimed/r primed)] × 100.
Evaluation of antibody responses
Human Her-2/neu-specific antibodies were detected by hHer-2/neu binding assay. Briefly, hHer-2/neu-expressing cells (hHer-2/CT26) were incubated with diluted serum from immunized mice for 1 hr and unbound free antibodies were then washed out. The cells were stained with an FITC-labeled goat anti-mouse IgG secondary antibody (BD Biosciences) for 30 min and hHer-2/neu-specific antibody binding was then measured by flow cytometry.
Therapeutic tumor models
The E6/E7-expressing TC-1 cell line (C57BL/6 background) (American Type Culture Collection [ATCC], Manassas, VA), the human Her-2/neu-expressing CT26 cell line (hHer-2/CT26) (Penichet et al., 1999), and the murine Her-2/neu-expressing CT26 cell line (mHer-2/CT26) (Ko et al., 2007) were used for mouse tumor models. In the human Her-2/neu tumor model, BALB/c mice were challenged intravenously or subcutaneously with 1–2 × 105 hHer-2/CT26 tumor cells and then administered the indicated APC-based vaccine on day 1 or day 3. In the E6/E7 tumor model, C57BL/6 mice were injected with 5–10 × 104 TC-1 tumor cells and then immunized on day 7 or day 10, when the tumor was palpable. In the tolerogenic tumor model, BALB/c mice were injected subcutaneously with 3 × 105 mHer-2/CT26 tumor cells. Treatment was then started with gemcitabine (Dong-A Pharmaceuticals, Seoul, Korea) on days 11 and 13, and followed by DC-based vaccines on day 14. Survival of the mice was observed and tumor growth was measured with calipers three times per week. Mice were killed for humanitarian reasons when they showed abnormal tumor-related symptoms or when the tumors exceeded a three-dimensional volume of 2500 mm3.
Preventive tumor model
BALB/c mice were immunized with a DC- or B cell-based vaccine on day 0, and 2 × 105 mHer-2/CT26 cells were then injected subcutaneously into the left flank on day 7. Tumor growth was measured with calipers three times per week.
Statistical analysis
The Kaplan–Meier method was used to determine the statistical significance of differences in survival time. We performed a log-rank test (Mantel–Cox) with SPSS 16.0K for Windows (SPSS, Chicago, IL). The Student t test was used to compare differences between two groups. To compare multiple groups, we used one-way analysis of variance (ANOVA) followed by Tukey's post hoc test. Values of p < 0.05 were considered significant at a 95% confidence interval.
Results
CFm40L enhances the antitumor effect of DC- and B cell-based vaccines
It has been reported that CFm40L enhances antigen-specific CD4+ T cell and CD8+ T cell immune responses when coupled with an adenovirus encoding β-galactosidase (β-Gal) (Pereboev et al., 2004). In this study, we used CFm40L to enhance the antitumor effect of an APC-based cellular vaccine transduced with an adenovirus encoding a tumor antigen. First, we applied this system to a DC-based vaccine, a generally used cellular vaccine, for the treatment of cancer. The therapeutic antitumor effect of this DC-based vaccine transduced with a CD40-targeted adenovirus was measured using a metastatic hHer-2/CT26 tumor model. Three days after tumor challenge, BALB/c mice were immunized with DCs transduced with AdHM-CFm40L (DC/AdHM-CFm40L) or AdHM (DC/AdHM). We found that DC/AdHM-CFm40L immunization significantly prolonged the survival of tumor-challenged mice compared with DC/AdHM (Fig. 1A). At the end point of the experiment, all of the living mice remained tumor free. These data indicate that CFm40L can enhance the antitumor effect of a DC-based vaccine.

Enhanced antitumor effect of DC- and B cell-based vaccine by CFm40L. (
B cells have been emerging as an alternative source of cellular vaccines because of their abundance in the blood and ease of preparation (Schultze et al., 1997, 2004; Chung et al., 2006; Kim et al., 2007, 2008). Thus, we also examined whether CFm40L could enhance the antitumor immune response induced by a B cell-based vaccine. In the metastatic hHer-2/CT26 tumor model, immunization with B cells transduced with AdHM-CFm40L (B/AdHM-CFm40L) significantly prolonged the survival of tumor-challenged mice compared with B cells transduced with AdHM (B/AdHM) (Fig. 1B). In addition, both DC/AdHM-CFm40L and B/AdHM-CFm40L efficiently inhibited tumor growth in the solid hHer-2/neu tumor model (Fig. 1C).
We also tested our CD40-targeted APC-based vaccines in the HPV16 E6/E7-expressing TC-1 tumor model. Immunization with B/AdE6E7-CFm40L significantly inhibited tumor growth compared with B/AdE6E7 (Fig. 2A), and DC/AdE6E7-CFm40L exhibited a greater antitumor effect than did DC/AdE6E7 (Fig. 2B). The antitumor immunity induced by B/AdE6E7-CFm40L was comparable to that induced by DC/AdE6E7-CFm40L. These data indicate that the antitumor immune response induced by APC-based vaccines can be significantly enhanced by targeting adenovirus to APCs via CD40, using CFm40L.

Enhancement of antitumor effect against HPV16 E6/E7-expressing tumor. (
CFm40L enhances adenovirus-mediated gene transfer to APCs
Given that the tumor antigen levels presented by APCs determine the outcome of antitumor immunity (Kurts et al., 1999; Spiotto et al., 2003), we tried to enhance the efficacy of adenoviral transduction by targeting CD40. Furthermore, we tested whether targeting adenovirus to APCs via CD40, using CFm40L, would increase tumor antigen delivery to DCs and B cells. To that end, DCs and B cells were transduced with 100 MOI of AdHM-CFm40L or AdHM, the optimal amount to use without cytopathic effect (Ribas et al., 1997). DCs transduced with AdHM-CFm40L expressed higher cell surface levels of Her-2/neu than those transduced with AdHM. Similarly, CFm40L also enhanced Her-2/neu gene delivery to B cells (Fig. 3A). Furthermore, CFm40L consistently enhanced the efficacy of adenovirus-mediated E6/E7 gene transfer to APCs, as E7 mRNA expression was increased in DCs and B cells transduced with AdE6E7-CFm40L compared with that in cells transduced with AdE6E7 (Fig. 3B). Collectively, targeting adenovirus to CD40 by means of CFm40L enhanced the adenovirus-mediated target gene transfer to both DCs and B cells.
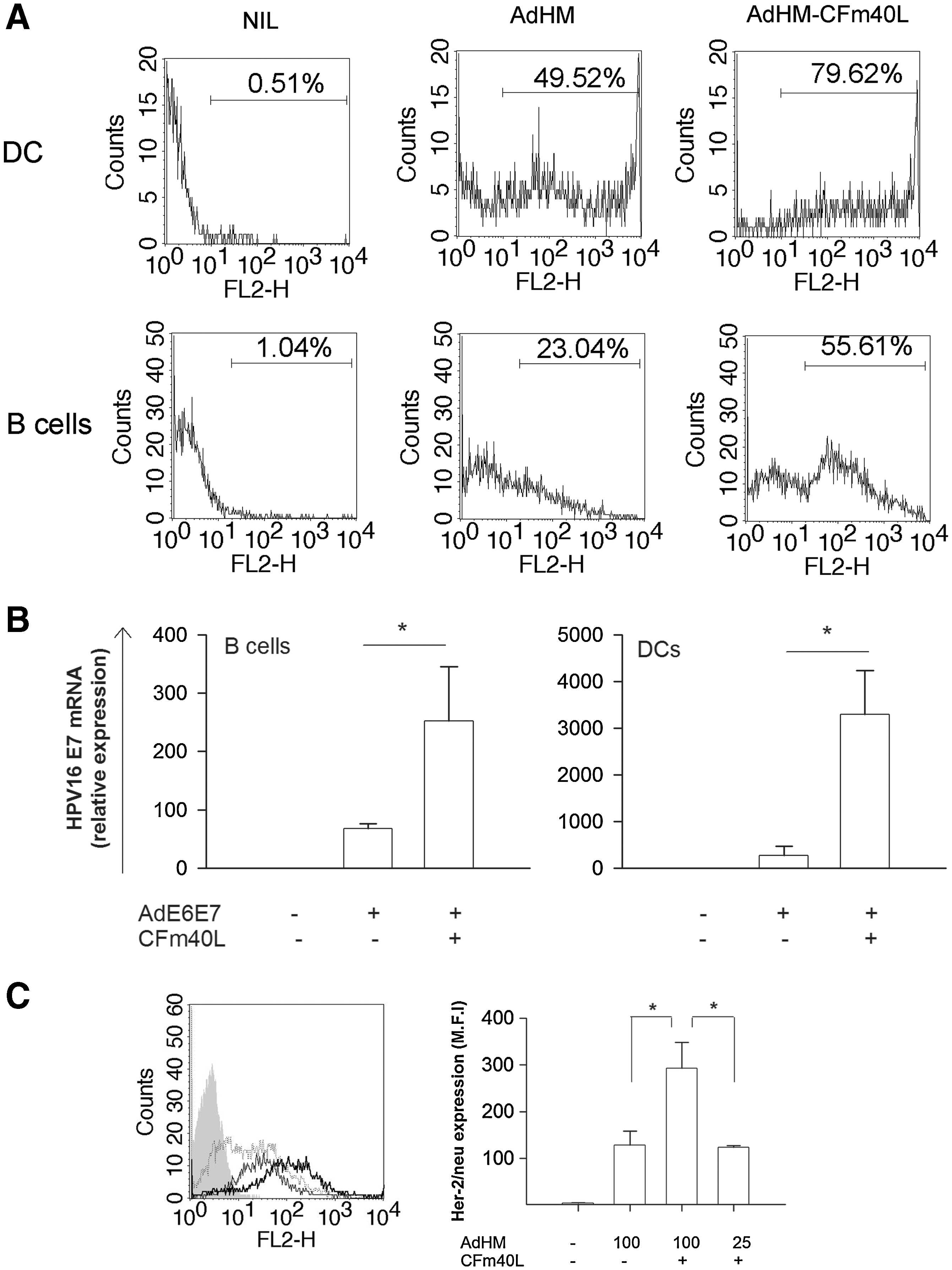
Enhancement of adenovirus-mediated gene transfer to APCs by CFm40L. (
We also evaluated whether CFm40L allowed the use of less adenovirus. Her-2/neu expression was similar in both 100 MOI AdHM- and 25 MOI AdHM-CFm40L-transduced B cells. However, B cells transduced with 100 MOI of AdHM-CFm40L expressed much more antigens than those with 25 MOI of AdHM-CFm40L (Fig. 3C). These data show that CFm40L allows the use of less adenovirus, but optimization of the adenoviral dose is necessary for the quantitative and qualitative control of antigen expression.
Ad-CFm40L-activated APCs
As CFm40L contains trimeric CD40L, the ligand can interact efficiently with CD40 and consequently activate APCs (Morris et al., 1999). Furthermore, it has been reported that adenoviral transduction promotes DC maturation (Lyakh et al., 2002; Mehrotra et al., 2004). Thus, we investigated the activation status of B cells and DCs after transduction with Ad-CFm40L (Fig. 4A). We first analyzed activation status based on the expression of surface molecules after incubating DCs and B cells with CFm40L, AdHM, or AdHM-CFm40L. Adenovirus slightly increased CD86 expression, and CFm40L remarkably increased CD86, CD80, and MHC class II molecule expression, on B cells and DCs. Collectively, adenovirus and CFm40L enhanced the immune-stimulatory abilities of DCs and B cells by upregulating costimulatory molecules and MHC class II molecules.
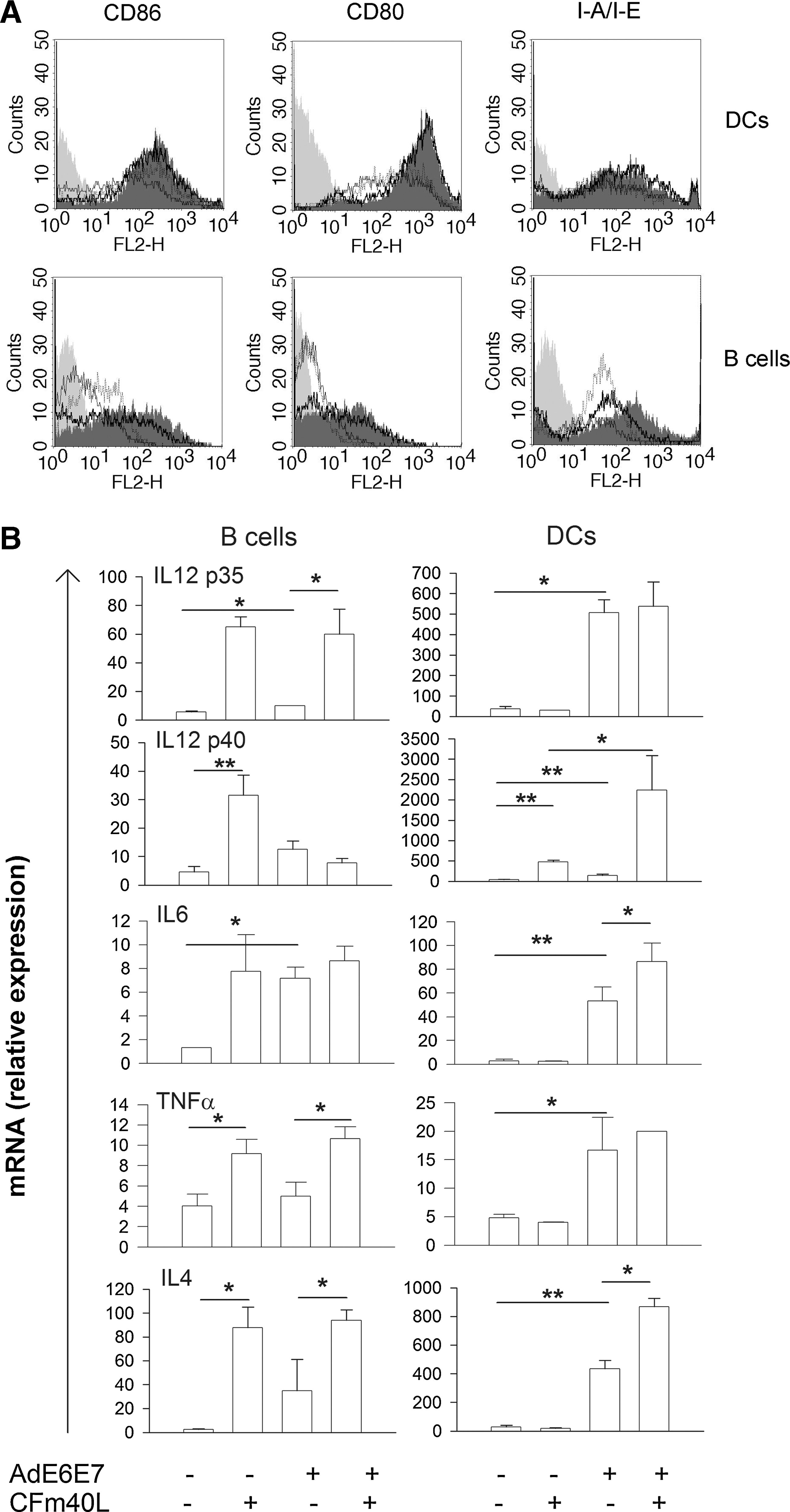
Activation of APCs by adenovirus-CFm40L. (
It has been reported that CD40L-activated DCs regulate T cell responses via the production of proinflammatory cytokines such as IL-12, IL-6, and TNF-α (Bleharski et al., 2001; Pereboev et al., 2004; Rethi et al., 2006; Xu et al., 2007). We asked whether this cytokine production could be altered by Ad-CFm40L transduction. To that end, DCs and B cells were incubated with CFm40L, AdE6E7, or AdE6E7-CFm40L, and cytokine levels were then measured (Fig. 4B). Increased cytokine production was observed after incubation with CFm40L and/or adenovirus. Specifically, CFm40L increased IL-12 p40, IL-12 p35, IL-6, TNF-α, and IL4 expression, and adenovirus increased IL-12 p40, IL-12 p35, IL-6, and IL-4 expression by B cells. Consequently, B cells transduced with Ad-CFm40L efficiently produced diverse cytokines such as IL-12 p35, IL-6, TNF-α, and IL-4. DCs transduced with Ad-CFm40L also produced increased cytokine levels. Cytokine production by DCs was induced mainly by adenovirus: however, DCs transduced with Ad-CFm40L produced greater levels of IL-12 p40, IL-6, and IL-4 than those transduced with adenovirus alone. The production of IL-12 p40 by DCs was synergistically increased by CFm40L and adenovirus. Taken together, these results show that adenovirus and CFm40L simultaneously activated DCs and B cells by upregulating the expression of molecules involved in antigen presentation and T cell priming and by regulating the cytokine production that regulates T cell immune responses.
CFm40L enhances antigen-specific CTL responses essential for tumor rejection
As it has been shown that not only the CTL response but also the antibody response is important in the rejection of Her-2/neu-expressing tumors (Whittington et al., 2009), we examined whether targeting adenovirus to APCs via CD40 would enhance Her-2/neu-specific CTL and antibody responses. High titers of human Her-2/neu-specific antibodies were observed in both B/AdHM- and B/AdHM-CFm40L-treated mice, with no significant difference in antibody levels (Fig. 5A). On the other hand, the percentage of human Her-2/neu-specific target cell lysis was significantly higher in mice vaccinated with B/AdHM-CFm40L (B/AdHM-CFm40L, 94.65 ± 0.21; B/AdHM, 69.59 ± 2.78; p < 0.05) (Fig. 5B). These results indicate that CFm40L preferably enhances the CTL responses induced by a B cell-based vaccine.
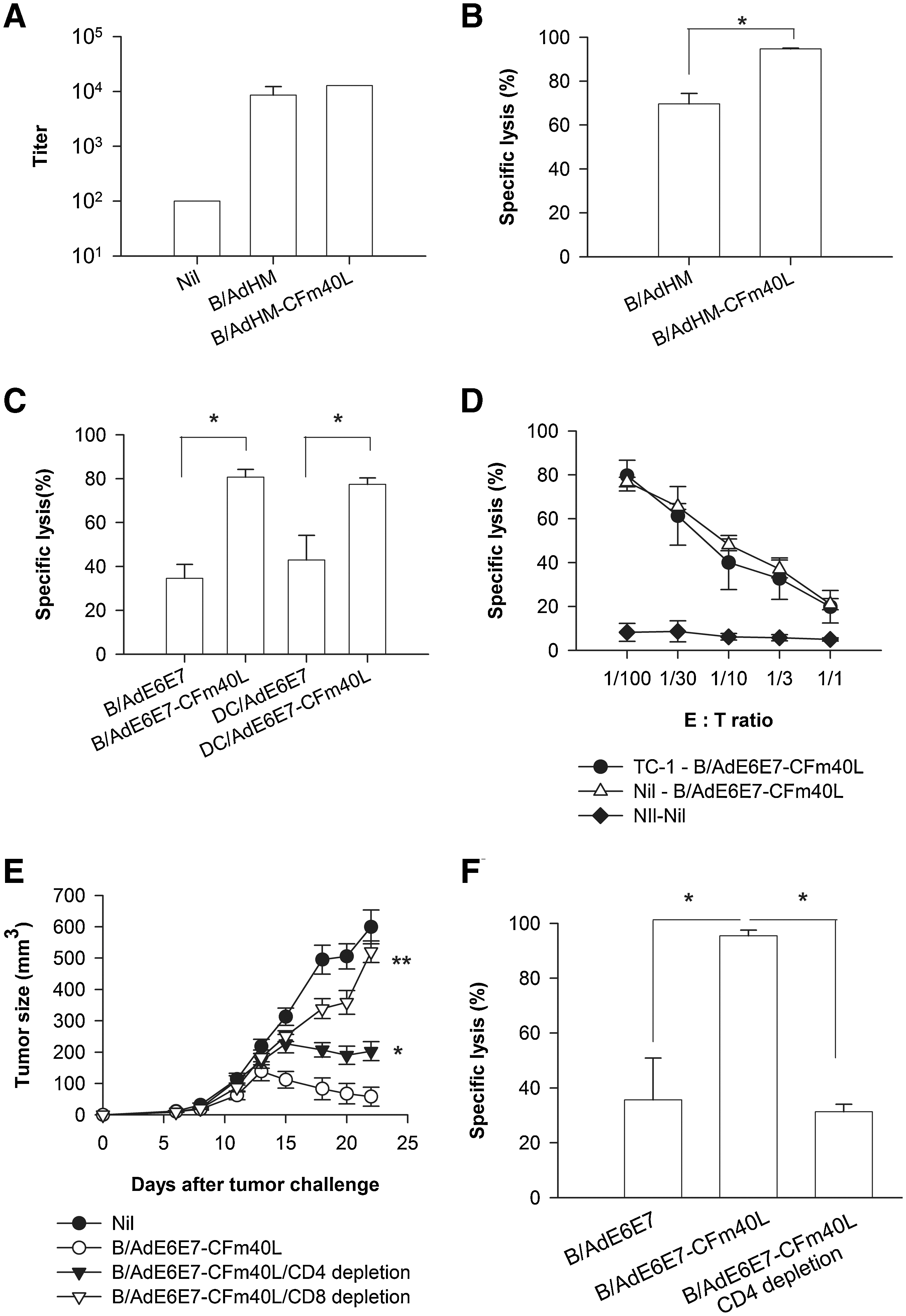
CFm40L-enhanced antigen-specific CTL responses essential for tumor rejection. (
Enhanced E6/E7-specific CTL activity was consistently observed on vaccination with DCs or B cells transduced with AdE6E7-CFm40L. The percentage of E6/E7-specific target cell lysis increased from 34.5 ± 3.1 to 80.7 ± 1.7% on vaccination with AdE6E7-CFm40L-transduced B cells and from 42.9 ± 5.6 to 77.4 ± 1.4% on vaccination with AdE6E7-CFm40L-transduced DCs (Fig. 5C). Furthermore, B/AdE6E7-CFm40L efficiently induced CTL responses in both tumor-challenged and nonchallenged mice (Fig 5D). To investigate which types of immune cells were involved in the elimination of tumors, CD8+ T cells or CD4+ T cells were depleted, using depleting antibodies. As shown in Fig. 5E, CD8+ T cell depletion abolished the antitumor effect induced by B/AdE6E7-CFm40L, whereas CD4+ T cell depletion only partially reduced it. Furthermore, B/AdE6E7-CFm40L could not induce a strong CTL response without CD4+ T cells (Fig. 5F). The partial rejection observed when CD4+ T cells were depleted might result from an insufficient CTL response, although we cannot rule out direct tumor cell killing by CD4+ T cells. These results indicate that both CD8+ T and CD4+ T cell responses are involved in TC-1 tumor rejection, but that the CD8+ T cell response is essential.
CFm40L-modified adenoviral transduction of DCs can overcome self-tolerance to murine Her-2/neu
Immunization of mice with DC-based vaccines engineered to express the xenogeneic tumor antigen human Her-2/neu and loaded with a natural killer T cell (NKT) ligand has been shown to induce immunity against syngeneic murine Her-2/neu (mHer-2/CT26) by inducing self-reactive CTLs (Ko et al., 2007). We asked whether DCs or B cells transduced with AdHM-CFm40L could overcome self-tolerance and induce mHer-2/neu-specific immune responses. CFm40L enhanced mHer-2/neu-specific CTLs by DC- or B cell-based vaccines (Fig. 6A). However, because of self-tolerance, mHer-2/neu-specific target cell lysis was relatively low considering the hHer-2/neu-specific target cell lysis (Fig. 5B). In addition, immunization with DC/AdHM-CFm40L inhibited mHer-2/CT26 tumor growth in a prevention model, but it was not dramatic (Fig. 6B). We tried to evaluate the therapeutic antitumor effect of a DC vaccine transduced with AdHM-CFm40L in this tolerance model. As the murine antigen induced self-tolerance, however, a single treatment with DC/AdHM-CFm40L could not inhibit tumor growth efficiently (data not shown). In a previous study (Ko et al., 2007), we observed that gemcitabine treatment not only reduced tumor burden by a direct cytotoxic effect but also eliminated immunosuppressive myeloid-derived suppressor cells (MDSCs) and thus contributed to an enhanced antitumor CTL response. In this study, therefore, we attempted to test a combined therapy of DC/AdHM-CFm40L vaccination and gemcitabine treatment. Mice with established mHer-2/CT26 tumors were treated with gemcitabine twice and immunized with DC/AdHM-CFm40L or with DC/AdHM as a control. Mice immunized with DC/AdHM-CFm40L after gemcitabine treatment showed significantly reduced tumor growth compared with those treated with gemcitabine and DC/AdHM or gemcitabine alone (Fig. 6C). These results indicate that CFm40L can enhance the antitumor effect of DC-based vaccines against tolerogenic tumors in combination with chemotherapy.
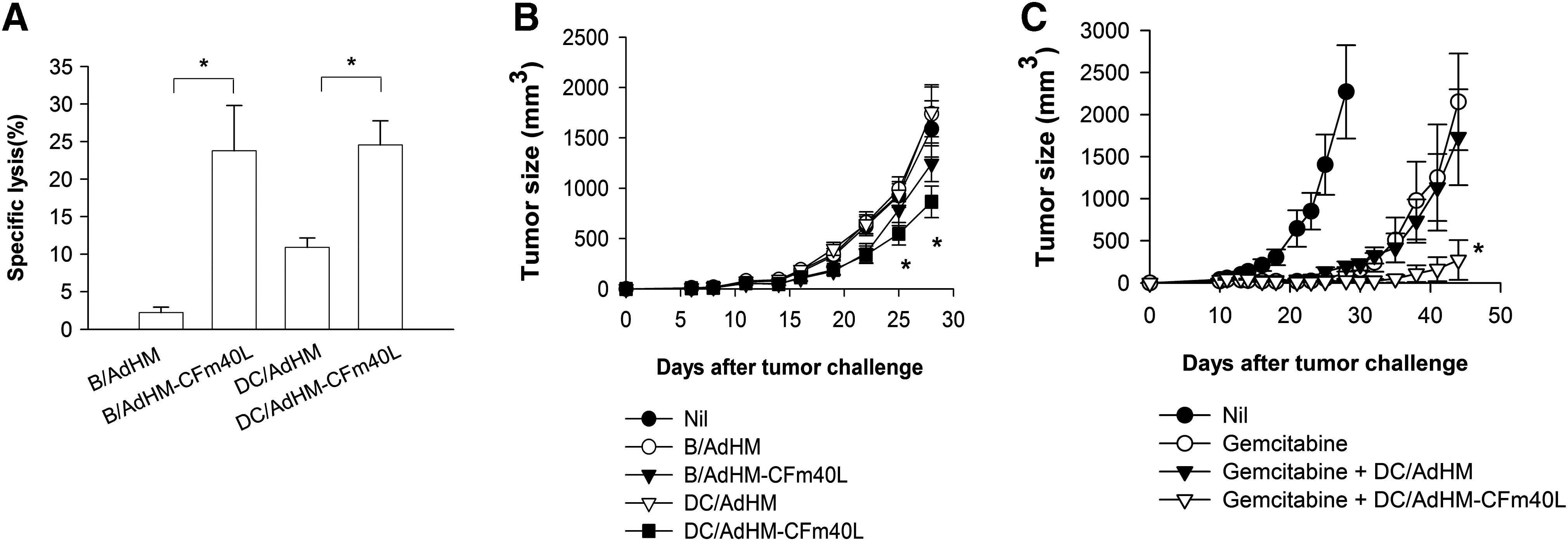
CFm40L-complexed adenovirus transduction of DCs can overcome self-tolerance to murine Her-2/neu. (
Discussion
DCs and B cells have been shown to be promising APC-based vaccine targets for the induction of antitumor immunity to control tumor growth (Kirk and Mule, 2000; Kim et al., 2007, 2008). Recombinant adenoviruses serve as an efficient means for antigenic gene delivery (Jenne et al., 2001). However, DCs and B cells are relatively resistant to adenoviral transduction because of their lack of surface expression of CAR (Rebel et al., 2000). In this study, we have shown that targeting recombinant adenovirus to APCs such as DCs or B cells via CD40, using an adapter molecule, CFm40L, enhances antitumor immunity. This is probably due to enhanced antigenic gene delivery to and activation of APCs. Our results are consistent with notions that CD40 ligation on DCs promotes their maturation, which in turn stimulates T cell activation (Rethi et al., 2006), and that CD40-activated B cells efficiently induce antigen-specific immune responses (Schultze et al., 1997).
It has been reported that targeting adenovirus to CD40 by means of bispecific antibodies enhances the efficacy of DC-based vaccines (Tillman et al., 2000). Despite the successes of this bispecific antibody-based adenovirus-targeting approach, the variability and instability of chemically conjugated antibodies highlight the need for the development of invariable and stable targeting molecules for adenoviral transduction. In addition, an adaptor molecule with efficient binding to both adenovirus and CD40 is needed. A recombinant adaptor protein, CFm40L, which contains genetically fused trimeric CAR and CD40L, is expected to mediate the enhancement of gene transfer efficacy and DC maturation.
The level of antigen presented by APCs determines how naive T cells respond to tumor antigens, and antigens must be expressed at sufficient levels to induce an immune response (Kurts et al., 1999; Spiotto et al., 2003). Notably, CFm40L enhanced the adenoviral transduction of both DCs and B cells, resulting in the expression of sufficient levels of tumor antigens for the induction of specific immune responses (Fig. 3). In addition, CFm40L allowed the use of less adenovirus for antigen delivery.
The activation of APCs is also important to induce antigen-specific immune responses, and inappropriate stimulation from APCs may induce anergic and regulatory T cells rather than effector T cells (Hawiger et al., 2001; Spiotto et al., 2003). Immature DCs and nonactivated B cells may induce antigen-specific tolerance (Sun et al., 2008). In this study, Ad-CFm40L enhanced the activation of DCs and B cells as indicated by the expression of cell surface markers such as CD80, CD86, and MHC class II molecules and the production of proinflammatory cytokines.
Of note, transduced DCs expressed greater levels of antigen (Fig. 3A and B) and costimulatory molecules and cytokines (Fig. 4) compared with transduced B cells. This is consistent with generally accepted evidence that DCs are professional APCs (Palucka et al., 2007). Our results, however, indicated that B cells, which may be less competent APCs compared with DCs (Schultze et al., 2004), can be used for the treatment of cancer when transduced with adenovirus complexed with CFm40L. The resistance against hHer-2/CT26 and TC-1 tumor growth induced by such B cells was comparable to that induced by DCs (Figs. 1C and 2B), suggesting that B cells can also be used for the treatment of cancer when transduced with adenovirus complexed with CFm40L. The use of B cells has great advantages for clinical applications as B cells are abundant in the blood and large numbers of B cells can be easily isolated as a homogeneous cell population (Schultze et al., 1997, 2004). Furthermore, the process for generating B cell-based vaccines is simple and fast, consisting of only two steps: the isolation of B cells from the blood and transduction with adenovirus. Thus, B cell-based vaccines could be used efficiently in the clinic.
Although therapeutic antitumor immunity against human Her-2/CT26 tumors was efficiently induced by Ad-CFm40L-transduced APC-based vaccines in a mouse model, these results may not apply to the natural conditions of tolerance against self-tumor antigens. Thus, we examined the strategy of using CFm40L in a tolerogenic murine tumor model containing a syngeneic murine Her-2/neu (mHer-2/CT26) tumor that mimicked human cancers (Ko et al., 2007). Although DCs and B cells transduced with AdHM-CFm40L induced higher mHer-2/neu-specific CTL responses than cells transduced with AdHM alone (Fig. 6A), it was not sufficient to eliminate already established tumor cells. When used in combination with gemcitabine, however, which reduced the tumor burden and eliminated myeloid-derived suppressor cells (MDSCs) (Ko et al., 2007), CFm40L was effective in overcoming self-tolerance (Fig. 6B). The synergistic effect of this combination therapy suggests that immunotherapy using APC-based vaccines can be more effective when combined with chemotherapy.
In summary, use of the CFm40L adapter protein overcame the limits of adenovirus as a tool for in vitro gene delivery in the generation of APC-based antitumor vaccines by targeting the adenovirus to CD40 instead of CAR. Moreover, Ad-CFm40L-transduced APCs showed enhanced immune stimulatory activity by upregulating costimulatory molecules and proinflammatory cytokines. As a consequence, Ad-CFm40L-transduced APC-based vaccines showed enhanced antitumor effects in diverse tumor models. Targeting adenovirus to CD40 on APCs via the CFm40L adapter molecule, therefore, offers a new strategy for antitumor therapy.
Footnotes
Acknowledgments
This study was supported by a grant from the National R&D Program for Cancer Control, Ministry of Health and Welfare (0720500), an NRL grant (R0A-2008-000-20113-0), and a WCU grant (R31-2008-000-10103-0) from the National Research Foundation of Korea (NRF). The authors thank Viromed for AdHM and Dr. Young-Chul Sung for the HPV16 E6/E7 gene.
Author Disclosure Statement
No competing financial interests exist.