
Abstract
Antisense-mediated exon skipping has proven to be efficacious for subsets of Duchenne muscular dystrophy mutations. This approach is based on targeting specific splicing motifs that interfere with the spliceosome assembly by steric hindrance. Proper exon recognition by the splicing machinery is thought to depend on exonic splicing enhancer sequences, often characterized by purine-rich stretches, representing potential targets for antisense-mediated exon skipping. We identified and functionally characterized two purine-rich regions located within dystrophin intron 11 and involved in splicing regulation of a pseudo-exon. A functional role for these sequences was suggested by a pure intronic DMD deletion causing X-linked dilated cardiomyopathy through the prevalent cardiac incorporation of the aberrant pseudo-exon, marked as Alu-exon, into the dystrophin transcript. The first splicing sequence is contained within the pseudo-exon, whereas the second is localized within its 3′ intron. We demonstrated that the two sequences actually behave as splicing enhancers in cell-free splicing assays because their deletion strongly interferes with the pseudo-exon inclusion. Cell-free results were then confirmed in myogenic cells derived from the patient with X-linked dilated cardiomyopathy, by targeting the identified motifs with antisense molecules and obtaining a reduction in dystrophin pseudo-exon recognition. The splicing motifs identified could represent target sequences for a personalized molecular therapy in this particular DMD mutation. Our results demonstrated for the first time the role of intronic splicing sequences in antisense modulation with implications in exon skipping-mediated therapeutic approaches.
Introduction
At present, because of improvements in direct sequencing and dosage methodologies, the dystrophinopathy detection rate via genomic DNA analysis is about 93–96%.
Furthermore, the implementation of high-throughput platforms has made feasible the recognition of a previously difficult-to-identify category of DMD mutations, represented by deep intronic mutations. These rare mutations often create novel splice sites, resulting in the inclusion of intronic sequences as a pseudo-exon within the mRNA. Work has described the modulation of two different DMD mutations, causing BMD and DMD phenotypes, by AON-mediated pseudo-exon skipping, demonstrating the feasibility of this approach for this class of mutations (Tuffery-Giraud et al., 2003; Béroud et al., 2004; Gurvich et al., 2008). Notably, a therapy based on pseudo-exon skipping could be particularly beneficial, as the resultant rescued dystrophin is a wild-type, not internally deleted protein.
Several examples have clearly demonstrated that deep intronic mutations may affect the pre-mRNA splicing process in a number of disease-associated genes (Highsmith et al., 1994; Metherell et al., 2001; Pagani et al., 2002).
Exon recognition is defined through consensus sequences at the 5′ splice site, the 3′ splice site and associated polypyrimidine tract, and the branch point sequence. The selection of an exon is further determined by RNA cis elements generally referred to as exonic or intronic splicing enhancers (ESEs or ISEs) and exonic or intronic splicing silencers (ESSs or ISSs).
The binding of splicing factors, such as serine/arginine-rich proteins (SR proteins), to sequences classified as splicing enhancers, and the subsequent recruitment of other essential splicing factors to the splice sites, result in exon inclusion into mRNA (Valcarcel and Green, 1996; Chandler et al., 1997; Mayeda et al., 1999; Smith and Valcarcel, 2000; Lam and Hertel, 2002; Zheng, 2004; Lewandowska et al., 2005; Buratti et al., 2006). As SR protein binding to ESEs is essential for exon inclusion, blocking ESEs with AONs would be expected to result in exon skipping (Sazani and Kole, 2003; Baralle and Baralle, 2005; Aartsma-Rus and van Ommen, 2007; Wilton et al., 2007; Solis et al., 2008).
Alternatively spliced exons contain more than one regulatory cis element in the exon and/or in the flanking introns, and inclusion or skipping of the exon is determined by the activity of several proteins associated with these elements (Smith and Valcarcel, 2000; Black, 2003). Tissue-specific splicing patterns are thought to be determined by subtle changes in the proportion of SR proteins present in various cell types (Caceres et al., 1994; Zhu et al., 2001; Qi et al., 2006) and by the tissue-restricted expression of trans-acting factors that specifically interact with intronic regulatory cis elements (ISEs/ISSs), as described for the recognition of the rat fibroblast growth factor receptor-2 (FGFR2) exon IIIc in mesenchymal cells (Seth et al., 2008).
In previous work we described a splicing mutation occurring in a family with X-linked dilated cardiomyopathy (XLDC) (Ferlini et al., 1998; Rimessi et al., 2005). This mutation determined the tissue-specific inclusion of an out-of-frame pseudo-exon (Alu-exon) into the dystrophin transcript with the coexistence of wild-type and mutated transcripts in skeletal muscle, and the exclusive presence of the aberrant transcript in the heart, giving rise to the XLDC phenotype (Gualandi et al., 2003; Cohen et al., 2004).
Here we show that Alu-exon recognition is strongly downregulated by the deletion of two predicted splicing motifs in cell-free splicing assays and by the competition with antisense molecules in cell cultures, underlining the importance of local sequence analysis for developing therapies to turn off activated pseudo-exons. Selective targeting of these sequences, using AONs, induced a partial restoration of wild-type splicing, showing that the XLDC mutation has the potential to be rectified by antisense gene therapy. This is the first report of XLDC pseudo-exon modulation by targeting both exonic and intronic splicing motifs, with obvious implications in designing AON-mediated therapies.
Materials and Methods
Minigenes for in vitro transcription
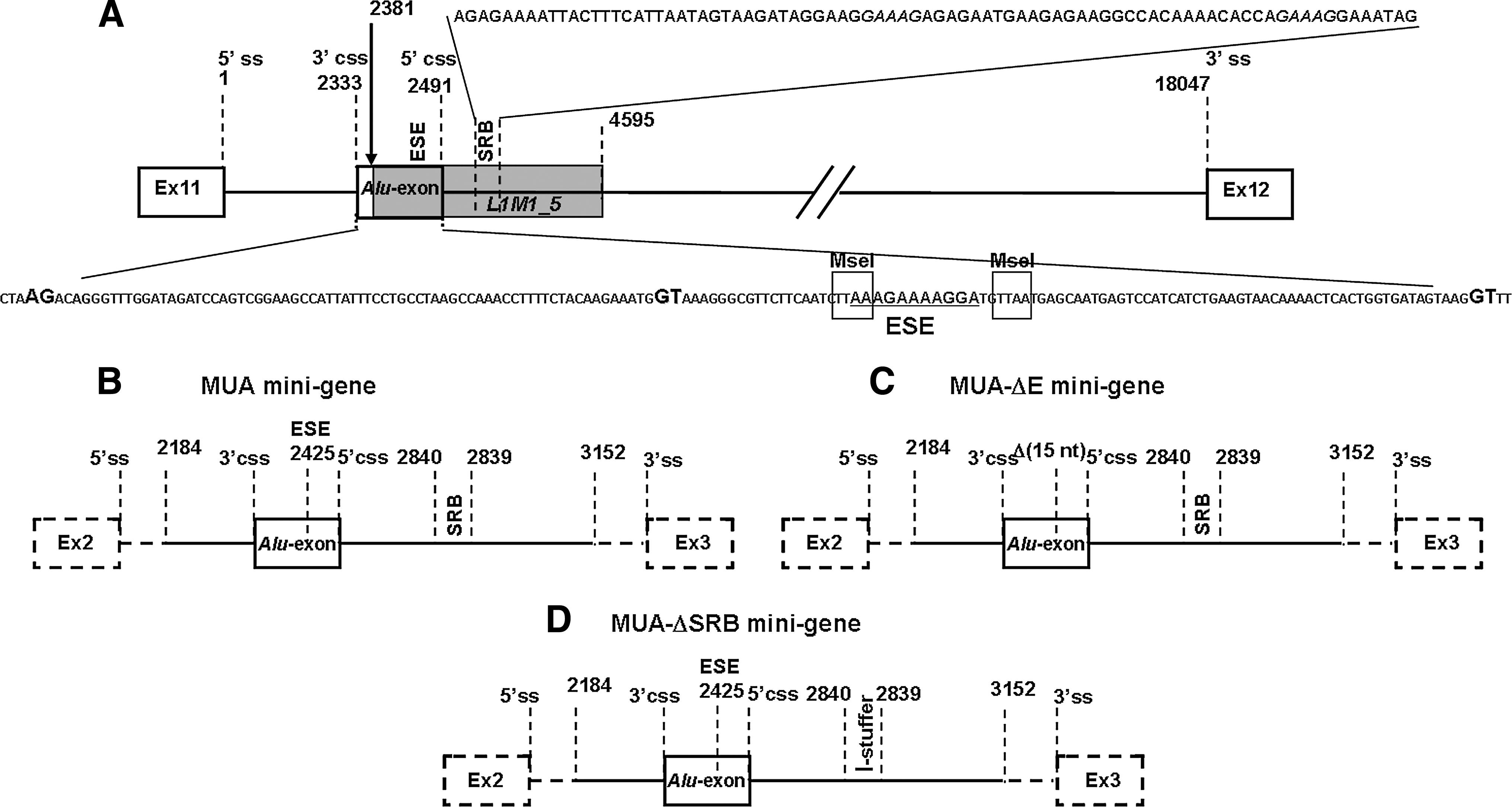
All the constructs used in this study were prepared by standard cloning techniques (Sambrook et al., 1989). The sequences of the oligonucleotides used for the experimental procedures are reported in Table 1. A graphic representation of the mutated dystrophin region is shown in Fig. 1A.
(
Primers modified at their 5′ end for cloning procedures.
The sequence and orientation of the inserts of all the recombinant plasmids (Gualandi et al., 2003) were verified by double-strand DNA sequencing. The entire sequence of each insert was determined in both directions, using fluorescent dideoxynucleotide triphosphates (Applied Biosystems, Foster City, CA) and an automated DNA sequencer (ABI PRISM 3130; Applied Biosystems).
Three minigenes were designed (Fig. 1B–D).
In vitro transcription and splicing assays
The templates for in vitro transcription reactions were obtained by PCR amplification of the minigenes, using the forward primer T7Ex2, which contains the T7 promoter sequence, and the reverse primer Ex3R (Table 1), as previously described (Gualandi et al., 2003).
In vitro splicing reactions were carried out according to previously reported procedures (Eperon et al., 2000), using HeLa cell nuclear extracts (Computer Cell Culture Centre, Seneffe, Belgium). The products of the splicing reactions were resolved by electrophoresis through a denaturing (8 M urea) polyacrylamide gel (5%) and visualized by exposure to Kodak BioMax X-ray film (Carestream Health, Rochester, NY), using an intensifying screen.
RT-PCR of splicing products
After autoradiography, slices of the dried gel containing RNA splicing products were excised and incubated overnight in sodium dodecyl sulfate (SDS) buffer at 4°C. The eluted RNA products were recovered by ethanol precipitation and used as templates for cDNA synthesis. Reverse transcription (RT) was performed with random hexanucleotide primers and the SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA) as previously described (Muntoni et al., 1995). cDNAs were amplified, using as primers the oligonucleotides Ex2F and Ex3R. These oligonucleotides map to sequence segments of exon 2 and exon 3, respectively, of the rabbit β-globin gene flanking the splicing constructs (Table 1).
The M3 transcript was characterized by sequencing the RT-PCR product obtained with oligonucleotides Ex2F and Ex3R (Table 1).
AON design and synthesis
AON design was based on in silico analysis of the dystrophin intron 11 mutated sequence, using both the ESEfinder algorithm (Cartegni et al., 2003) and Human Splicing Finder tool (Desmet et al., 2009) to identify regulatory motifs involved in exon definition, and the M-FOLD program (Zuker, 2003) to predict mRNA secondary structure (Aartsma-Rus et al., 2006b, 2009) (Table 2).
Abbreviation: AON, antisense oligoribonucleotide.
The synthesized AONs contain a full-length phosphorothioate backbone and 2′-O-methyl-modified ribose molecules. Oligonucleotides synthesis was carried out on an ÄKTA oligopilot plus 10 DNA/RNA synthesizer (GE Healthcare Life Sciences, Piscataway, NJ) as described (Rimessi et al., 2009).
Myogenic cell cultures and AON transfection
Primary human fibroblasts from the XLDC patient were isolated from a skin biopsy (obtained after informed consent for research purposes, Ethical Approval No. 9/2005). Cells were grown in high-glucose Dulbecco's modified Eagle's medium (GIBCO DMEM; Invitrogen), supplemented with 20% fetal bovine serum (GIBCO FBS; Invitrogen) and antibiotic–antimycotic solution (Sigma-Aldrich, St. Louis, MO). Myogenesis was induced by infection with an adenovirus serotype 5 (Ad5)-derived, E1A-deleted adenoviral vector carrying the MyoD gene as previously described (Aartsma-Rus et al., 2004). Myotubes obtained after 7–10 days of culture in differentiation medium (2% FBS) were transfected with AONs (100 nM) in the presence of polyethylenimine (ExGen 500; MBI Fermentas, Woburn, MA) (2 μl/μg of AON) as transfection reagent, according to the manufacturer's instruction. For ESEN2 and SRBN3, 50 and 75 nM concentrations were also tested.
Immunofluorescence analysis
Forty-eight hours after AON treatment, myotube cultures grown onto coverslips were rapidly washed with phosphate-buffered saline (PBS) and fixed with cold methanol for 7 min. Samples were saturated with 4% bovine serum albumin (BSA)–PBS solution for 30 min and double-labeled with mouse monoclonal antibodies against desmin or developmental myosin heavy chain (Novocastra Laboratories, Newcastle upon Tyne, UK), diluted 1:10 and 1:60, respectively, or with polyclonal antibody against dystrophin H300 (diluted 1:100; Santa Cruz Biotechnology, Santa Cruz, CA). After several washings with PBS, samples were incubated for 1 hr with fluorescein isothiocyanate (FITC)/tetramethylrhodamine isothiocyanate (TRITC)-conjugated secondary antibodies (diluted 1:100; Dako, Carpinteria, CA). The slides were mounted with ProLong Antifade reagent (Molecular Probes/Invitrogen, Eugene, OR) and observed with an Eclipse 80i fluorescence microscope (Nikon Instruments, Melville, NY).
RNA analysis
Forty-eight hours after transfection, total RNA was isolated from myotube cultures (RNeasy kit; Qiagen, Valencia, CA) and reverse-transcribed into cDNA, using a high-capacity cDNA reverse transcription kit (Applied Biosystems) with random primers. RT-PCR was performed on β-actin (primer sequences available on request) to verify cDNA synthesis and on dystrophin (10F, 5′-tggaagctcctgaagacaagtc-3′; and M12, 5′-gattctggagatccattaaaactct-3′) to analyze the relative amount of skipped/not-skipped transcripts (399 and 558 bp, respectively).
To precisely quantify both the percentage of skipping of the Alu-exon and the amount of dystrophin transcript, we developed exon-specific real-time assays detecting the “aberrant” exon and human dystrophin exons 10 and 12. These exons were chosen because they are not involved in spontaneous alternative splicing events in humans. Real-time assays on exons 10 and 12 were performed as reference to quantify the amount of physiological pseudo-exon skipping and percentage of induced pseudo-exon skipping in treated cells compared with untreated cells (internal reference). The same exons, exons 10 and 12, were used as target to quantify the dystrophin transcript level with β-actin transcript (β-actin gene; Applied Biosystems) as reference, in AON-treated/not-treated XLDC patient myogenic cells, in comparison with myogenic cells from a healthy donor (cutaneous fibroblasts, MyoD induced). Real-time assays were based on TaqMan MGB technology (Applied Biosystems), and primer and probe sequences were designed with Primer Express software (Applied Biosystems; primer and probe sequences are available on request). The amount of target sequences with respect to internal references (represented by the adjacent dystrophin exon) and to an appropriate endogenous control (β-actin gene) was evaluated by the comparative C
t method with respect to the untreated control (ΔΔC
t method) (Applied Biosystems User Bulletin #2) [Applied Biosystems. (2001). User Bulletin #2: ABI PRISM 7700 Sequence Detection System. (Applied Biosystems, Foster City, CA). Available at
Results
Cell-free splicing assays
To functionally characterize the two purine-rich regions contained in the Alu-exon, three splicing constructs were set up (Fig. 1). The MUA minigene contains the rearranged XLDC genomic region of dystrophin intron 11, including the Alu-exon, the two cryptic splice sites, the putative branch point, and the intronic purine-rich region, named the SRB motif (Fig. 1A and B). MUAΔE and MUAΔSRB represent deletion constructs.
The MUA-ΔE minigene, obtained from the MUA minigene, lacks 15 nucleotides containing the purine-rich exonic motif (ESE), and maintains the Alu-exon, the two cryptic splice sites, the putative branch point, and the SRB motif (ISE) (Fig. 1A and C).
The MUA-ΔSRB minigene was generated by replacing the SRB region (about 90 nucleotides long) of the MUA minigene with a neutral sequence of similar length from phage γ in order to avoid length-related artifacts (Fig. 1D).
All the constructs were assayed in in vitro splicing assays.
The MUA minigene in vitro splicing assay shows the rabbit β-globin canonical products (B1 and B2) detected after 30 min of incubation, a short time for processing in vitro a large transcript (1372 bp). The 437-bp fragment (M2) corresponding to a splicing product incorporating the Alu-exon between rabbit β-globin exons 2 and 3 was detected after 1 hr and its relative abundance increased with time (Fig. 2A).

(
The MUA-ΔE minigene splicing assay showed, in addition to the β-globin canonical splicing products, a low-abundance novel 344-bp transcript (M3), visible at 2 and 3 hr of incubation time (Fig. 2A). Sequence analysis of this fragment revealed that it represents a splicing product incorporating a novel 66-bp exon defined by the activation of two different splice sites: a 3′ cryptic splice site (not recognized by any splice site prediction tools), located 4 bp downstream from the 3′ splice site (41% score, as defined by the BDGP Splice Prediction tool, at
The comparison of the parental MUA minigene with the deleted MUA-ΔE in different splicing assays demonstrated that the deletion of the ESE motif abolished the Alu-exon definition (Fig. 2A).
To test the involvement of the SRB motif in Alu-exon recognition, we assayed the splicing behavior of the MUA-ΔSRB minigene. In addition to the β-globin canonical products, only a splicing product referred to as M4 was detected. M4 corresponded to an aberrant β-globin transcript containing exon 2 joined to two copies of exon 3 (Fig. 2B). The deletion of the SRB region therefore abolished the Alu-exon inclusion.
Immunofluorescence analysis of MyoD-transformed fibroblasts
Because muscle biopsy suitable for cell culture was not available, patient skin-derived fibroblasts were converted into myogenic cells by infection with a replication-defective adenovirus encoding MyoD.
Immunofluorescence analysis of desmin and myosin, two proteins specifically expressed in muscle tissue, was performed to define the differentiation stage of the MyoD-transformed fibroblasts. We detected expression of desmin and myosin in patient-derived myogenic cultures, therefore confirming myogenic conversion (Fig. 3).
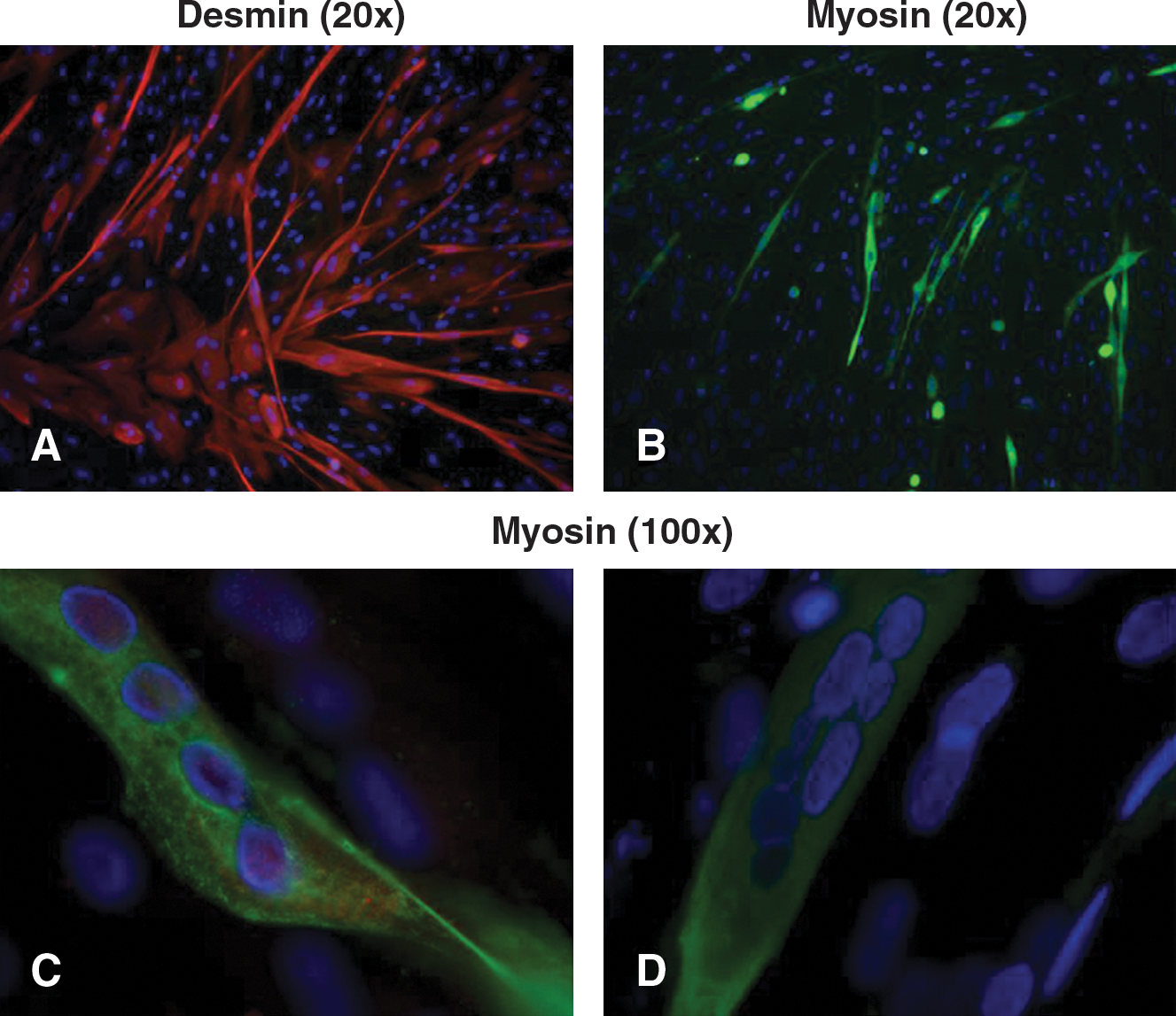
Immunofluorescence characterization of MyoD-infected fibroblasts. To assess the myogenic differentiation induced by infection with recombinant adenoviral vector carrying the MyoD gene, samples were double-labeled with monoclonal antibodies against desmin (
Dystrophin expression was detected both in AON-treated and untreated patient-derived myogenic cells, correctly localized at the sarcolemma, resembling the XLDC patient's skeletal muscle behavior. However, it was not possible to accurately quantify increased expression of the protein in AON-treated cells (Fig. 4).

Immunofluorescence analysis of dystrophin in patient myogenic cells. Double-labeling for developmental myosin heavy chain (
Splicing modulation by AONs in patient-derived myogenic cells
An analysis performed with the Human Splicing Finder tool (Desmet et al., 2009) revealed that the two regulatory sequences (ESE and ISE) correspond to overlapping enhancers (Tra2β and 9G8 motifs) and silencers (hnRNPA1 motifs), potentially explaining the tissue-specific splicing pattern. AONs directed at predicted ESE/ISE motifs were evaluated for their potential to induce targeted Alu-exon skipping at a concentration of 100 nM. Cultured patient myogenic cells were transfected with the various designed AONs (Table 2) and analyzed for Alu-exon skipping.
RT-PCR amplification of AON-treated or untreated patient-derived cells, using oligonucleotides on dystrophin exons 10 and 12, generated a mixture of two products: one corresponding to the wild-type transcript and a second, larger fragment containing the cryptic exon. Comparison of the relative abundance of the two products in treated and untreated cells demonstrated that AONs specifically targeting the ESE motif (ESEN1 and ESEN2) induced an increased ratio of the correct transcript, whereas AONs directed at the ISE domain appeared less effective (data not shown). Sequence analysis confirmed the correct exon 11–exon 12 junction (data not shown).
AON-induced skipping efficiency was estimated by exon-specific real-time assay (ESRA) of the Alu-exon, using both adjacent dystrophin exons (exons 10 and 12) and β-actin as references, in treated compared with untreated cells (Fig. 5A). The percentage of exon skipping varied considerably, with a consistent increase when using ESEN1 and ESEN2 (10.9 and 11.06%, respectively), whereas AONs directed against the ISE motif were less effective (5% skipping with SRBN3, 1.8% with SRBN4, 2% with SRBN5, 1.2% with SRBV1, and 0.8% with SRBV2); furthermore, SRBN1 and SRBN2 were unable to induce any detectable skipping (data not shown).
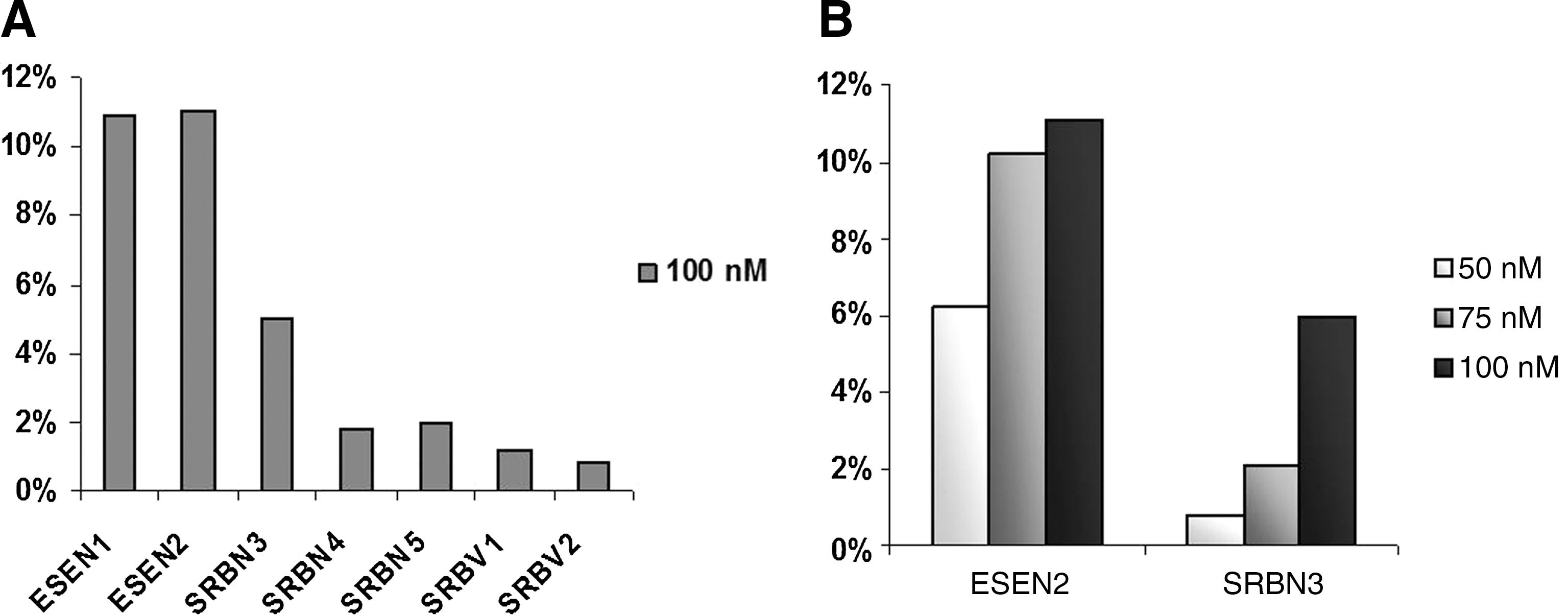
Real-time RT-PCR quantification of Alu-exon skipping. (
The two more effective AONs, ESEN2 and SRBN3, worked at almost all concentrations tested in excluding the Alu-exon from the mature transcript. The effect was shown to be concentration dependent, with approximately 6.22% induced skipping at 50 nM, 10.24% at 75 nM, and 11.08% at 100 nM (ESEN2) and approximately 0.82% at 50 nM, 2.08% at 75 nM, and 6% at 100 nM (SRBN3) (Fig. 5B). By treating patient-derived cells with various combinations of these two AONs, an additive action was excluded (data not shown).
Importantly, treating patients' cells with effective AONs resulted in a 2- to 3-fold increase in dystrophin transcription level (as detected by ESRA), with respect to myogenic cells from healthy donors, considered an added value due to the low basal level detected in these cells (20% of control cells).
Discussion
We identified and functionally characterized two purine-rich regions located within dystrophin intron 11 and putatively involved in splicing regulation of a pseudo-exon that causes an XLDC phenotype. The splicing behavior of the dystrophin transcript, because of the large pure intronic deletion, resulted in the inclusion of this aberrant pseudo-exon in the dystrophin transcript (Gualandi et al., 2003). Skeletal muscles can produce both wild-type and out-of-frame transcripts, whereas cardiac tissue produces only the transcript including the Alu-exon, therefore accounting for dystrophin full deficiency (Ferlini et al., 1998).
To address both the pseudo-exon splicing regulation and its propensity for antisense-mediated exon-skipping we set up first a cell-free splicing assay using constructs with artificial deletions of the putative exonic or intronic splicing motifs, and then we modulated these motifs with a large number of AONs in myogenic cells derived from the XLDC patient.
Our studies demonstrated that the two predicted splicing motifs are actually involved in the pseudo-exon recognition and therefore their targeting by AONs induces a reduction of Alu-exon incorporation.
One of the two identified motifs consists of an exonic 11-bp purine stretch identified by comparative analysis with other known ESEs, which revealed only two different nucleotides between the Alu-exon ESE and the well-characterized SMN1 exon 7 ESE motif (insertion of two A's in boldface; SMN1, AAAGAA
Eperon and colleagues reported the characterization in mouse cells of a splicing silencer located within the Alu-exon (Nasim et al., 2003). They hypothesized that this motif may act differently in the heart and in skeletal muscle, therefore accounting for the tissue-specific expression of the XLDC mutation in this family, emphasizing the complexity of tissue-specific exon choice known to involve multiple cis elements and trans factors (Tarn, 2007).
The second splicing sequence (SRB) we identified represents an intronic GA-rich stretch located downstream from the 5′ cryptic splice site of the alternative Alu-exon. Deletion of the SRB region in the minigene abolished Alu-exon inclusion, suggesting it could act as an ISE sequence.
The bioinformatic analysis (Desmet et al., 2009) revealed that, indeed, the two regulatory sequences, Alu-exon ESE and SRB, correspond to overlapping enhancers (Tra2β and 9G8 motifs) and silencers (hnRNPA1 motifs), suggesting that the level of expression of these key proteins in various tissues could explain the tissue-specific splicing pattern observed in the XLDC patient, as in general described in many reports (Barnard et al., 2002; Tran et al., 2003; Fischer et al., 2004; Qi et al., 2006).
The enhancer activity of these two splicing sequences was also confirmed by the results we obtained by AON modulation in myogenic cells. Only targeting the Alu-exon ESE succeeded in inducing a significant percentage (10–12%) of exon skipping, therefore confirming that this ESE could represent an effective target for inhibiting Alu-exon recognition.
When targeting the intronic splicing enhancer with AONs, we again obtained increased skipping of the Alu-exon (6%), once more suggesting a possible cooperative action between the two splicing sequences.
Dystrophin pseudo-exon modulation by AONs has been described (Gurvich et al., 2008; Madden et al., 2009). In contrast to the present work, in both cases the AON treatment was performed in myogenic cells derived from muscle biopsies and targeting exonic sequences.
To our knowledge this is the first example of both pseudo-exon modulation in MyoD-transformed fibroblasts and AON-mediated exon skipping targeting an ISE. In fact, in the absence of XLDC patient muscle cells we were able to reproduce, in cells from a skin biopsy, the same dystrophin splicing pattern we observed in the patient's skeletal muscles, and therefore used these cells for testing AONs versus both ESE and ISE sequences. ISE sequence targeting represents a novelty; in fact, these motifs have not been subjected to the same extensive analysis as ESEs, and our current understanding of ISEs and the mechanisms by which they exert their effects is still incomplete.
The levels of AON-induced skipping we obtained in cell cultures were too low when considered from a therapeutic perspective; however, our results open a new option for AON designing for DMD exon skipping. In fact, to date only exonic sequences or splice site consensus sequences have been tested as AON targets, and in some cases with no effect, leading to the definition of poorly or even unskippable exons (Errington et al., 2003; Wilton et al., 2007). In those cases, searching for intronic splicing motifs as putative AON targets could represent an alternative solution.
In addition, we demonstrated that the dystrophin mRNA level increased 2- to 3-fold in response to AON treatment in our patient's myogenic cells, suggesting that AON-induced pseudo-exon skipping, although enhancing mRNA stability, may increase the abundance of wild-type transcript.
In conclusion, we have demonstrated that both an exonic and an intronic splicing region act as enhancers for pseudo-exon recognition, and that their modulation by AONs can reduce Alu-exon incorporation within the dystrophin transcript. Development of pseudo-exon skipping therapies represents a type of personalized medicine directed at individual patients with private mutations.
Interestingly, although it is not surprising that the deletion/targeting of an ESE can strongly affect the exon definition process, the effect on splicing we observed after deletion of the ISE region or its targeting by AONs is less obvious. As a consequence, it may be relevant to carefully consider the flanking intronic regions when designing exon-skipping AONs on the basis of criteria that have been widely described (Aartsma-Rus et al., 2009); in fact, both their efficiency and efficacy may greatly vary depending on the intronic mutation breakpoints, and consequently on the loss/maintenance of intronic splicing regulatory sequences. Considering our results, we think that this issue deserves to be further studied.
Footnotes
Acknowledgments
This work was fully supported by Telethon Italy Foundation (grants GGP5115 and GUP0711 to A.F.).