
Abstract
Classical gene therapy for cystic fibrosis has had limited success because of immune response against viral vectors and short-term expression of cDNA-based transgenes. These limitations could be overcome by delivering the complete genomic CFTR gene on nonintegrating human artificial chromosomes (HACs). Here, we report reconstruction of the genomic CFTR locus and analyze incorporation into HACs of three P1 phage-based and F factor bacteria-based artificial chromosomes (PACs/BACs) of various sizes: (1) 5A, a large, nonselectable BAC containing the entire wild-type CFTR locus extending into both adjacent genes (296.8-kb insert, from kb −58.4 to +51.4) containing all regulators; (2) CGT21, a small, selectable, telomerized PAC (134.7 kb, from kb −60.7 to + 2) containing a synthetic last exon joining exon 10, EGFP, exon 24, and the 3′ untranslated region; and (3) CF225, a midsized, nonselectable PAC (225.3 kb, from kb −60.7 to +9.8) ligated from two PACs with optimized codons and a silent XmaI restriction variant to discriminate transgene from endogenous expression. Cotransfection with telomerized, blasticidin-S-selectable, centromere-proficient α-satellite constructs into HT1080 cells revealed a workable HAC formation rate of 1 per ∼25 lines when using CGT21 or 5A. CF225 was not incorporated into a de novo HAC in 122 lines analyzed, but integrants were expressed. Stability analyses suggest the feasibility of prefabricating a large, tagged CFTR transgene that stably replicates in the proximity of a functional centromere. Although definite conclusions about HAC-proficient construct configurations cannot be drawn at this stage, important transfer resources were generated and characterized, demonstrating the promise of de novo HACs as potentially ideal gene therapy vector systems.
Introduction
Because CF is a recessive disorder, a single copy of the normal CFTR gene is sufficient to achieve functional CFTR levels that avoid CF, making CF an attractive candidate disease for gene therapy. The airway epithelium is the major target organ, as lung disease contributes mainly to morbidity and mortality in CF patients. Moreover, the therapeutic gene should target the cells that normally express CFTR, preferably at physiological levels. It is expected that such physiological and stable CFTR expression levels inside defective cells, or correction of even a fraction of epithelial cells, could alter pathological epithelial physiology, thus being of clinical benefit (Dorin et al., 1996; Ramalho et al., 2002).
CF gene therapy, however, has failed until now because of multiple technical problems, among them gene silencing of cDNA constructs and lack of stability. Thus, for successful gene therapy, with persistent, tissue-specific expression of the transgene, delivery of the complete genomic DNA locus including native regulatory and promoter elements should be achieved. This in turn also involves other technical difficulties, such as construct size. The primary transcript of the CFTR gene is ∼189 kb long (Rommens et al., 1989) and comprises 27 exons. The adjacent genes, GASZ (germ line-specific protein with ankyrin repeats, sterile α motif, basic leucine zipper) and CORTBP2 (widely expressed cortactin binding protein-2), show different nuclear localization in human cells, depending on their differently regulated expression (Zink et al., 2004; Sadoni et al., 2008), and therefore sequences of these genes are unlikely to belong to the chromatin domain that regulates CFTR. Because the distance between the genomic regions of the primary transcripts of these adjacent genes is 283 kb, the size of the functional CFTR locus should be between 189 and 283 kb. The gene order and exon structure around the CFTR locus is highly conserved in vertebrates (Sadoni et al., 2008), bringing into question why this order was maintained for more than 500 million years in the absence of gene regulatory constraints. It is presently not known whether all intronic and extragenic sequences of the CFTR locus are required for normal gene expression or play a role in locus stability.
To avoid random integration into the host chromosomes and allow stable inheritance during somatic cell divisions, our goal here is to achieve incorporation of the complete CFTR genomic locus into a human artificial chromosome (HAC). This requires additional genetic elements, the most important being a functional centromere. HACs based on either centromeric α-satellite DNA (i.e., long arrays of tandem repeats >80 kb) as the only human component in a circular P1 phage-based artificial chromosome (PAC), or on linear, telomerized α-satellite DNA have been shown to faithfully replicate and segregate during mitosis for many cell divisions in the absence of selection (Ebersole et al., 2000; Grimes et al., 2001).
In addition to a functional centromere, the vectors to be transfected into CFTR-expressing epithelial cells should also carry telomeres and should show correct splicing from the de novo formed HAC for correct expression of the transgene. P1- and F factor-based bacterial artificial chromosomes (PACs/BACs) show high stability, have a relatively large insert capacity, and yield a large amount of DNA per culture volume, making them ideal for cloning large genomic sequences (Shizuya et al., 1992; Ioannou et al., 1994). With the current sequence information available for most BAC and PAC clones, these artificial chromosomes have become a popular resource to generate artificial chromosome-based transgenes.
The de novo formation of HACs after transfer of naked DNA molecules is a poorly understood process of DNA assembly, concatemerization, and chromatinization leading to novel, individual chromosomes in the recipient cells. Cotransfections of gene loci and centromeres are usually inefficient, but in combination with intact DNA preparations can represent a workable strategy to characterize the function of a cloned DNA fragment and determine its suitability for the further prefabrication of an HAC construct. Some of the de novo assembled structures may contain all transferred sequences in a suitable composition to constitute a functional HAC. This should include open chromatin regions for the regulated expression as well as a specialized open chromatin domain and replicating portions of heterochromatin for faithful centromere formation (Nakano et al., 2008). Active centromeres are marked by the histone H3 variant CENP-A, and need to be protected from adjacent gene expression.
We have previously reported the genomic PAC construct CGT21 containing about one-half of the CFTR gene locus and a tagged last exon, and demonstrated that it was stably propagated in lung sarcoma cells, where it was expressed and correctly spliced (Laner et al., 2005). Thus, the next logical step was to generate a tagged genomic CFTR construct carrying all 27 exons and flanking regulatory sequences for incorporation into an HAC. We describe herein ligation of such a PAC construct, termed CF225, using previously characterized DNA preparations of two resource PACs. A third resource to study incorporation of a large version of a CFTR locus on an HAC was BAC clone 5A containing the entire wild-type (wt) sequence of a previously described, 310 kb-sized insert of yeast artificial chromosome (YAC) 37AB12 (Anand et al., 1991). This was shown to generate full levels of copy number-dependent expression and to correct the detected features of the phenotype in CF null mice, and has been recombined into a preexisting minichromosome and expressed in Chinese hamster ovary (CHO) cells (Manson et al., 1997; Vassaux et al., 1997; Auriche et al., 2002).
To achieve incorporation of the CFTR locus into HACs, we cotransfected these three cloned CFTR loci with centromere-proficient α-satellite constructs and analyzed their expression. To our knowledge, this is the first report of the successful de novo formation of an HAC containing the entire CFTR locus.
Materials and Methods
PAC/BAC clones covering the CFTR locus
Details of the isolation of clones carrying the CFTR locus from PAC library RPCIP704 (Ioannou et al., 1994), the sequencing procedure, and primers are available online (Ramalho et al., 2004). The ends of PAC CF1 were mapped by pulsed-field gel electrophoresis (PFGE) and amplified with primers In9F and T7 (3′) or directly sequenced with PAC end primer SP6 (5′). The ends of PAC CF6 were mapped with primers P77 and CF12R (5′) and primers aMF and P86 in a long PCR (3′). For construction of the intermediate construct CF1-Met, primers MetF and MetR (Table 1) were used for the V470M exchange by site-directed mutagenesis in a plasmid containing an intron 9 and exon 10 portion during construction of CGT21, and primers E10XcF and CF10NMR (Table 1) for the introduction of the silent XmaI variant and intronic NotI site for PAC ligation. Introduced primers and PCR products were sequenced in the resulting clones. Construct CGT21 (EMBL/GenBank accession number BN000167), and the centromere construct B2T8, containing a 190 kb-sized α-satellite array of chromosome 7 in vector pTAT-BS (17 kb), or TTE1 containing a 116 kb-sized α-satellite array of chromosome 5 in vector pTT (26 kb), are described elsewhere (Laner et al., 2005). BAC 5A was generated from a circularized YAC stuffed with a BAC vector in yeast cells. Intact BAC DNA was purified from yeast cells in agarose plugs and electroporated into DH10B (recA-deficient Escherichia coli strain of large insert BACs/PACs) cells, resulting in the master culture EC100 cCFTR 5A. Insert ends were mapped in two single cell-derived sublines 5A a and b, using primers contYAC and 5′CF (5′) and primers 3′CF and prim2. PCR products were sequenced and compared to the human genome sequence (build 37.1) using blastn at NCBI. The expected size of the insert was confirmed by pulsed-field gel analysis in both subclones.
Modified primers.
Escherichia coli growth and agarose plug preparation
Escherichia coli DH10B strain [F mcrA Δ(mrr-hsdRMS-mcrBC) (Φ80dlacZΔM15) ΔlacX74 deoR recA1araD139 Δ(ara-leu)7697 galU galK rpsL (SmR) endA1 λ nupG] was grown in LB broth or agar medium. PAC clones were selected with kanamycin (30 μg/ml). Telomerized PAC clones B2T8 and TTE1 were selected with both kanamycin (30 μg/ml) and ampicillin (50 μg/ml), and CGT21 with both plus tetracycline (4 μg/ml). BAC 5A was selected with chloramphenicol (12.5 μg/ml).
For large-scale growth simulation, single-cell subclones 2, 3, and 5 of the master culture of CF225 were established on day 1, grown in 50-ml LB cultures at 37°C, and tested for sequence-tagged site (STS) content (see Results) with follow-ups during subsequent growth phases, indicating full stability. For the prolonged growth periods, the subclones were grown in 1 liter of rich, buffered LB medium at 30°C, from which agarose plugs containing ∼1015 cells were prepared on day 5. Subsequently, 1 ml of subclones 2 and 3 was transferred to 1 liter of fresh medium every other day, resulting in a theoretical number of ∼1021 and ∼1027 bacteria on days 9 and 13, respectively, when agarose plugs were prepared.
Preparation of intact DNA
Long DNA preparations in agarose plugs were carried out according to the protocol of Smith and colleagues (1988) supplemented by purification steps to either remove linear yeast chromosomes or E. coli fragments and damaged DNA fractions from the circular DNA preparations as described (Schindelhauer and Cooke, 1997).
Construction of CF225
In-gel restriction digestion and dephosphorylation
For partial digestion of CF1-Met PAC clones, 1 ml of NotI buffer with 150 U of enzyme (New England BioLabs, Ipswich, MA) was added to 7 agarose plugs (∼20 μg) whereas for total digestion of CF6, 600 U of enzyme in 500 μl of reaction buffer with 1× bovine serum albumin (BSA) was added directly onto a slice containing ∼20 plug equivalents (∼100 μg in 2 ml). The plugs were left overnight at 0°C. The next day, another 200 U of NotI was added directly on the CF6 slice. Digestions were carried out for 1 hr at 37°C for the CF1-Met plugs and for 4 hr at 37°C in a wet chamber for the CF6 plugs. The partial digestion of the CF1-Met PACs was stopped on ice by adding 10 vol% of 0.5 M EDTA, pH 7.9, and later NDS buffer (0.5 M EDTA, pH 9; 1% N-lauroylsarcosine) containing proteinase K (10 μg/ml), incubated at 55°C overnight, whereas CF6 was immediately dephosphorylated as follows. After removal of NotI buffer, the CF6 plug slice was washed with bidistilled water, placed on ice, and incubated with 460 μl of 5× calf intestine phosphatase (CIP) buffer containing 20 μl of CIP (New England BioLabs), which was directly added onto the plug slice and incubated for 2 hr. Incubation for 30 min at 37°C was stopped on ice, using 250 μl of 0.5 M EDTA, pH 7.9. CIP was inactivated overnight with NDS buffer containing proteinase K (10 μg/ ml) at 55°C in a humid chamber. After equilibrating in 0.5× TAE, the slice was loaded on a 1% agarose pulsed-field gel.
PFGE separation and DNA isolation
After a 20-hr run at 6 V/cm with a switch time of 1–30 sec in 0.5× TAE at 12°C (CHEF DRII; Bio-Rad, Hercules, CA), the PAC DNA bands were cut from the gel without UV exposure, and kept at 0°C. DNA was recovered from gel slices by electroelution with a BioTrap BT1000 (Biometrics, Schleicher & Schuell, Dassel, Germany) placed in a CHEF DRII or III PFGE chamber in 0.5× TAE.
Ligation and electroporation
The ligation reaction was carried out with 200 μl of eluate, 1× T4 DNA ligase buffer, and 20 μl of T4 DNA ligase (New England BioLabs), at 12°C, overnight. Reaction mixtures without ligase were used as controls. Four microliters of the ligation reaction was added to 5 μl of electrocompetent E. coli DH10B cells (ElectroMAX DH10B cells; Invitrogen, Carlsbad, CA) in a volume of 80 μl of bidistilled water and electroporated (Gene Pulser II; Bio-Rad) at 1.2–1.4 kV, 100 Ω, and 25 μF using 0.1-cm gap cuvettes (Bio-Rad) chilled on ice. Warm SOC medium (500 μl) was immediately added to the cuvette and the content was transferred to sterile 10-ml white cap tubes containing 5 ml of LB medium for 1 hr of growth at 37°C with moderate shaking. Spun bacteria were spread on LB agar containing kanamycin at 30 μg/ml (Sigma, Munich, Germany), and incubated for a minimum of 24 hr at 37°C.
Escherichia coli screening
Individual colonies were picked into 30 μl of TTE buffer (0.01% Triton X-100, 20 mM Tris-HCl [pH 8], 2 mM EDTA [pH 8]), heated for 1 min at 95°C, and spun for 15 min at 10,000 rpm. Two microliters of supernatant was used in a final volume of 50 μl for a PCR of 30 cycles with an annealing temperature of 55°C with primer pair CFi10fus/R7 (Table 1). Two microliters of ligation reaction was used for the positive control. Aliquots of the PCR product were run on a 1% standard plus 1% low melting point agarose gel.
Very long PCR
Agarose plug sections with purified, intact PAC/BAC template of ∼10 μl were added to a total reaction volume of 50 μl containing tuning buffer (1× Eppendorf ), 0.5 mM dNTPs, 0.5 μl Mg(OAc)2, 0.2 mM primers, 2 U of Taq polymerase, and 2 U of TripleMaster enzyme mix (Eppendorf ). The program has an initial denaturation at 92°C for 30 sec, 6–9 cycles of denaturation for 12 sec, annealing at 60–64°C for 1 min, and elongation at 66°C for 15–45 min (20–60 kb), and a final extension at 66°C for 5 min. Ten to 20 μl of the gellike product (0.2% LMP agarose) is mixed with 10 μl of loading buffer (Ficoll) and analyzed on a pulsed-field gel.
Generation of stable HT1080 lines
Linearized DNA was isolated with appropriate restriction enzymes (New England BioLabs). NotI was used for TTE1 and CGT21, I-SceI for B2T8, and SalI for BAC 5A and CF225 inserts. PFGE separation, excision without UV illumination, and electroelution was as described (Laner et al., 2005) with the modification of placing the BioTrap elution chamber in a CHEF DRII apparatus.
Cell culture
HT1080 cells (pseudodiploid, epitheloid fibrosarcoma of the lung) were grown in Dulbecco's modified Eagle's medium (DMEM; PAA Laboratories, Pasching, Austria) supplemented with 10% fetal calf serum (FCS; PAA Laboratories), 1% Glutamine 100× (GIBCO, Karlsruhe, Germany), and 2% (v/v) penicillin–streptomycin (GIBCO) at 37°C and 5% CO2.
Lipofection
Ten-centimeter tissue culture plates containing ∼50% confluent HT1080 cells were washed with phosphate-buffered saline (PBS; PAA Laboratories). For each plate, 6 μl of Lipofectamine 2000 reagent (Invitrogen) and 294 μl of Opti-MEM I (GIBCO) were mixed and incubated for 5 min at room temperature.
Variable volumes of DNA eluate were gently mixed with Opti-MEM, resulting in a volume of 300 μl, which was gently added to the Lipofectamine/Opti-MEM mixture. The tube was turned twice and incubated for 20 min at room temperature. The solution was directly added onto PBS-rinsed cells, using wide-bore plastic Pasteur pipettes to avoid DNA shearing. Plates were incubated at 37°C for 12 hr and the cells were washed with medium and incubated for 1 day without selection. Medium supplemented with blasticidin S (BS, 4 μg/ml; InvivoGen, Toulouse, France) was added on day 3 and changed every other day.
Clone expansion and isolation
Transfected plates were screened by bright-field and fluorescence microscopy with an Axiovert 10 (Zeiss, Oberkochen, Germany) in a dark room equipped with a 100-W Hg lamp and filters for blue excitation and green detection of enhanced green fluorescent protein (EGFP). Individual BS-resistant cell clones were isolated with cloning rings, and expanded in 12-well dishes, 25- and 75-cm2 flasks for PCR screening, growth on and off selection, DNA extraction, RNA extraction, freezing, and fluorescence in situ hybridization (FISH) analyses. DNA for PCR screening was extracted with lysis buffer (0.1 M Tris [pH 8.3], 5 mM EDTA, 0.2 M NaCl, 0.2% sodium dodecyl sulfate [SDS], proteinase K [10 μg/ml]) and ethanol precipitation.
Expression analysis and sequencing
RT-PCR
Total RNA was isolated from confluent BS-resistant cells grown in T25 flasks, using TRIzol reagent (Invitrogen) according to the manufacturer's protocol, with additional DNase digestion (Ambion, Austin, TX). The reverse transcription (RT) reaction was also performed according to the Invitrogen protocol, using a SuperScript III one-step RT-PCR kit. Primer sequences used for RT-PCR (polymerase chain reaction with a reverse-transcribed cDNA template) are shown in Table 1. The efficiency of RNA extraction and the RNA levels in the various samples were controlled by RT-PCR with 1 μl of each RNA sample to amplify a β-actin fragment of 385 bp, using primers AcF and AcR (Table 1). Primers A1R/C16D were used to amplify a 1584-bp product of wt CFTR and primers A1R/GFP1-AL for a 1668-bp product specific for CGT21. Primers B3F/C16D were used to amplify a 391-bp product indicating correct splicing, and a minor product of 208 bp indicating skipping of exon 9, or primers B3F/GFP1-AL resulting in a 474-bp product specific for CGT21, also showing a minor band of 291 bp indicating exon 9 skipping. B3F/C16D were also used for analyzing a silent XmaI variant present in transgene CF225 as follows. To distinguish CF225 and endogenous CFTR transcripts in the respective lines, 15 μl of each RT-PCR was digested in a 25-μl reaction with 20 U of XmaI, resulting in 310- and 81-bp fragments derived of the transgene (and a reduction of the minor 208-bp band to 121 bp from exon 9 skipping of the transgene). Fragments were analyzed on 1% standard plus 1% low melting point agarose gels and images were registered on an UV Gene Genius bioimaging system digital analyzer (Syngene, Cambridge, UK). RT-PCR products corresponding to the main transcript (without exon 9 skipping) were cut from agarose gels, purified, and sequenced.
FISH and immuno-FISH analyses
Fluorescence in situ hybridization (FISH) was carried out as described (Laner et al., 2004) after 30 days on and off BS selection. After initial screening, a minimum of 20 (mostly >30) metaphases were analyzed for each growth phase. The probes used were rsf for the vector sequence including the BS marker (Laner et al., 2005) labeled by PCR with biotin-16-dUTP (Roche, Mannheim, Germany) and Cy3.5 avidin, nick-translated PAC inserts CF1 (biotin-16-dUTP, Cy3.5) and CF6 (digoxigenin-11-dUTP, fluorescein isothiocyanate [FITC]), nick-translated 2.6 kb EcoRI repeats excised of PAC B2 containing the cen17 α-satellite array (digoxigenin-11-dUTP, FITC), and a PCR-generated, nick-translated probe E1 of vector TTE1 (diethylaminocoumarin [DEAC]), hybridizing to cen5 and α-satellite arrays on chromosomes 1 and 19. Signals were visualized with an Axiovert 200 microscope (Zeiss, Oberkochen, Germany) and digitally captured. Triple-color FISH and immuno-FISH analyses were carried out as described. Briefly, for immuno-FISH, CENP-A antibodies (Valdivia et al., 1998) were bound to unfixed, cytospun chromosomes and stained with Cy3.5 (red) before fixation, followed by fixation and FISH analysis with probes E1 (DEAC, pink) and the CFTR region of PAC CF6 (DIG, green).
Results
Construction of the fusion PAC CF225 from characterized resource clones
Here, we aimed at constructing a tagged version of the entire genomic CFTR gene cloned in a P1 phage- or F factor-based artificial chromosome suitable for large-scale, high-quality DNA preparation. In addition to the previously described construct CGT21 carrying a tagged half-locus (Laner et al., 2005) and BAC 5A with the YAC-derived wt insert (Anand et al., 1991), we constructed PAC CF225 carrying the human CFTR gene with all exons and introns plus regulatory sequences by ligating two PACs, CF1-Met (i.e., with M at the M470V locus) and CF6, each containing roughly half of the CFTR gene and flanking regions (Fig. 1). PAC CF1-Met carries an insert running from −60.7 kb upstream from the start of translation in exon 1 to intron 10, and PAC CF6 carries an insert running from intron 10 to +9.7 kb (relative to the end of translation) of downstream DNA. Because of the choice of the resource PACs, both introns 9 and 10 are substantially shortened by 5.1 and 27.1 kb, respectively (Fig. 1A). As a consequence, the described DNase I-hypersensitive sites (DHS) in intron 10 (McCarthy and Harris, 2005) are excluded from locus CF225.
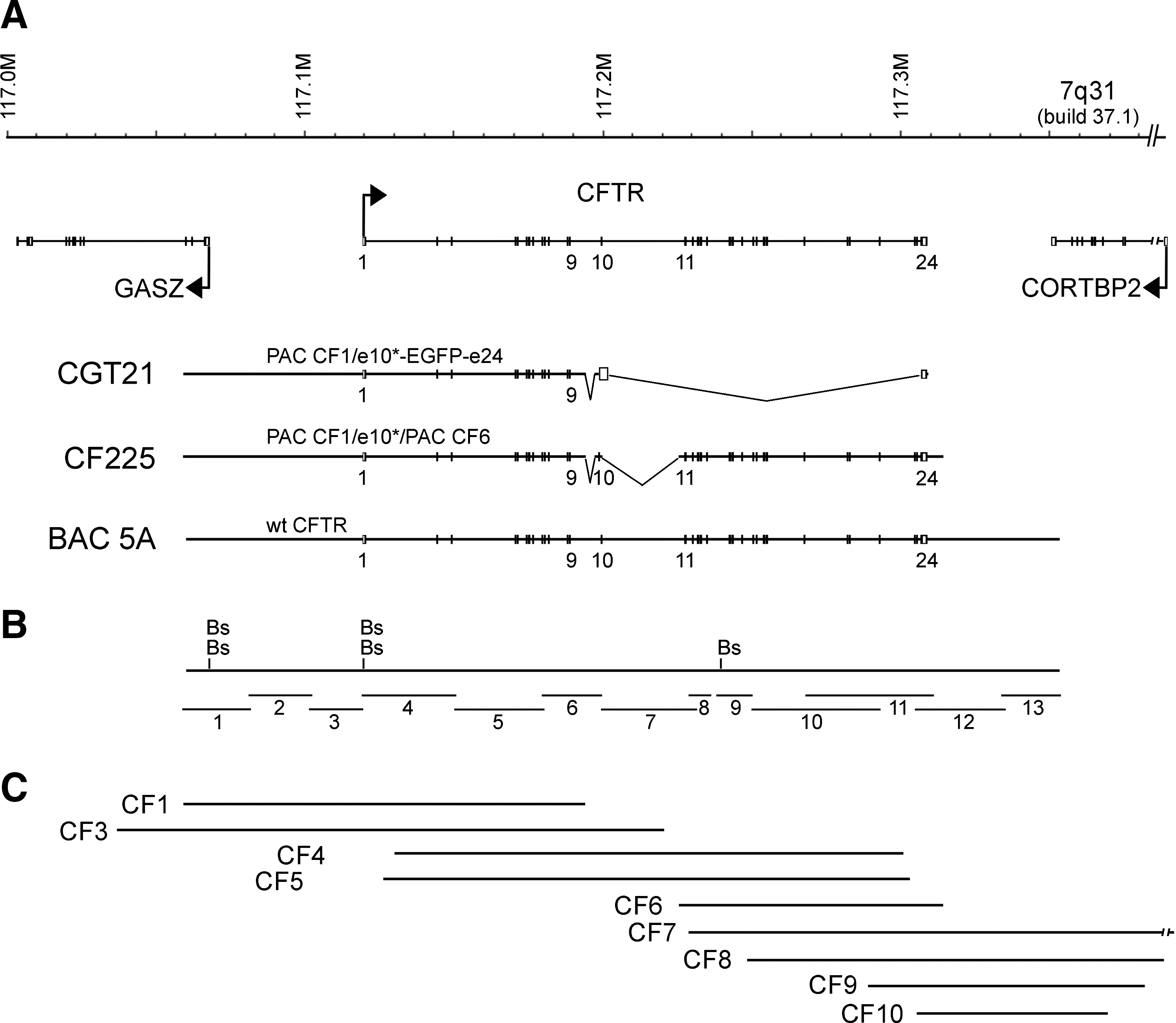
Schematic of the CFTR locus, constructs and resource clones drawn to scale. (
The nine PAC clones, CF1–CF10 (Fig. 1C), covering the human CFTR gene locus (a resource from the Human Genome Project) were analyzed here for exon content by PCR. Because approximately 1 in 25 white individuals carries a CFTR mutation, and functionally relevant polymorphisms exist, we sequenced all exons and splice junctions of the resource PACs before construction. PCR primers and sequencing data are summarized online (Ramalho et al., 2004) and sequences are available under EMBL/GenBank accession numbers AJ574939–AJ575055. We found that PACs CF1–CF5 contained the common splice variant (TG)11T7 described to result in a small proportion of an alternatively spliced product lacking exon 9 (Cuppens et al., 1998; Ramalho et al., 2002), but mostly producing a normal transcript. Exon 10 of PACs CF3–CF5 was found to encode valine at amino acid position 470, which we exchanged to methionine during construction of CF1-Met PAC, giving rise to a 1.7-fold higher chloride conductance activity (Cuppens et al., 1998). Construction of CF1-Met PAC required a number of engineering steps. In short, we used here again a stored DNA preparation of a cloning intermediate based on PAC CF1, which was used for the construction of CGT21, a synthetic half-CFTR locus that was shown to be functional and that expressed the intended M470 variant and tag sequences (Laner et al., 2005). Using the silent XmaI restriction variant introduced into exon 10 downstream of M470, the EGFP/exon 24 portion of CGT21 was replaced by the missing exon 10 and flanking intronic sequences by ligating a PCR fragment of this region from PAC CF3, resulting in PAC CF1-Met. PAC ligation with the exon 10 fragment resulted in three clones (CF1-Met10-43/-44/-54) containing the novel exon 10 cloning junction as assessed by PCR before agarose plug preparation.
Amplification by PCR with primer pairs In9F/P77-B (spanning exon 10), In9F/MetR (spanning from intron 9 to part of exon 10), In9F/CF10R, and CF10F/CF10R (both amplify the entire cloned exon 10 sequence, as well as wild-type upstream and downstream sequences; see primers in Table 1) showed that sizes of the PCR products were as expected for the three clones. Sequencing of the cloned exon 10, determined with primers In9F/P77-B, confirmed the right orientation and the presence of the corrected V470M polymorphism, and that the sequence in all three clones had no PCR-derived mutation. Restriction analysis of the In9F/P77-B PCR product revealed the expected bands corresponding to the synthetic restriction sites XmaI and NotI. Digestion of one-tenth of an agarose plug from all three CF1-Met clones with restriction enzymes NotI and/or BssHII also showed the expected fragment sizes of 9.1, 52.1, and 91 kb (BssHII) and 8, 15, 52.1, and 76 kb (BssHII and NotI), respectively. In addition, the expected band sizes were shown for all clones by a semiquantitative, low cycle number, long-range PCR (LR-PCR) using reactions 2–6 (see Table 2, Fig. 1B, and Fig. 2B) covering the entire CFTR sequence cloned in PAC CF1-Met (see map of CGT21 in Fig. 1A), including the 5.1 kb-sized reduction of intron 9 in reaction 6 (Fig. 2B). These data indicate that the clones contained the complete insert without rearrangement. As the 31.8-kb band of subclone CF1-Met10-43 (lane 3 of reaction 5 in Fig. 2B; compare with Table 2) was faint compared with the products of the other bacterial subclones, possibly indicating a change in some of the bacteria used for plug preparation, the corresponding clone was not further used.
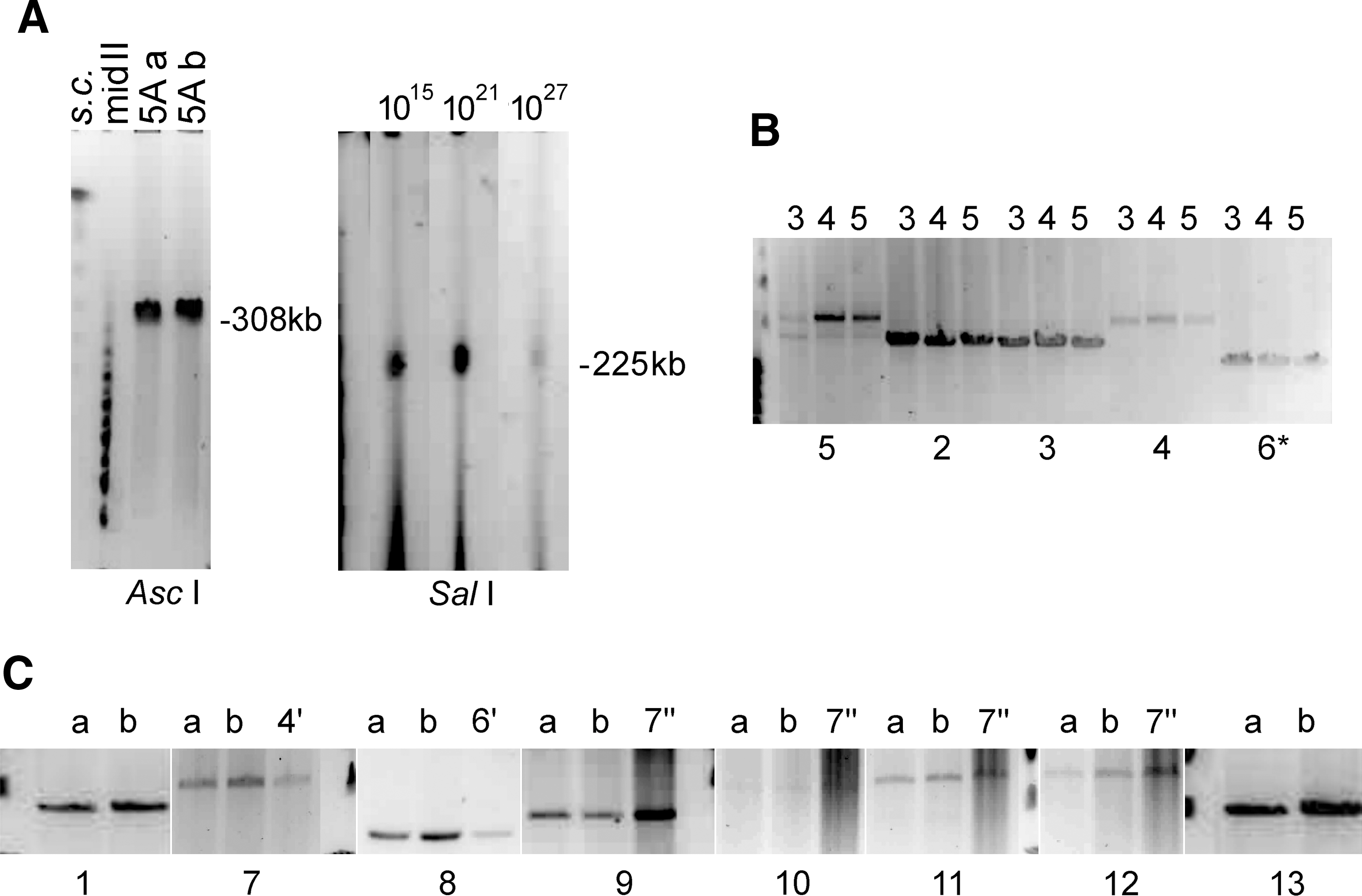
Stability analysis of the CFTR locus cloned in BACs and PACs, using size mapping of restriction fragments and LR-PCRs on pulsed-field gels (ethidium bromide stained). (
See Fig. 1 and Fig. 2B and C.
Product with other than wt size.
After confirming that the CFTR sequence cloned in PAC CF1-Met had the same structure as the original PAC CF1, the next step was to fuse CF1-Met PAC with the insert of PAC CF6, which carries the rest of the CFTR genomic sequence with correct exon and intron junction sequences (Ramalho et al., 2004). The cloning strategy was as follows: (1) partial digestion of CF1-Met PAC (clone 44) with NotI, total digestion of CF6 with NotI followed by dephosphorylation to suppress circularization; (2) separation by pulsed-field gel electrophoresis (PFGE) of NotI restriction products and excision of the bands without UV exposure; (3) electroelution of the DNA fragments from the gel slices and mixture at an ∼1:1 (CF1-Met/CF6) ratio; (4) T4 DNA ligation; (5) electroporation into E. coli DH10B; and (6) PCR screening of KanR colonies with primers CFi10fus/R7 (Table 1) specific for the fusion region between the two PACs. One clone, later designated CF225, was found to be positive for the fusion and for STSs of exons 4 and 12 of both PACs.
Analysis of the cloning stability of CF225 and 5A
Two single E. coli cell-derived clones of BAC 5A were prepared in agarose plugs and subjected to LR- PCRs 1–13 covering the entire locus (Table 2 and Fig. 1), which resulted in identical bands in all sections, indicating stability. During initial growth of the obtained CF225 PAC master culture, 12 single E. coli cell-derived subclones were plated and analyzed by PCR to assess cloning stability. Nine subclones contained the eight tested PCR STSs covering the locus at positions −18 kb (CF-18F/R), −1 kb (CF-1F/R), exon 3 (CF3F/R), exon 10 corrected/fusion (CFi10fus/R7), exon 11 (CF11F/R), exon 17 (CF17bF/R), exon 21 (CF21F/R), and the poly(A) region (CFaF/R; primer sequences are described in Table 1). To further analyze cloning stability, three single cell-derived cultures of PAC CF225 were grown for various time periods simulating a potential final yield of 1015, 1021, and 1027 E. coli cells, each containing up to ∼10 completely replicated copies of the construct (Ioannou et al. 1994). A DNA yield of a 1015 cell preparation represents a normal laboratory scale required for functional testing. The 1021 scale may represent the upper range required for extensive testing in a multicenter gene therapy trial. Larger cell numbers could perhaps represent the range of a continuous production of a DNA-based drug for the clinical setting.
Restriction digestion of plug material from the three single cell-derived colonies of CF225 showed an identical insert size of ∼225 kb (SalI) (Fig. 2A) and identical BssHII fragments of the predicted sizes of 96.5, 83.4, 52.1, and 9.1 kb (data not shown), indicating high overall cloning stability of the locus. For the large locus 5A, restriction analysis of plug material from two single cell-derived subcolonies, a and b (each ∼1015), showed the expected AscI-linearized size of ∼308 kb (Fig. 2A) and identical BssHII patterns of the bands predicted to be 128.6 and 52.1 kb (within the locus), and ∼112.2, ∼9.7, and ∼5 kb (including BAC vector portions) in both cases (data not shown), indicating both stability of the internal structure and the presence of the promoter regions, as expected. Altogether, these data indicate a faithful and stable propagation of the CFTR loci in both PAC CF225 (225.3 kb insert) and BAC 5A (296.8-kb insert). CGT21 stability was previously demonstrated (Laner et al., 2005).
Structural analysis of cloned CFTR loci
LR-PCR
The CFTR loci cloned in the resource PACs and the YAC-derived BAC 5A are from independent sources. To analyze the stability of these cloned CFTR loci, the entire genomic regions were analyzed in detail by LR-PCR spanning the whole CFTR locus. For this analysis, a minimum of 13 overlapping PCR products (except for a ∼3-kb gap between reactions 8 and 9 in intron 14) were generated (Fig. 1B and Fig. 2). Primer pairs and sizes of these 13 and other redundant products used to confirm the structure and internal order within the clones from the different sources, or to analyze end products, are given in Table 2. The PCR products showed identical fragment sizes for all genomic regions from the various clones (Fig. 2 and Table 2). Two exceptions were found within phosphate-bound CG dinucleotide (CpG)-rich regions close to the BssHII restriction sites in the promoter region of CFTR and a region of low complexity within intron 14, both representing sequences that amplify inconsistently. Analysis of these regions revealed either short side products, which were identical in both BAC subclones studied (Table 2), or resulted in failure of amplification in all resource clones (3-kb gap in intron 14). In addition, both regions do not differ from the expected fragment sizes in a BssHII restriction analysis. Thus, the long PCR and pulsed-field restriction analysis demonstrates the presence of the expected structure and a lack of internal rearrangements in clones of both resources. These data indicate that the entire CFTR gene can be stably cloned in yeast and in E. coli. Moreover, recloning of the YAC insert into the BAC vector via yeast recombination did not introduce internal rearrangements or deletions. On occasion, deletions have been observed under prorecombinatorial conditions in E. coli expressing RecE and RecT in the PAC host DH10B, which is RecA negative for the stable propagation of large inserts (Shizuya et al., 1992; Ioannou et al., 1994), as reported for a repetitive region 50 kb 3′ to the β-globin locus (Imam et al., 2000). In addition, electroporation of an intact DNA preparation of BAC 5A apparently did not result in a deletion, as has frequently been observed with circularized YAC/BACs >250 kb (Cocchia et al., 2000). Overall, these structural analyses indicate high cloning stability of the CFTR locus in E. coli, suggesting that a genomic DNA of clinical use can be produced in sufficient quantity and quality.
End sequencing of loci CF225 and 5A
To determine the ends of CF225, LR-PCRs were carried out with primer pairs SP6/CF1-5R (5′ end) and CF6-4F/SP6 (3′ end) (Tables 1 and 2). These were found to amplify 605- and 265-bp products (Fig. 3E), respectively. SP6 hybridizes to a sequence present on both sides of the ligated PAC vector backbone of CF225. The amplified fragments were sequenced and compared to the human genome build 37.1 using blastn at the National Center for Biotechnology Information (NCBI). The reconstructed CFTR locus runs from nucleotide position −60651 relative to the start of translation to nucleotide position +9767 relative to the end of translation. Both ends coincide with a Sau3AI site, in agreement with the partially digested genomic DNA cloned in the BamHI site of the PAC vector pCYPAC2N in library RPCIP704 (Ioannou et al., 1994).
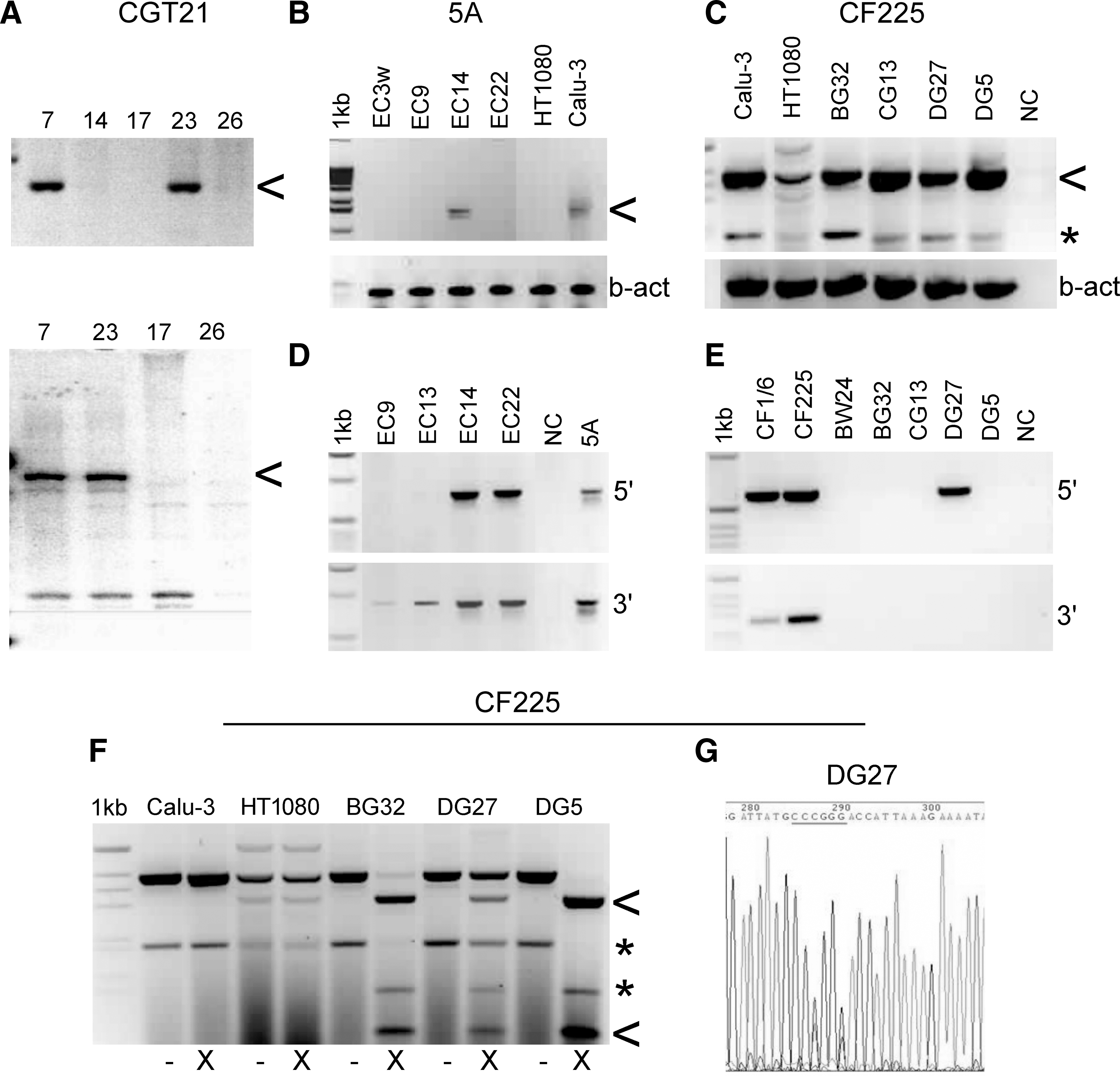
Expression of CFTR constructs and analysis of locus ends. (
As a result of the cloning procedure, CF225 has two deletions within introns 9 and 10, representing regions that were not covered by the genuine PACs CF1 (intron 9) and CF6 (intron 10), and were omitted by reconstructing exon 10 and its flanking intron sequences. The resulting 5.1-kb deletion in intron 9 was already known from the functional analysis of CGT21 (Laner et al., 2005) not to affect expression and correct splicing in this region. To precisely locate and determine the extent of those deletions, PCRs were carried out with primer pairs In9F/C16D (intron 9/exon 10) and CFi10fus/CF11R (intron 10/intron 11) (Table 1), which generate DNA fragments of 735 and 773 bp, respectively. The PCR products were sequenced and compared against published human BAC sequences using blastn at NCBI. In9F hybridizes to nucleotide positions 73794–73816, and C16D to nucleotide positions 79552–79529 on the CFTR genomic sequence (relative to the start of translation). The deletion in intron 9 is located from nucleotide position 74148 to 79204 and is 5058 bp long. CFi10fus hybridizes to nucleotide positions 80085–80105, and CF11R to nucleotide positions 107929–107952. The deletion in intron 10 runs from nucleotide positions 80281 to 107408 and is 27,128 bp long. DNA from HT1080 cells served as a control for the absence of amplification from wt CFTR loci (data not shown).
To determine the ends of the large locus of BAC 5A, PCRs were carried out with primer pairs ContYAC/5′CF (5′ end) and 3′CF/Prim2 (3′ end) (Table 1), which were found to amplify ∼0.8- and ∼0.9-kb products, including 735 and 558 bp of the insert, respectively (Fig. 3D). The amplified fragments were sequenced and blasted against human genome build 37.1. The YAC-derived CFTR locus in BAC 5A runs from nucleotide position −58351 relative to the start of translation to nucleotide position +51381 relative to the end of translation. Both ends coincide with an EcoRI site, in agreement with the partially digested genomic DNA ligated to the arms of the YAC vector pYAC4 (Anand et al., 1991).
Cotransfection experiments
To analyze the functional incorporation of the obtained CFTR loci in a de novo formed human artificial chromosome (HAC), we employed simple cotransfection experiments with linearized DNA components. HAC formation by cotransfection is not efficient, but has successfully been used to incorporate an HPRT gene into a de novo formed HAC (Grimes et al., 2001). In addition, it is advantageous not to prefabricate a fixed composition regarding the size of the locus, the orientation with respect to the centromere, and the type of centromere included. Indeed, cotransfection of the HAC components separately may to some extent increase the flexibility of the assembly of an HAC, which may adopt some rules of how stable structures need to be composed. On the other hand, the ongoing repair and recombination processes required to generate a stable genetic entity may by chance alter the input DNA. Thus, cotransfections represent a workable tool to initialize HAC formation studies.
Construct CGT21 (Fig. 1) was cotransfected with construct B2T8 (for both see Laner et al., 2005) carrying a centromere-competent α-satellite array of 190 kb derived from chr17, which was cloned in the telomerized PAC vector pTAT-BS (17 kb) (Ebersole et al., 2000) and transfected as an ∼200-kb construct. Cell lines 1–7 were transfected with the telomerized, ∼151 kb-sized construct CGT21 alone, which contains a BS-selectable marker, resulting in integration. Cell line 7 showed expression of the tagged transgene by RT-PCR (Fig. 3A), and sequencing of the entire coding region demonstrated correct expression and splicing, and the presence of the improved 470M variant (the analyzed cDNA sequence is available at EMBL/GenBank under accession number AY299332; Laner et al., 2005). From 24 cotransfected lines, numbered 8–32, 13 lines were screened by FISH. Cen17-based HACs lacking CFTR sequences were observed in four lines, and three lines showed integration of the vector probe rsf only. Four lines did not allow a consistent detection of the transferred constructs, and two lines showed HACs with CFTR and cen17 sequences. Only HAC line 23 showed expression of the CGT21 transgene and persistence still after 30 and 50 generations without selection. This was demonstrated by RT-PCR analyses specific for the transgene, using primers B3F and GFP1-AL, amplifying a 474-bp product between exons 8 and 10 (synthetic portion) (Fig. 3A, top), or primers A1R and GFP1-AL, generating a 1668-bp product from exon 1 to the synthetic exon 10 (Fig. 3A, bottom). Despite correct sequences and expression, the EGFP tag did not result in visible green fluorescence under the microscope, as previously described for integrated lines (Laner et al., 2005), suggesting that a functional CFTR/EGFP fusion protein is not sufficiently accumulating in HT1080 cells. FISH analyses of line 23 after 30 days of growth with BS selection, and after 30 and 50 days without selection (approximating 80 generations in total), revealed that the HACs contained a stable structure consisting of cen17 and CF1 sequences (Fig. 4A). However, at later culture stages, an integration of HAC material in a host chromosome was detected. This integrated material showed truncation products with separated CFTR and cen17 signals with both probes, in addition to an HAC. Attempts to select out a single cell-derived subline only containing an HAC with CFTR and cen17 sequences were not successful after expanding four sublines, two isolated and expanded on BS, and two off selection.
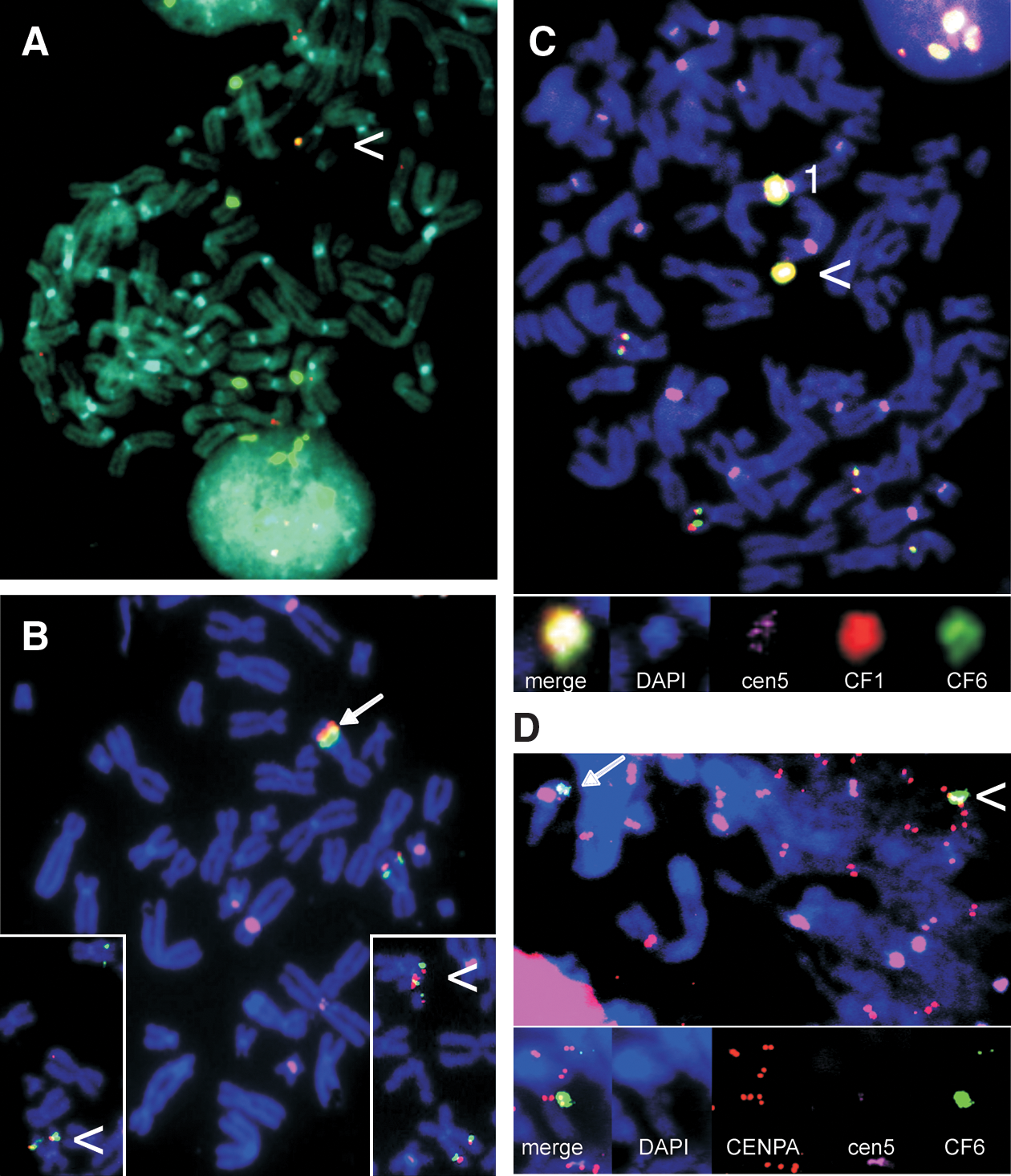
FISH-based HAC detection and analysis of centromere chromatin, using immuno-FISH. (
The ∼300-kb insert of BAC 5A was cotransfected with the 116 kb-sized, centromere-proficient α-satellite array of chromosome 5, termed “E1” (Laner et al., 2004, 2005), which was cloned in the tetratelomeric PAC vector pTT (26 kb), resulting in construct TTE1 (142 kb). Construct TTE1 consistently results in about two-thirds green clones, when using a stored preparation with two-thirds of the molecules having an EGFP marker (data not shown). The EGFP marker is prone to deletion from the vector because of its location between two canonical telomere repeats, and the lack of an antibiotic marker gene within this fragment of the vector precluding its selection against deletion. Nevertheless, once a good DNA preparation has been characterized, a high rate of green fluorescent clones can be obtained. Cotransfection of the 133 kb-sized TTE1 fragment containing a duplicated BS-selectable marker gene and the EGFP marker, with the unselected, ∼300-kb fragment of BAC 5A resulted in 24 green clones. Three independent green lines (EC9, EC14, and EC22) were found to be positive for both ends of the 5A locus in a PCR screening (data not shown). After expansion for an additional 30 days off BS selection, lines EC14 and EC22 stably contained both ends (Fig. 3D). In a FISH screen, EC9 and EC22 showed either signals for cointegration of both vector and CFTR, or of vector, centromere, and CFTR in an endogenous chromosome. Cell line EC14 showed a free HAC in addition to an integration of the HAC material into chromosome 1, which was present on and after 30 days off selection (Fig. 4C). The integrated centromere at a distance of ∼20 Mb from the endogenous chr1 centromere was inactivated and did not bind CENP-A. In contrast, the free, de novo formed HAC showed CENP-A in a combined immuno-FISH analysis (Fig. 4D), indicating the formation of a functional centromere in the vicinity of the CFTR locus on the free, de novo formed HAC. Completeness of the CFTR locus on the HACs is suggested by the presence of the ends, as confirmed by PCR and by RT-PCR analyses using primers A1R and C16D, which amplify a 1584-bp product from exon 1 to exon 10 (Fig. 3B). Although these showed various levels at different culture phases and between experiments, increased amounts of wt sequences were present in line EC14, compared with the weaker expression of the endogenous loci in the HT1080 cells. Expression appeared still increased after 30 days of culture (approximating 30 generations) in the absence of selection. Expression of the transgene was further supported by detection of the 470V polymorphism of the BAC by sequencing the RT-PCR product of line EC14. In contrast, sequencing of a genomic PCR product of exon 10 of the endogenous loci in an HT1080 control line, using primers CF10F/CF10R, revealed that it was homozygous for 470M (data not shown). The presence of multiple complete loci is also suggested from the FISH signals of both locus halves on the HACs, which appear to be organized into multiple domains arranged around the comparably tiny centromeric portion, like the leaves of a flower, each domain containing CF1 and CF6 sequences in a triple-color FISH analysis (see Fig. 4D inset, captured under low-exposure conditions to avoid blooming).
Four rounds of colipofection of the 225-kb insert of PAC CF225, with the characterized preparation of construct TTE1 (133 kb fragment), resulted in 185 (white and green) cell clones, 122 of which were expanded and screened by PCR with primers CFi10fus/R7 (Table 1), specific for CF225. Five individual cell clones, BW24, BG32, CG13, DG27, and DG5, were positive for the exon 10 junction region, indicating that only 1 in ∼25 cell clones was cotransfected with both CF225 and TTE1 DNA. Although the ratio of intact molecules per liposome preparation was not further assessed in the four individual transfer experiments resulting in the 122 analyzed clones, there is no obvious explanation for this low cotransfection efficiency. Other cotransfections >100 kb regularly approached efficiencies of 1 in ∼3–10 clones when equimolar DNA preparations were used under similar conditions, regardless of whether one or both components carried the BS marker. Successful cotransfections leading to HAC formation were possible at higher efficiency even if one component lacked telomeric repeats, as was the case here for the 5A and CF225 inserts. Nevertheless, the cotransfections of CF225 still represented a workable means of selecting out stable clones, allowing an initial functional assessment of the locus not carrying a selectable marker.
RT-PCRs were carried out with primers B3F and C16D (Table 1), generating a spliced product of 391 bp between exons 8 and 10, which represents a mixture of products from endogenous CFTR genes of the HT1080 cell line and the transgene loci. RNA/cDNA preparations were controlled with β-actin primers (Fig. 3C). All lines showed various levels of CFTR expression after 30 days off selection. To distinguish between endogenous and transgene expression, the RT-PCR products were digested with XmaI cutting the engineered exon 10 from CF225 into two fragments of 310 and 81 bp (Fig. 3F). In four cell lines, various proportions of the CFTR transcript resulted from the transgene, which demonstrated expression levels of the transgene above wt background in most cases, and showed correct splicing (Fig. 3F). Cell line BW24 did not express the transgene (data not shown). The expressing cell lines and parental HT1080 cells were further analyzed by sequencing of the RT-PCR products with the same primers and primer CFc3F (Table 1), demonstrating that all lines contained both the 470M polymorphism and the synthetic XmaI variant in exon 10, confirming transgene origin. Figure 3G shows a section of the electropherogram from sequencing of line DG27, evidencing both the XmaI site (CCCGGG) expressed from CF225 and the wt sequence (CCTGGC) from the HT1080 loci, both encoding wt amino acids 499P and 500G (corresponding to silent exchanges).
To check the integrity of construct CF225 in the clonal cell lines, PCRs were carried out with primer pairs SP6/CF1-5R for the left vector/CFTR junction of 605 bp, and CF-4F/SP6 for the right CFTR/vector junction of 265 bp (Table 1). Of the five cell lines that were positive for the exon 10 junction PCR, line DG27 has kept the left end of CF225, confirmed by sequencing the PCR product. All five lines were negative for the right end junction PCR (Fig. 3E) and for two other LR-PCRs extending approximately 2 and 3 kb from the right end into the CFTR locus (data not shown), suggesting that 3′ DNA of CF225 was lost in all four expressing lines. Loss of locus ends may have occurred during integration or subsequent propagation.
Triple-color FISH analyses of these cell lines after 30 days of growth on and 30 days off BS selection revealed either integration of the CF225 locus into host chromosomes, or integration and truncation in all five clonal cell lines. No HACs carrying the CFTR locus were observed. Clonal line DG27 showed a stable cointegration close to the endogenous CFTR gene on chr7 (arrow in Fig. 4B), which was positive for CF1, CF6, and rsf signals on and off selection. Weak signals for E1 were regularly visible at the site of integration. Line BG32 revealed a distal/telomeric integration into a chromosome (non-7), which was positive for CF1, CF6, and rsf, but not for E1 signals, indicating that the α-satellite DNA was either not cotransfected or lost. Cell line DG5 showed integration of CF1, CF6, and E1 signals in a distal position of chr19q, and line CG13 showed integration of CF1 and CF6 portions in the p arm of a metacentric chromosome (non-7) accompanied by truncation. The truncated portion containing the endogenous centromere and signals for CF1 and CF6 was stably maintained whereas the p arm portion positive for signals E1 and CF6, but not CF1, was frequently lost. In cell line BW24 only CF6 sequences were detected on a small truncated chromosome. Overall, we conclude that CF225 and the E1 centromere did not efficiently form a stably replicating structure together. Instead, rare stable clones were selected out that contained at least the BS marker and various portions of locus CF225 lacking the very 3′ end, which nevertheless showed expression of the tagged exon 10 sequence in four of five lines obtained.
Discussion
CF gene therapy, with delivery of the correct CFTR gene to cells, promises a great benefit for all CF patients, irrespective of the type of mutation. However, classical gene therapy to cystic fibrosis has had limited success due to immune response or short-term transgene expression of cDNA delivered by viral vectors or cationic lipids due to silencing. These limitations could be overcome by delivering the complete CFTR gene with relevant regulatory regions within its genomic context on nonintegrating human artificial chromosomes. Thus, for gene therapy success, a stable and functional CFTR locus containing all exons, introns, and regulatory elements should be inserted into a human artificial chromosome (HAC) vector that can be delivered to cells.
CGT21, a genomic construct containing about one-half of the CFTR locus from which expression and correct splicing was demonstrated (Laner et al., 2005), is a functional gene with 10 exons encoding a nonfunctioning CFTR protein derivative. Because of its reduced size and the tagged exon, such a construct can ease certain transfer studies. Another version, CGT20, with opposite orientation with respect to the BS marker, and basic PAC version CG2 are also available (our unpublished results).
Here, we aimed at producing and analyzing the stable incorporation of the entire CFTR locus (including all exons and introns, and regulators) on a de novo HAC, which could be used for gene therapy. The de novo approach based on the transfection of “naked” DNA molecules to the cells of the patient offers a technically feasible strategy toward long-term gene therapy, provided sufficient numbers of large constructs can be produced in a functional form and delivered.
We describe the successful generation of a large construct containing the CFTR locus with a reduced size (PAC CF225), constructed from characterized CFTR PAC resources, with a functionally optimized polymorphism and a silent restriction variant in exon 10, and also used the 296.8 kb-sized BAC 5A covering the entire wt CFTR locus with all regulators (see below). For reconstructing the tagged CFTR locus, we adopted a technically simple approach based on a “rare cutter” (NotI) restriction site present at the PAC vector boundary, thus allowing “conventional” cloning on a genomic scale, followed by electroporation of the entire CFTR fusion PAC into E. coli DH10B cells. To our knowledge, cloning of PAC fragments of this size, which is based on intact DNA preparations, represents a novel method by which to assemble genomic loci.
To define the CFTR locus, we used our previous data, describing that the human CFTR gene and its adjacent genes localize individually and differentially to distinct nuclear regions, according to their transcriptional activity. This suggests that the principal CFTR regulatory information is contained within the limits of the two adjacent genes, that is, <283 kb (Zink et al., 2004; Sadoni et al., 2008). In addition to developmentally regulated alternative 5′ exons (Mouchel et al., 2003) numerous DNase I-hypersensitive sites (DHSs) have been described and analyzed within this region (McCarthy and Harris, 2005; Ott et al., 2009). Most of these are included in CF225, namely DHSs that lie at −20.9 kb (to the start of translation); in introns 1, 2, 3, 11, 16, 17a, 18, 20, and 21; and 3′ to the gene at +5.4, +6.8, +7, and +7.4 kb relative to the end of translation. DHSs not present in CF225 likely belong to the adjacent genes, or are in the reduced region of intron 10, including the DHS that has been shown to be active in intestinal cells (Ott et al., 2009), and DHSs at +15.6, +17.2, and +20.1 kb (to the end of translation). The latter are placed in the vicinity of the start position of an ∼17-kb transcript mapped in opposite direction of the CFTR gene (source: human genome build 37.1 at the NCBI), running in from the intergenic region between CFTR and CORTBP2 toward the end of the CFTR transcript.
Similar transcripts in the opposite strand can be found in the 3′ region of many genes, for example, the genes FOXP2, MDFIC, CAV2, CAV1, MET, and CAPZA2, to name just a few in the vicinity of the CFTR locus (our unpublished observation). The common presence of such 3′ transcripts suggests an important genetic function, for example, in gene regulation, formation of a chromatin domain, or stability.
By cotransfecting CF225 and TTE1, we obtained five BS-resistant cell lines that, however, did not show de novo formation of HACs. All obtained integrated cell lines have lost the 3′ end of CF225 DNA, which could include loss of DHS at +5.4, +6.8, +7, and +7.4 kb. Although these DHSs are potentially absent and the ones at +15.6 kb and downstream are not in the construct, it was possible to detect the XmaI-specific, correctly spliced transcript in RT-PCR products from four of five cell lines, albeit at various levels relatively to the endogenous CFTR genes of HT1080.
Because of the poor cotransfection efficiency observed with PAC CF225, it remains unknown whether the CF225 locus is complete and replication-competent (autonomously) when ligated to a centromere. It seems possible that additional 3′ sequences, such as an origin of replication, or chromatin barriers separating gene expression from centromere function, could be required on a prefabricated HAC. PAC CF225 contains three NotI sites at both locus ends and in intron 10, which is 27 kb reduced in size compared with the wt locus. Thus, the possibly critically short 3′ end, or the lacking DHS in intron 10 can easily be altered by cloning suspected regulatory regions into the NotI sites, representing an interesting system to verify expression regulation and stability on an HAC in suitable cell or animal models.
To study incorporation of the three distinct versions of the E. coli-cloned CFTR locus into de novo HACs, colipofection experiments were carried out in HT1080 cells. A workable number of 24 and 25 clones, respectively, revealed de novo formation of a stable HAC when using either the short, BS-selected construct CGT21 (151 kb, linearized with I-SceI) and the BS-selected cen17 construct B2T8 (∼200 kb, linearized with I-SceI), or when using the long, nonselected wt locus 5A (∼300-kb SalI insert) and the BS-selected cen5 construct TTE1 (133 kb, linearized with NotI). In contrast, the more elaborate analysis of 122 clones from the cotransfection of the midsized, nonselected locus CF225 (225 kb, SalI insert) with the BS-selected cen5 construct TTE1 (133 kb, linearized with NotI) did not reveal HAC formation. Only five integrated lines positive for CF225 were obtained. Although the numbers are not sufficient to conclude that CF225 is less HAC competent than 5A, it is tempting to speculate that CF225 is prone to instability. Although the larger 5A construct has kept both locus ends in 2 individual cell lines (of 25 analyzed lines, which represent fewer than 25 individual lines because double-picked lines were excluded when they have been identified), including an integrated and the HAC line, all 5 integrated lines of the midsized locus CF225 (of 122 lines) have lost the 3′ end of the locus. Nevertheless, the specific RT-PCR analysis demonstrated that CF225 is expressed in four of five cell lines, and that the introduced XmaI variant in exon 10 can be used to distinguish between transgene and endogenous transcripts. It is possible that CF225 underwent a 3′ trimming process to generate a stable structure, which may have reduced the cotransfection efficiency and thus HAC incorporation.
The HAC present in cell line EC14 was highly stable on and off BS selection and contained the entire wt CFTR locus. Moreover, the cen5 α-satellite DNA present on the HAC bound CENP-A, indicating formation of an active centromere. Accordingly, the HAC in line EC14 represents the first de novo formed human artificial chromosome carrying the entire CFTR locus. Cell line EC14 showed expression of the CFTR locus above the endogenous background on and off selection, suggesting that the functional locus was transferred. Unambiguous proof of expression from the CFTR genomic construct that localizes in the vicinity of the active centromere is not possible at present, because high copy numbers are likely to be present on the HAC, resulting in a greater distance to the centromere of some copies. Moreover, HAC material integrated into chr1 could also contribute to expression.
We previously demonstrated that PAC-cloned, >100 kb-sized human α-satellite arrays with ∼99% identical 2-kb repeats did not result in internal recombination or deletion during a prolonged growth period of 2× ∼400 E. coli generations in a colony-replating experiment (Schindelhauer and Schwarz, 2002). Here, we provide extensive structural data, including a set of LR-PCRs covering the CFTR loci from various cloning sources, that indicate stability. Moreover, we could confirm cloning stability after prolonged growth periods in fluid culture, which would be required to test and produce an HAC-based DNA therapeutic product. Overall, the cumulative data presented here suggest that the construction of a prefabricated CFTR-HAC based on the defined resource clones is feasible. These results are thus promising for the development of HAC-based therapy.
Footnotes
Acknowledgments
The authors thank Sebastian Beck for RT-PCR advice and Sónia Pedro and Ana Cardoso for RT-PCR product sequencing, Ulrich Zissler for PAC end sequencing, Steffi Simon for setting up large-scale growth simulations, Michael Speicher and Burgis Cleve for access and support with the FISH microscope, Manuel Valdivia for the kind gift of CENP-A antibodies, Christina Auriche for BAC handling, and Angelika Schnieke for excellent working conditions, a supportive atmosphere, and hospitality at her chair. This work was financed by WZW/TUM for student bench fees, two grants of the Forschungsgemeinschaft Mukoviszidose (D.S.), Vaincre la Mucoviscidose grants (D.S. and A.L.), two grants of the Friedrich Baur Stiftung (D.S.), Deutsche Forschungsgemeinschaft (D.S.), and multiannual funding from BioFIG (FCT, Portugal and FEDER/EU), the European Community (QLK3-CT-2002-02119) and partially Fondazione Italiana per la ricerca sulla fibrosi cistica and Istituto Pasteur-Fondazione Cenci Bolognetti, Sapienza University (F.A.). L.R. was a guest scientist employed by La Sapienza University, Rome (Dottorato in Biologia Cellulare e dello Sviluppo). C.B. is the recipient of doctoral fellowship SFRH/BD/17912/2004 (FCT, Portugal).
Author Disclosure Statement
No competing financial interests exist.