
Abstract
Adeno-associated viral (AAV) vectors are the most efficient in vivo gene transfer tools for gene therapy applications. Efforts have been made to translate encouraging results in small animal models to human patients. However, the need for large quantities of vector for clinical application remains a great challenge. Developing novel AAV vectors with enhanced infectivity may reduce the high vector dose requirement for many applications such as gene therapy for muscular dystrophy. Selective mutation of AAV capsid surface-exposed tyrosine (Y) is a novel strategy to improve transduction efficiency. AAV6 has been considered one of the most robust muscle gene delivery vehicles. Here, we hypothesize that AAV6 transduction efficiency can be further enhanced by mutating surface Y to phenylalanine (F). We found that mutants AAV6-Y445F and AAV6-Y731F, especially the former, achieved more efficient gene transfer than the original AAV6 after intramuscular administration to mice. Expression of both firefly luciferase and alkaline phosphatase reporter genes increased up to 8-fold and DNA copy numbers in muscle increased up to 6-fold. Our results suggest that tyrosine-mutant AAV6 vectors may represent powerful tools for testing muscle gene therapy in animal models and potentially in humans.
Introduction
AAV serotype 2 (AAV2) was the first AAV tested for gene transfer applications (Samulski et al., 1982). Many studies have indicated that AAV2 can direct persistent and relatively strong gene expression in striated muscles (Xiao et al., 1996; Li et al., 2003; High, 2004). However, the restricted tissue tropism, low efficiency of gene transfer, slow onset of gene expression, and highly prevalent preexisting immunity in human populations have hampered the successful clinical development of AAV2-based vectors (Gao et al., 2005). To overcome such drawbacks of rAAV2 vectors, many investigators have begun to explore either naturally occurring (Gao et al., 2002) or artificially modified (Muzyczka and Warrington, 2005; Yang et al., 2009) novel AAV capsids, which are the major determinant of tropism and transduction efficiency.
For artificially modified AAVs, the novel tyrosine-mutant AAVs stand out. The tyrosine-mutant AAV2 vector was first described by Zhong and colleagues (2008). The idea was originated from the understanding of factors affecting AAV transduction. The efficiency of AAV transduction depends on the efficiency at each step of AAV infection: binding, entry, vial trafficking, nuclear entry, uncoating, and second-strand synthesis (Daya and Berns, 2008). Among them, inefficient AAV trafficking (Hauck et al., 2004) and second-strand synthesis (Ferrari et al., 1996) have been identified as rate-limiting factors. It was discovered that epidermal growth factor receptor protein tyrosine kinase (EGFR-PTK) signaling negatively affects AAV2 vector transduction by priming capsid ubiquitination and proteasomal degradation (Zhong et al., 2007). EGFR-PTK also phosphorylates tyrosine residues on AAV2 capsids. It was hypothesized that mutations of the surface-exposed tyrosine residues might allow the vectors to evade phosphorylation and subsequent ubiquitination and, thus, prevent proteasome-mediated degradation. Indeed, mutations of the surface-exposed tyrosine in AAV2 vectors led to dramatic improvement of transduction efficiency both in vitro and in vivo (Zhong et al., 2008). Considering the limitations of AAV2 for future clinical application, testing alternative tyrosine-mutated AAV serotypes may have additional translational implications.
To date more than 11 AAV serotypes (Wu et al., 2006) and more than 100 naturally occurring primate AAV variants have been reported (Gao et al., 2005). For those reported AAVs, AAV6 was demonstrated to have superior ability to transduce skeletal muscles, with greater than 500-fold higher transduction efficiency than AAV2 vector when the vector was delivered intramuscularly (Blankinship et al., 2004). In this study, we compared the transduction efficiency of tyrosine-mutant AAV6 vector versus the original AAV6 vector. The identification of more potent AAV6 vectors will allow for improved transduction efficiency at a lower dose. This will lead to a favorable safety profile and ease the burden on vector production.
Materials and Methods
Recombinant AAV stock
The original AAV6 packaging plasmid, and the original AAV8 and AAV9 packaging plasmids, were gifts from J. Wilson and G. Gao (Bish et al., 2008). Tyrosine-mutant AAV packaging plasmids were generated by site-directed mutagenesis as described for AAV2 tyrosine mutants (Zhong et al., 2008). The cis plasmid pcis.RSV.AP has been described previously (Yue et al., 2008) and the plasmid pcis-cmv-luciferase was reported elsewhere (Wang et al., 2008). Recombinant AAV vectors were generated by triple plasmid transfection using pcis.RSV.AP, adenoviral helper plasmid (pHelper; Stratagene, La Jolla, CA), and the respective packaging plasmids (all carrying the AAV2 rep gene). For the luciferase vectors, viral stocks were generated by PEG precipitation followed by double CsCl gradient purification. For the alkaline phosphatase (AP) vectors, viral stocks were purified through three rounds of CsCl2 ultracentrifugation. After two changes of dialysis in HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid] buffer at 4°C for 48 hr, viral genome copy titer was determined by quantitative PCR and slot blot.
In vivo gene transfer
All animal experiments were approved by the local institutional animal care and use committee. For the luciferase studies, 6- to 8-week-old male ICR mice were purchased from Taconic (Hudson, NY). Various doses of AAV vectors (3 × 1010 vector genomes [VG]/muscle for the low-dose group, and 3 × 1011 VG/muscle for the high-dose group) were directly injected into tibial anterior (TA) and gastrocnemius (Gas) muscles. For AP studies, 5-week-old male C57BL/6 (BL/6) mice were purchased from Jackson Laboratory (Bar Harbor, ME). AAV vector was directly injected into the middle belly of the TA muscle at a dose of 5 × 108 vector genomes (VG) of AAV particles (30 μl) per muscle.
Optical imaging study
For live animal imaging studies, mice were injected with luciferase substrate (firefly
Luciferase activity assay
Tissues (25–100 mg) were lysed and homogenized in luciferase lysis buffer (0.05% Triton X-100, 0.1 M Tris-HCl [pH 7.8], 2 mM EDTA). The homogenized lysate was extensively vortexed and spun down at 4°C for 2 min. The supernatant was used for luciferase activity analysis. The analysis was performed according to a previously described protocol (Yu et al., 2009), and the luciferase assay kit (cat. no. E1501) was purchased from Promega (Madison, WI).
AP expression analysis
Six weeks after AAV injection, mice were killed and TA muscles were collected. Half of each TA muscle was embedded in O.C.T. medium for in situ histochemical staining. Eight-micrometer muscle sections were heated at 65°C for 45 min to inactivate endogenous heat-labile AP according to a published protocol (Ghosh et al., 2007). NIH ImageJ software (National Institutes of Health, Bethesda, MD) was used to quantify the AP-positive region, expressed as a percentage, for each muscle section. The remaining half of the TA muscle was snap frozen in liquid nitrogen. AP activity in muscle lysate was determined with a StemTAG alkaline phosphatase activity assay kit (Cell Biolabs, San Diego, CA) as reported previously (Yue et al., 2008).
Genome copy determination
For the AP studies, genomic DNA was extracted from the same tissue lysate used for the AP activity assay, using a salting-out method after overnight proteinase K digestion. Viral genome copy number was determined with a 7900HT fast real-time PCR system (Applied Biosystems, Foster City, CA) as described previously (Yue et al., 2008). For the luciferase studies, total DNA was extracted with a kit purchased from Qiagen (DNeasy blood and tissue kit, cat. no. 69506; Qiagen, Valencia, CA). Vector copy number was determined with a 7300 real-time PCR system (Applied Biosystems). TaqMan assays for endogenous control of the cystic fibrosis transmembrane conductance regulator (CFTR) gene (for the AP study) and the glucagon gene (for the luciferase study) were developed to normalize vector copy numbers.
Statistical analysis
Data are presented as means ± standard error of mean. Comparison among groups was performed using SPSS software (SPSS, Chicago, IL). p < 0.05 was considered as statistically significant.
Results and Discussion
To study the performance of tyrosine-mutant AAV6 vectors, we used two different reporter genes, the firefly luciferase gene driven by the ubiquitous cytomegalovirus (CMV) promoter (Wang et al., 2008; Yu et al., 2009) and the human placental alkaline phosphatase (AP) gene driven by the ubiquitous Rous sarcoma virus (RSV) promoter (Yue et al., 2008). Similar to approaches described elsewhere (Zhong et al., 2008), site-directed mutagenesis was performed to generate surface-exposed single tyrosine-mutant AAV6 vectors, AAV6-Y445F and AAV6-Y731F. In both mutants, the corresponding surface-exposed tyrosine (Y) residue was replaced with phenylalanine (F).
We generated AAV6 and tyrosine-mutant AAV6 vectors containing the firefly luciferase reporter gene: AAV6-Luc, AAV6-Y445F-Luc, and AAV6-Y731F-Luc. The viruses were purified by PEG precipitation followed by double CsCl gradient purification. The vector titer generated from tyrosine-mutant plasmids was similar to that of the original AAV6 packaging plasmid, which indicated that modification of tyrosine to phenylalanine did not affect their packaging capacity. We then tested the functionality of tyrosine-mutant AAV6 vectors in vivo. For muscle-directed gene therapy, intramuscular injection of AAV vector is a valuable approach to achieve robust transgene expression in skeletal muscle. We therefore used this method to examine the performance of tyrosine-mutant AAV6 vectors. The mice were divided into three groups, with three mice in each group. For each mouse, we delivered a relatively lower dose to the left leg (3 × 1010 VG/injection site in a 50-μl volume, two sites for each leg) and a 10-fold higher dose to the right leg (3 × 1011 VG/injection site in a 50-μl volume, two sites for each leg). The two injection sites were located in the middle of the tibialis anterior (TA) and gastrocnemius (Gas) muscles. Three weeks after vector delivery, the live mice were subjected to optical imaging to examine luciferase intensity. As shown in Fig. 1A, the luciferase signal of the right leg (high dose) was stronger than that of the left leg. More importantly, the luciferase signal of mice injected with the tyrosine-mutant vectors (AAV6-Y445F and AAV6-Y731F) was much stronger than that of the mice injected with the original AAV6 vector. We then killed the mice and carefully dissected the TA and Gas muscles. Half the muscle tissue was subjected to luciferase activity assay, and the other half was used for DNA extraction and quantification of vector copy numbers. The luciferase activity assay showed that the tyrosine-mutant AAV6 vectors resulted in 8- to 10-fold higher luciferase activity than that of the original AAV6 vector in both the low-dose and high-dose groups (Fig. 1B) (p < 0.05). Quantitation of real-time PCR indicated that there were more genome copy numbers (4- to 6-fold more) in the tyrosine-mutant groups than in the original AAV6 group, particularly in the high-dose groups (Fig. 1C) (p < 0.01).
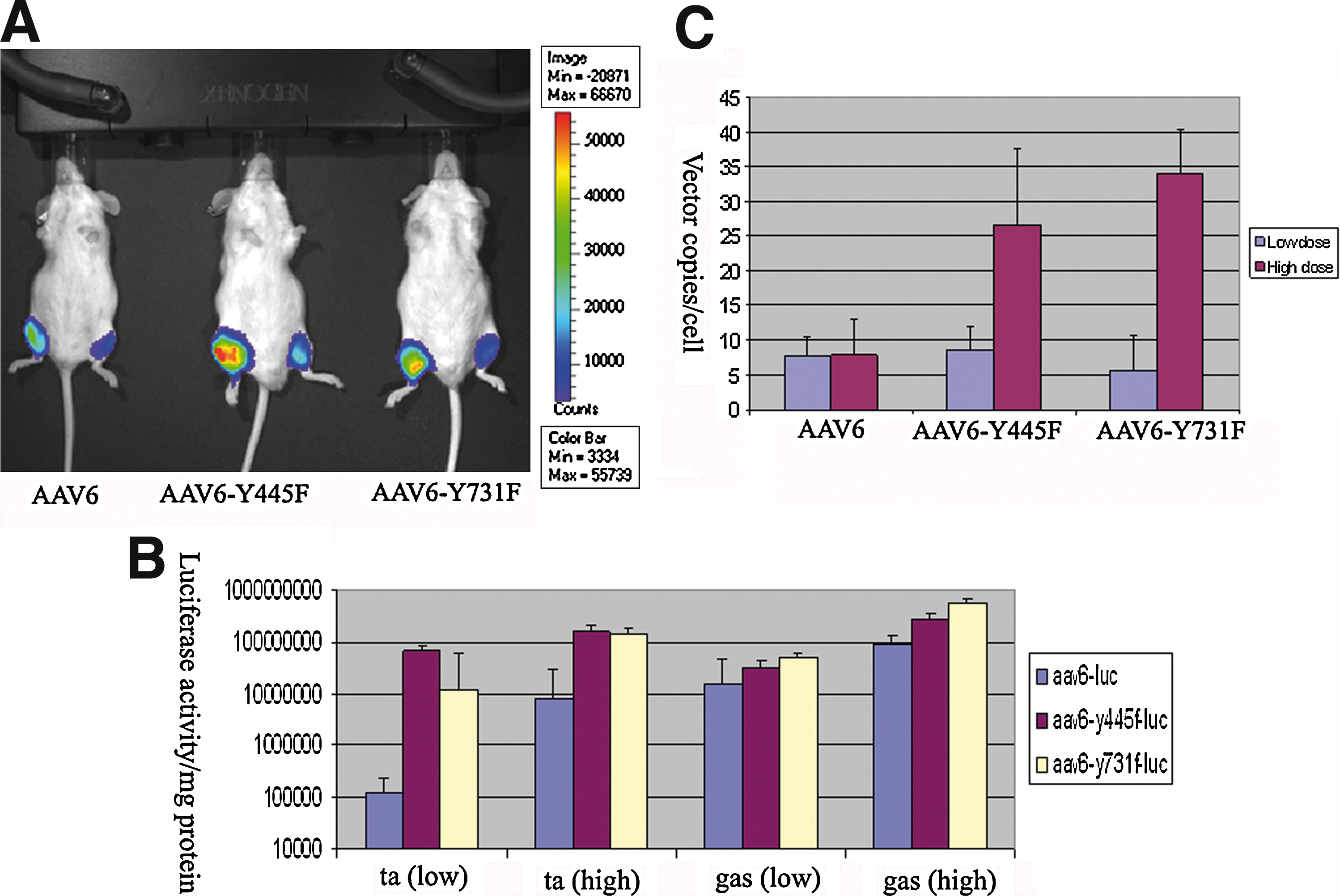
Tyrosine-mutant AAV6-Luc vectors mediate more robust expression in muscle via intramuscular injection.
To further test the tyrosine-mutant AAV6 vectors, we used a different reporter gene, the human placental alkaline phosphatase (AP) gene. AAV vectors were purified by three rounds of isopycnic ultracentrifugation in CsCl gradient in a different laboratory and the experiments were performed independently from those of the luciferase studies. This time, we used much lower vector doses. AAV vector was directly injected into the middle belly of the tibialis anterior (TA) muscle at a dose of 5 × 108 VG of AAV particles (30 μl) per muscle. Six weeks after AAV injection, mice were killed and the TA muscles were collected. Half of each TA muscle was embedded in O.C.T. medium for in situ histochemical staining, and the remaining half of the TA muscle was snap frozen in liquid nitrogen for AP activity assay and vector copy number analysis. For in situ histochemical staining, 8-μm cryosections were heated at 65°C for 45 min to inactivate endogenous heat-labile AP, and the staining were performed according to a published protocol (Ghosh et al., 2007). The results are displayed in Fig. 2A. The tyrosine-mutant AAV6-Y445F vector showed the highest AP expression, followed by the tyrosine-mutant AAV6-Y731F and the original AAV6 vector. On quantification (from three independent locations of the TA muscle including the proximal end, middle belly, and the distal end), AAV6-Y445F, Y731F-AAV6, and the original AAV6 resulted in 87.5 ± 3.1, 66.2 ± 3.9, and 41.6 ± 4.4% AP-positive areas, respectively (Fig. 2B). We then analyzed AP activity with the StemTAG alkaline phosphatase activity assay kit (Cell Biolabs) as described elsewhere (Yue et al., 2008). Consistent with the histochemical staining results, tissues injected with the tyrosine-mutant AAV6-Y445F vector displayed the highest AP activity (88.3 ± 22.3 μM/μg), followed by tissues injected with tyrosine-mutant AAV6-Y731F (39.8 ± 7.5 μM/μg); the original AAV6 vector resulted in only 9.4 ± 1.3 μM/μg AP activity (Fig. 2B). Last, we extracted total DNA from those samples and performed quantitative PCR to determine vector copy numbers. As expected, both tyrosine-mutant groups (AAV6-Y445F and AAV6-Y731F) demonstrated a significant higher copy number of AAV genomes than did the original AAV6 vector group (Fig. 2B, p < 0.05).
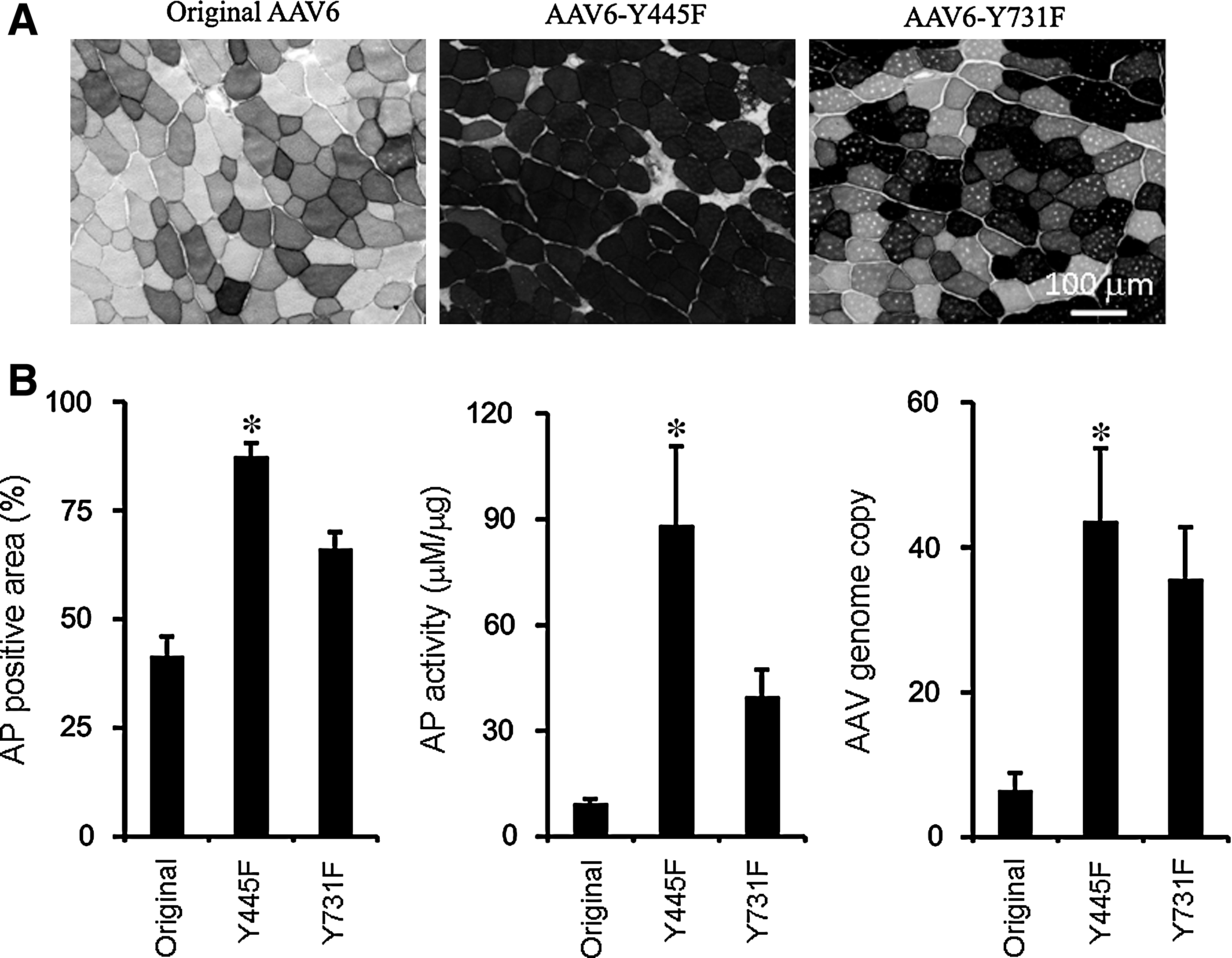
Analysis of alkaline phosphatase (AP) expression and AAV genome copy after local injection of original and tyrosine-mutant AAV6 vectors carrying the AP gene.
In summary, consistent results were obtained from two independent laboratories using two different reporter genes, the firefly luciferase gene and the AP gene. Collectively, the tyrosine-mutant AAV6 vectors mediated much stronger transgene expression than did the original AAV6 vector after direct muscle injection. Further, AAV6-Y445F appeared to lead to better transduction than AAV6-Y731F. Both AAV6-Y445F and AAV6-Y731F demonstrated significant higher vector copy numbers in injected muscles. Our data suggest that mutating the surface tyrosine residues may represent a valid strategy to significantly enhance the transduction efficiency of AAV serotypes other than AAV2.
One of the drawbacks for AAV vectors is that it is often necessary to use large amounts of viral particles to achieve sufficient levels of transgene expression (Zhong et al., 2008). This presents a particular challenge for large animal studies and human clinical applications. In these cases, up to 1015 VG particles are required even for a single experiment. It is costly and labor-intensive to produce large quantities of AAV vectors. Our results here suggest that we may be able to achieve a similar expression level with an at least severalfold lower dose were we to package the therapeutic expression cassettes in a tyrosine-mutant capsid. This would not only ease the burden of vector production, but would also add to the safety of AAV gene therapy because of the reduced vector load.
Our luciferase and AP reporter gene studies suggest that there were more vector genome copy numbers in the muscles injected with tyrosine-mutant AAV6 vectors compared with wild-type AAV6-injected muscles. The exact molecular mechanisms remain to be investigated. Nevertheless, we speculate that it may involve less vector degradation and/or improved intracellular trafficking. In the case of the AAV2 vector, the selective mutation of surface-exposed tyrosine bypasses the ubiquitination step, significantly enhancing intracellular trafficking and entry of the viral genome into the nucleus (Zhong et al., 2008). It is possible that modulation of the AAV6 capsid may also evade phosphorylation, ubiquitination, and subsequent proteasome-mediated degradation, thus improving intracellular trafficking and nuclear transport of viral particles.
Footnotes
Acknowledgments
This research was supported in part by grant 8187368876 from the Roche Foundation for Anemia Research, a research grant from the Fanconi Anemia Research Fund, and institutional research grant IRG-01-188-04 from the American Cancer Society (to L.Z.), and Public Health Service grants R01 EB-002073, R01 HL-065770, HL-076901, and P01 DK-058327 (Project 1) from the National Institutes of Health (to A.S.). G.R.J. was supported in part by an “Overseas Associate Fellowship-2006” from the Department of Biotechnology, Government of India. X.X. was supported by NIH grants AR056394 and AR45967. D.D. was supported by NIH grant AR49419 and by an MDA grant.
Author Disclosure Statement
No competing financial interests exist.