
Abstract
In 1971 Judah Folkman proposed the concept of antiangiogenesis as a therapeutic target for cancer. More than 30 years later, concept became reality with the approval of the antivascular endothelial growth factor (VEGF) monoclonal antibody bevacizumab as a first-line treatment for metastatic colorectal cancer. Monoclonal antibodies and small molecular drugs are the most widely applied methods for inhibition of angiogenesis. The efficacy of these antiangiogenic modalities has been proven, in both preclinical and clinical settings. Although angiogenesis plays a major role in wound healing, hypoxia, and in the female reproductive cycle, inhibition of angiogenesis seems to be a relatively safe therapeutic option against cancers, and has therefore become a logical arena for a wide range of experimentation. The twentieth century has shown the boom of gene therapy and thus it has been applied also in the antiangiogenic setting. This review summarizes methods to induce antiangiogenic responses with gene therapy and discusses the obstacles and future prospects of antiangiogenic cancer gene therapy.
Introduction
The ultimate goal in antiangiogenic cancer therapies is to starve the tumor by inhibiting the tumor vessel growth, thereby preventing the supply of nutrients and oxygen (Ellis and Hicklin, 2008). The most important mediator of angiogenesis, vascular endothelial growth factor (VEGF), and its receptor tyrosine kinase signaling have remained the centerpiece of angiogenesis studies (Ferrara, 2002; Ferrara et al., 2003). The main candidates currently employed are neutralizing antibodies against VEGFs and their receptors and tyrosine kinase inhibitors (RTKI). Since the approval of bevacizumab for colorectal cancer, several antiangiogenic drugs have demonstrated clinical efficacy in human cancers whereas many others have shown preclinical benefits. Bevacizumab has shown clinical efficacy in combination with chemotherapy in metastatic breast, non-small cell lung, and renal cell cancers (Sandler et al., 2006; Escudier et al., 2007; Miller et al., 2007) and was approved by the U.S. Food and Drug Administration (FDA). It was also approved for progressive glioblastoma multiforme (GBM) by the FDA. Oral small-molecular RTKIs sorafenib and sunitinib, targeting VEGF and platelet-derived growth factor (PDGF) receptor tyrosine kinases, have also been approved for metastatic renal cell cancer. Sorafenib is also approved as a monotherapy for hepatocellular cancer (Kerbel, 2008), whereas sunitinib is approved for gastrointestinal stromal tumors (Ebos et al., 2009). Angiostatin and endostatin are potent endogenous inhibitors of angiogenesis (O'Reilly et al., 1994; O'Reilly, 1997) and have demonstrated antiangiogenic properties, ranging from reduced tumor growth to improved survival in various animal models (Ma et al., 2002; Peroulis et al., 2002; Garkavtsev et al., 2004). However, the clinical trials have not been as promising (Kulke et al., 2006; Kurup et al., 2006).
The field of gene therapy has matured, with several new gene therapy-based cancer products in clinical studies. One of the main themes in cancer gene therapy is inhibition of angiogenesis. Gene therapy tries to introduce in the target tissue a gene/cDNA/short hairpin RNA (shRNA) that expresses therapeutic compounds leading to a treatment effect. With gene therapy it is possible to target treatments to influence tumors only locally, thereby reducing unwanted side effects caused by systemic administrations.
Mechanisms of tumor growth and angiogenesis
First, sprouting angiogenesis is by far the most important mechanism of vascular growth in tumors. However, some tumors do have the ability to obtain vascular supply through other mechanisms. Tumors, such as GBM, possess the ability to migrate along with the existing blood vessels, which is termed vessel cooption (Holash et al., 1999), and have been demonstrated to be able to initiate tumor growth independent of angiogenesis (Sakariassen et al., 2006). Glomeruloid angiogenesis is seen in a wide variety of cancers including high-grade gliomas, where tumor cells obtain their vascular supply from several closely associated microvessels (Dome et al., 2007). Bone marrow-derived endothelial progenitor cells are incorporated into the primary and metastatic tumors from the circulation and are believed to contribute to vasculogenesis; that is, de novo formation of new blood vessels (Duda et al., 2006). However, their role in tumor neovascularization has been questioned (Purhonen et al., 2008) and there seems to be great variation in the bone marrow dependence of tumor angiogenesis, ranging from negligible (De et al., 2003) to significant (Duda et al., 2006), emphasizing the fact that angiogenic responses differ among tumor types, organ site, and animal model studied. Development of new blood vessels by septal formation inside existing blood vessels is termed intussusception, which has been described in brain metastatic models, even though its contribution in primary tumor development is unclear (Carmeliet and Jain, 2000). Vasculogenic mimicry means the existence of vascular channels composed of tumor cells and basement membrane, but devoid of endothelial cell lining (Maniotis et al., 1999; Folberg et al., 2000; McDonald et al., 2000). In spite of having several mechanisms to obtain vascular supply, sprouting angiogenesis is considered the most important for solid tumor growth (Carmeliet, 2005).
Mediators of angiogenesis and role of VEGF and VEGF receptor
VEGF was first described as a vascular permeability factor (Senger et al., 1983). The VEGF family consists of VEGF-A (VEGF), VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF) (Roy et al., 2006), of which VEGF-A, VEGF-B, and PlGF are mainly angiogenic factors, whereas VEGF-C and VEGF-D are also important in lymphangiogenesis. Apart from VEGF, several other mediators are important in angiogenesis and are described in detail in Table 1.
The VEGF receptor (VEGFR) family consists of three tyrosine kinase receptors, VEGFR-1 (FLT-1), VEGFR-2 (FLK-1/KDR), and VEGFR-3 (FLT-4), that are involved in angiogenesis and lymphangiogenesis. VEGF binds both VEGFR-1 and VEGFR-2 (Terman et al., 1992; Ferrara et al., 2003), but VEGFR-2 is the main receptor involved in signal transduction through activation of Raf/Mek/Erk (Takahashi et al., 1992, 1999) and phosphatidylinositol (PI)-3 kinase–Akt pathways, leading to integrin-mediated cell adhesion, growth, proliferation, migration, survival, and increased permeability of endothelial cells (Ferrara et al., 2003; Kerbel, 2008). VEGFR-2 is expressed on endothelial cells as well as on circulating, bone marrow-derived endothelial progenitor cells (Kerbel, 2008).
VEGF may contribute to tumor vasculogenesis and angiogenesis by recruiting VEGFR-1+ and/or VEGFR-2+ hematopoietic progenitors and VEGFR-2+ endothelial progenitor cells from the bone marrow (Murdoch et al., 2008), in spite of the controversies about their exact contribution to tumor growth in patients (Kerbel, 2008).
Expression of both VEGF and its receptors, especially VEGFR-1, by cancer cells suggests that VEGF can act as a direct autocrine growth factor for cancer cells (Kerbel, 2008). This expression is believed to promote survival of these cells via “intracrine” mechanisms (Lee et al., 1992, 2007). Expression of VEGFR-1 and VEGFR-2 on cancer cells can also contribute to their proliferation and metastasis (Dias et al., 2000, 2001; Fragoso et al., 2006).
Intratumoral blood vessels are dilated, tortuous, and aberrant in nature (Deane and Lantos, 1981a,b) with increased permeability (Takano et al., 1991), leading to elevated interstitial fluid pressure within the tumors (Boucher and Jain, 1992; Jain, 2003). This is attributed mainly to the increased VEGF levels within the tumors (Senger et al., 1983, 1990), through its action on VEGFR-2 (Ferrara and Bunting, 1996; Ferrara and Davis-Smyth, 1997), generating immature dysfunctional vessels (Jain, 1998; Jain et al., 2007). Increased vascular permeability, along with the impaired lymphatic drainage, results in equilibrium of oncotic and hydrostatic pressures between tumor microvasculature and the interstitial space (Boucher and Jain, 1992). The increased leakiness of the vessels hinders chemotherapeutic delivery by causing a stasis of blood flow (Netti et al., 1996; Baish et al., 1997). This further leads to a vasogenic edema that contributes to the pathophysiology of tumors, especially in brain (Berkman et al., 1993; Strugar et al., 1994; Zagzag, 1995; Takano et al., 1996). Overexpression of VEGF also results in upregulation of membrane type-1 matrix metalloproteinase (MMP), MMP-2, and MMP-9, promoting invasiveness of tumors (Munaut et al., 2003). Figure 1 describes the role played by VEGF and VEGFR in tumor development and progression.
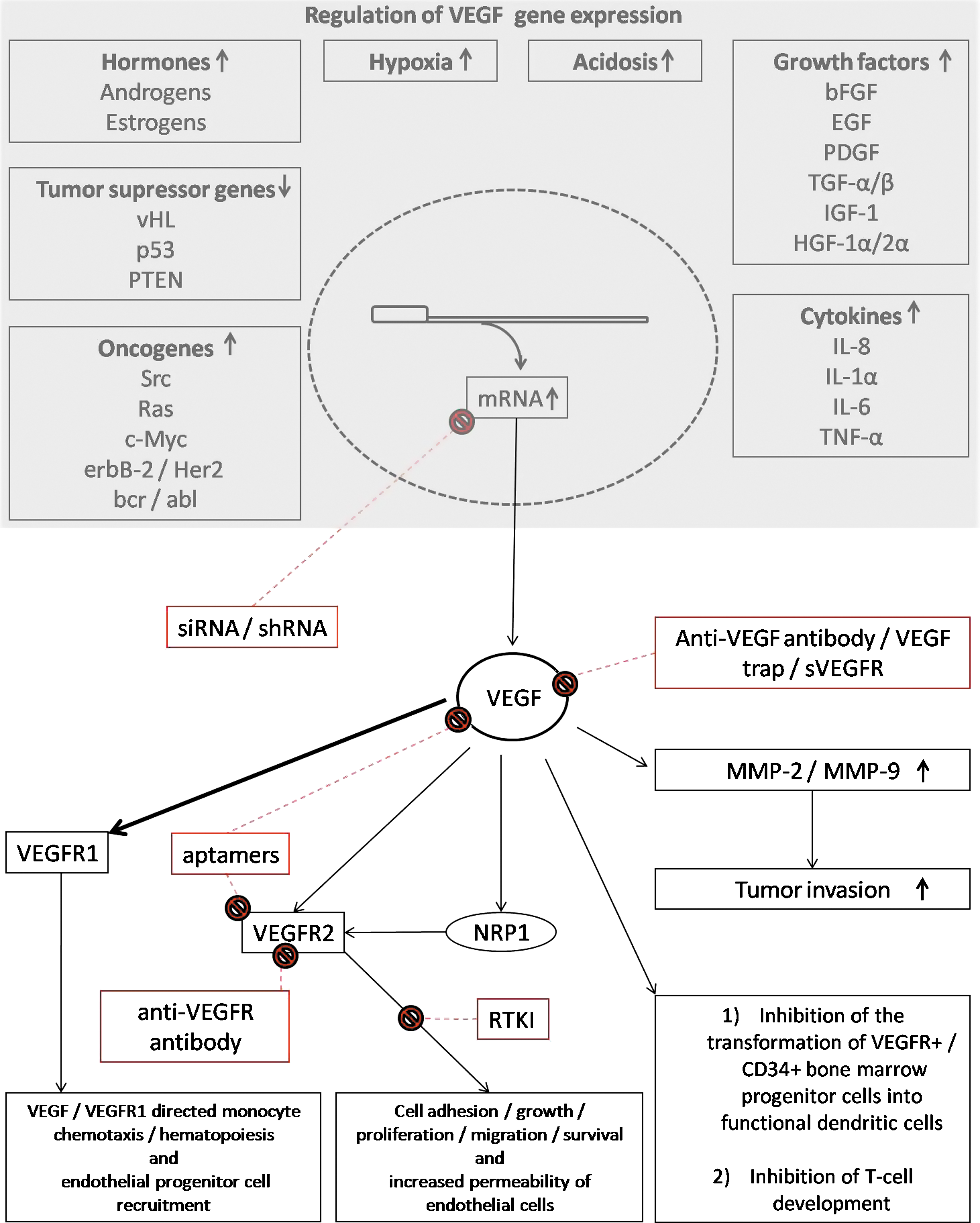
Regulation of vascular endothelial growth factor (VEGF) gene expression, and the role of VEGF and VEGF receptor (VEGFR) in tumor development and progression. Possible sites of intervention are indicated in red. bcr/abl, breakpoint cluster region/Abelson murine leukemia viral oncogene homolog; EGF, epidermal growth factor; erbB-2/Her2, erythroblastic leukemia viral oncogene homolog-2/human epidermal growth factor receptor-2; bFGF, basic fibroblast growth factor; HGF, hepatocyte growth factor; IGF, insulin-like growth factor; IL, interleukin; MMP, matrix metalloproteinase; NRP, neuropilin receptor; PDGF, platelet-derived growth factor; PTEN, phosphatase and tenascin homolog on chromosome 10; RTKI, receptor tyrosine kinase inhibitor; shRNA, short hairpin RNA; siRNA, small interfering RNA; Src, sarcoma viral oncogene homolog; sVEGFR, soluble VEGFR; TGF, transforming growth factor; TNF, tumor necrosis factor; vHL, von Hippel-Lindau disease gene; ↑, increase in gene expression; ↓, decrease in gene expression.
Immune system and tumor angiogenesis
VEGF also plays an important role in making the immune system nonresponsive to growing tumors (Gabrilovich et al., 1998; Oyama et al., 1998; Ohm and Carbone, 2001). VEGF is known to bind to VEGFR-1 on CD34+ bone marrow progenitor cells, decreasing their ability to differentiate into functional dendritic cells (DCs) (Gabrilovich et al., 1996; Kusmartsev and Gabrilovich, 2002; Dikov et al., 2005), which contributes to the reduction in the DC population in patients with cancer (Gabrilovich et al., 1998; Ohm and Carbone, 2002). VEGF is also believed to directly affect T cell development (Ohmoto et al., 1999; Ohm et al., 2003). Myeloid-derived suppressor cells once recruited into the tumors have been shown to suppress T cell- and natural killer (NK) cell-mediated antitumor immunity, contribute to tumor angiogenesis by secreting MMP-9, enhance the tumor-promoting type 2 response in tumor-associated macrophages, and possibly differentiate into endothelial-like cells (Murdoch and Finn, 2000; Murdoch et al., 2008). Likewise, monocytes have the capability to transdifferentiate into endothelial cells under sustained angiogenic stimulation (Kuwana et al., 2006). Angiopoietin receptor TIE2-expressing monocytes are possibly recruited into hypoxic areas of the tumors by angiopoietin-2 expressed by tumor cells as well as associated endothelial cells, and are believed to contribute to tumor angiogenesis. There is evidence suggesting that neutrophils, eosinophils, mast cells, and DCs can influence angiogenesis once recruited into tumors of various cancer models (Murdoch and Finn, 2000; Murdoch et al., 2008). VEGF, basic fibroblast growth factor (bFGF), granulocyte-macrophage colony-stimulating factor (GM-CSF), angiopoietin-1, and insulin-like growth factor-1 (IGF-1) are also implicated in the recruitment of endothelial cell precursors (Jain, 1998; Jain et al., 2007).
Major limitations of current antiangiogenic therapies
With the exception in renal cell cancer, the survival benefit gained by antiangiogenic therapies in clinical practice is usually a few months at the expense of high cost and side effects, and the tumor shrinkage or dormancy produced has not been remarkably significant (Verheul and Pinedo, 2007; Kerbel, 2008; Paez-Ribes et al., 2009). In most cases only progression-free survival was increased, with no major impact on overall survival. Transient response is lost by the development of resistance with increased tumor aggressiveness. It has also been observed that a certain cohort of patients does not respond to these therapies from the beginning (Bergers and Hanahan, 2008; Paez-Ribes et al., 2009). However, it must be emphasized that the therapeutic benefits seen with current antiangiogenic agents are comparable to those of chemotherapeutic agents but with far fewer toxic side effects.
The production of antiangiogenic monoclonal antibodies and proteins is costly and difficult, needing tedious purification, characterization, and validation processes. Technical problems related to their physical properties are not uncommon and the production capacities are limited with forecasted shortcomings in the future. Because of their cytostatic nature and limited serum half-life these drugs need to be administered at constant high doses over a prolonged period of time; sometimes even as repeated intravenous infusions needing hospitalization and specialized care, with consequent administration-related adverse effects, toxic side effects, and immunogenicity, at a high cost to the patients and/or to the health care systems (Kirsch et al., 2000; Puduvalli and Sawaya, 2000; Samaranayake et al., 2009).
Most of the current antiangiogenic agents are intrinsically selective in their action on the VEGF pathway but are not specific. This property renders additional therapeutic benefits in some occasions at the expense of added adverse effects (Bergers and Hanahan, 2008). Hypertension, proteinuria/renal dysfunction, thrombosis, and hemorrhage are the most common adverse effects seen with systemic administration of current antiangiogenic therapies (Shibuya, 2008). It must be emphasized that many of the side effects of synthetic small molecular inhibitors of angiogenesis are still largely unknown.
Studies have revealed several mechanisms by which resistance can develop to current antiangiogenic therapies. Upregulation of alternative proangiogenic pathways leading to revascularization of tumors, protection of tumor vasculature by recruitment of proangiogenic inflammatory cells and increased pericyte coverage of vessels, enhanced tumor invasiveness, vessel cooption, and metastasis are some of the evasive resistance mechanisms that will ultimately contribute to the failure of these therapies. On the other hand, some tumors may demonstrate intrinsic resistance to these therapies because of their stage of progression, previous treatments, genetic constitution, and tumor microenvironmental conditions (Bergers and Hanahan, 2008).
Several preclinical studies have demonstrated that antiangiogenic therapy made some tumor cells more aggressive, invasive, and metastatic, even employing alternative mechanisms for angiogenesis, such as vessel cooption (Rubenstein et al., 2000; Kunkel et al., 2001; Leenders et al., 2004; Gomez-Manzano et al., 2008). Host responses to systemic VEGF inhibition are probably responsible for most of these effects. The lack of pericyte coverage is believed to be the reason for increased tumor intravasation and metastasis, especially during rebound vascularization after antiangiogenic therapy, when the new vessels have poor pericyte coverage. Lack of pericyte coverage not only increases metastatic potential but may also reduce vascular normalization and entry of chemotherapeutic agents. On the other hand, pericytes are known to provide survival signals and to protect the endothelium, leading to treatment resistance, warranting their destruction. Alternatively, activation of preexisting invasion programs, triggered by the microenvironmental circumstances, could explain the invasiveness (Bergers and Hanahan, 2008; Paez-Ribes et al., 2009). Second, tumor growth was accelerated with some of these antiangiogenic therapies especially in treatment-free intervals and after discontinuation of therapy, emphasizing the fact that the systemic host response to VEGF inhibition may contribute to the evasion of treatment response and influence the progression of metastatic lesions (Ebos et al., 2009; Paez-Ribes et al., 2009).
Gene Therapy to Inhibit Tumor Angiogenesis
Gene therapy can overcome some of the problems mentioned previously, with its ability to produce the therapeutic agent at high concentrations over a desired period of time at the local site, thereby reducing unwanted systemic adverse effects (Dell'Eva et al., 2002). As a one-time therapeutic application, gene therapy could be more economical when compared with other pharmaceuticals (Morishita, 2004). Local administration of antiangiogenic gene therapy has the advantage of overcoming natural barriers for conventional drugs, such as the blood–brain barrier. The low toxic profile of gene therapy products makes them good candidates to be combined with cytotoxic chemotherapies for an enhanced therapeutic effect. Antiangiogenic cancer gene therapy has demonstrated promising results in preclinical studies in many types of cancers (Chen et al., 2001). Antiangiogenic cancer gene therapy has so far been limited to preclinical studies. Tumor-directed and systemic gene therapy approaches have been successful in delivering this therapy, using both viral and nonviral gene delivery methods. Both inhibition of proangiogenic pathways as well as stimulation of antiangiogenic pathways have been employed with success.
Inhibition of proangiogenic pathways
In the majority of preclinical experiments antiangiogenic gene therapy strategies directed at inhibiting proangiogenic pathways have targeted VEGF–VEGFR signaling pathways. Platelet-derived growth factor receptor (PDGFR), FGF/FGF receptor (FGFR), TIE2, hypoxia-inducible factor-1α (HIF-1α), and hepatocyte growth factor (HGF) are the other main proangiogenic pathways targeted. Most studies have used a single antiangiogenic gene therapy whereas some have combined several antiangiogenic gene therapies and a few have combined them with chemotherapy and other gene therapy strategies. One option to target the VEGF–VEGFR signaling pathways is to deliver soluble forms of VEGFRs (sVEGFRs) that lack the transmembrane domain and are thus incapable of initiating intracellular signal transduction cascades. Soluble receptors can hamper intracellular signaling by forming dominant-negative heterodimers with transmembrane VEGFRs. Furthermore, because of the high affinity for VEGF, especially sVEGFR-1 can sequestrate VEGF, making it unavailable for the transmembrane receptors. sVEGFR-1 has indeed been employed successfully in combination with various viral vectors (Hasumi et al., 2002; Mae and Crystal, 2002; Sallinen et al., 2009) and nonviral vectors (Mori et al., 2000; Ohlfest et al., 2005) in a variety of cancer models (Kong et al., 1998; Takayama et al., 2000; Mahasreshti et al., 2001; Hasumi et al., 2002; Mae and Crystal, 2002; Sallinen et al., 2009).
The routes of administration have been diverse, including intratumoral (Kong et al., 1998), intramuscular (Takayama et al., 2000; Yoshimura et al., 2004; Takei et al., 2007), intravenous (Mahasreshti et al., 2003; Mae et al., 2005), intratracheal (Kong et al., 1998), intraperitoneal (Mori et al., 2000; Sako et al., 2004), intranasal (Mae et al., 2005), and pleural (Mae and Crystal, 2002) in vivo gene delivery as well as ex vivo gene transfer (Shiose et al., 2000; Ye et al., 2004), depending on the target disease model and the vector used. For example sVEGFR-1 administered intravenously to syngeneic murine colon carcinomas in the spleen with liver metastases retarded the growth of both primary tumor and liver metastases, but the lung metastases were inhibited only by intratracheal vector administration. In the same model intratumoral gene therapy suppressed tumor growth in a subcutaneous model (Kong et al., 1998). Whether intravenously administered viruses are really able to reach the tumor before being inactivated by the blood components remains controversial. The intravenous route has also been shown to cause more severe side effects. Adenovirus-mediated sVEGFR-1 impaired the growth of ovarian cancer in mouse models and improved survival (Mahasreshti et al., 2001), but the same vector administered intravenously localized to the liver, leading to hemorrhage, toxicity, and reduced survival of the animals (Mahasreshti et al., 2003).
The therapeutic outcome with sVEGFRs is attributed to the inhibition of tumor angiogenesis, growth, metastasis, ascites formation, and increases in survival and apoptosis. Some studies have demonstrated synergism with chemotherapies such as 5-fluorouracil (5-FU) (Zhang et al., 2005) and cisplatin (Gao et al., 2007) whereas others exhibited an improved outcome with a combination of other antiangiogenic gene therapies such as angiostatin–endostatin fusion protein (Ohlfest et al., 2005) and sVEGFR-2 and − 3 (Sallinen et al., 2009). AAV-mediated delivery of sVEGFR-1/-2 fusion protein gene (sVEGFR-1/R-2) demonstrated significant antitumor efficacy in a murine glioma model, but tumor progression was seen later, suggesting the presence of escape mechanisms (Harding et al., 2006). The same therapy in murine colon carcinoma and melanoma models led to an increase in the number of activated CD4+ and CD8+ tumor-infiltrating effector T cells and activated DCs, while reducing the numbers of regulatory T cells. On the other hand, tumors modified to overexpress VEGF had a higher number of regulatory T cells in the tumor-infiltrating lymphocyte population (Li et al., 2006), suggesting that blockade of VEGF improves antitumor immunity.
The other sVEGFR employed is sVEGFR-2. Again, there are multiple studies that have evaluated its efficacy with several different vectors and in many tumor models (Pin et al., 2004; Jin et al., 2005; Reinblatt et al., 2005; Lyons et al., 2007). Ex vivo gene delivery has been the method of choice in most of the studies. Reduced tumor growth, tumor volume, angiogenesis, ascites formation, and metastasis were seen as the main effects of therapy. Combination with oncolytic adenovirus therapy and interleukin (IL)-4 improved the outcome whereas adding IL-12 reduced it. A comparative study revealed that adenovirus-mediated sVEGFR-2 gene therapy is far superior in inhibiting the growth of preexisting murine cancer models compared with adenovirus-mediated angiostatin, endostatin, and neuropilin gene therapies (Kuo et al., 2001). However, adding adenoviruses expressing sPDGFRβ or TIE2-Fc had no added therapeutic benefit, suggesting that targeting multiple antiangiogenic pathways is needed only when one is not blocked adequately (Kuhnert et al., 2008). However, combination of adenovirus-mediated sVEGFR-1, −2, and −3 in a mouse ovarian cancer xenograft model resulted in reduced tumor growth, vascularity, and ascites formation compared with the individual gene therapies, suggesting the superiority of antiangiogenic gene therapy combinations (Sallinen et al., 2009).
VEGF inhibition has also been achieved by anti-sense VEGF constructs (Saleh et al., 1996a,b) and VEGF small interfering RNA (siRNA) delivery (Im et al., 1999; Sasaki et al., 1999) and by expressing a dominant negative VEGFR-1 mutant (Heidenreich et al., 2004) and VEGFR-2 (Millauer et al., 1994, 1996; Machein et al., 1999). Intratumoral injection of a plasmid vector encoding VEGF siRNA reduced tumor vascularity but failed to stop tumor growth in a glioma xenograft model. However, retrovirus-mediated stable transduction of the same cell line with VEGF siRNA and IL-4 gene completely prevented the growth of tumors (Niola et al., 2006). Malignant glioma cells transfected with dominant-negative HIF-1α and siRNA against HIF-1α reduced ex vivo tumor growth and proliferation but not microvessel density, suggesting that mechanisms other than antiangiogenesis are responsible for the effects, in spite of reducing VEGF secretion (Jensen et al., 2006).
Stimulation of antiangiogenic pathways
Endogenous inhibitors of angiogenesis and their combinations, and cytokines have been commonly used to stimulate antiangiogenic pathways in cancer gene therapy.
A naturally occurring type XVIII collagen by-product, endostatin is a known inhibitor of VEGF and bFGF. Both nonviral vectors (Chen et al., 1999; Ding et al., 2001; Li et al., 2003; Luo et al., 2005a,b; Zheng et al., 2005) and viral vectors (Chen et al., 2000; Wen et al., 2001; Tai et al., 2003; Subramanian et al., 2005) were employed to treat various cancer models (Yoon et al., 1999; Sauter et al., 2000; Jin et al., 2001; Calvo et al., 2002; Pulkkanen et al., 2002; Luo et al., 2005a,b, 2006). The main therapeutics effects seen were in survival, tumor growth, angiogenesis, and apoptosis. Apart from these, some studies revealed an inhibition of tumor metastasis (Yoon et al., 1999; Subramanian et al., 2006) and vessel cooption. In spite of being effective in murine models some failed to demonstrate efficacy in human cancer xenograft models (Jin et al., 2001). The effect seen during the treatment of preinvasive cancers implicated the impact of therapy on the so-called angiogenic switch (Calvo et al., 2002). A paraendothelial action of endostatin through binding to integrin α5β1 was suggested to prevent peritoneal dissemination (Yokoyama et al., 2007). The route of gene delivery varied from those in previous studies, making direct comparisons difficult. Systemic adenovirus administration gave endostatin concentrations varying between 936 ng/ml (Sauter et al., 2000) and 1.34 μg/ml (Wen et al., 2001). Serum endostatin levels peaked 3 days after intratumoral gene therapy but dropped to almost half by day 7 (Li et al., 2004). An important observation was that the effect of intravenous adenoviral vector administration was lost after some time, because of anti-adenoviral antibodies (Jin et al., 2001). Moreover, high-dose intravenous administration of adenoviral vectors resulted in adverse effects such as weight loss, bleeding, and death of the animals (Wen et al., 2001). On the other hand, a single intramuscular administration of AAV vector gave sustained secretion of endostatin for up to 9 weeks (Subramanian et al., 2005). A study employing ex vivo transduction demonstrated that therapeutic efficacy persisted even when the percentage of transduced cells was only 25% (Yoon et al., 1999). Some studies demonstrated possible synergism with radiotherapy (Luo et al., 2005a,b; Zheng et al., 2005), chemotherapy (Subramanian et al., 2006), and other gene therapy strategies such as GM-CSF (Tai et al., 2003) and adenovirus-mediated herpes simplex virus thymidine kinase/ganciclovir (HSV-tk/GCV) suicide gene therapy (Pulkkanen et al., 2002).
Angiostatin is also a naturally occurring plasmin degradation product that binds and inhibits endothelial cell proliferation and migration, and induces their apoptosis. Stable transduction of liver cancer cells with angiostatin gene suppressed tumor growth (Ishikawa et al., 2003; Schmidt et al., 2006) in murine models but failed to inhibit tumor angiogenesis, suggesting that multifactorial effects of gene therapy can contribute to antitumor effects (Schmidt et al., 2006).
Several attempts have been made to combine different angiogenic inhibitors. Introduction of endostatin and angiostatin genes separately (Scappaticci et al., 2001; Isayeva et al., 2007a,b) or as a fusion protein gene (Hampl et al., 2001; Hajitou et al., 2002; Raikwar et al., 2005) and combination of the angiostatin kringle 1–3 gene with endostatin (Kim et al., 2004; Kim and Park, 2005) are some examples. Combinations had a better therapeutic effect compared with individual therapies and in some studies fusion protein gene therapy was superior to the combination of the two genes (Scappaticci et al., 2001). Adding paclitaxel (Isayeva et al., 2007b) and adenovirus encoding the soluble form of endothelium-specific receptor tyrosine kinase TIE2 gene therapy (Raikwar et al., 2005) enhanced the efficacy in some studies. Reduced tumor invasiveness and metastasis were observed in these studies (Kim et al., 2004).
Intramuscular delivery of plasmid DNA encoding vasostatin (N-terminal domain of calreticulin) gene inhibited tumor angiogenesis and growth, and increased survival and tumor cell apoptosis in tumor-bearing mice (Xiao et al., 2002).
Marrow-derived mesenchymal stem cells were ex vivo modified by AAV vectors to express interferon (IFN)-α; when systemically introduced into mice bearing metastatic melanoma, tumor growth and angiogenesis were reduced and survival was improved (Ren et al., 2008). The same therapy with lentiviral vector in a prostate cancer model gave similar results (Persano et al., 2009).
Treatment of established orthotopic and disseminated neuroblastomas with AAV-mediated IFN-β restricted tumor growth, increased survival, and reduced the expression of VEGF and bFGF with marked antiangiogenic effects, which were further enhanced by adding low-dose cyclophosphamide (Streck et al., 2005).
Intratumoral injection of IL-12 gene therapy, using Semliki Forest virus (SFV) vectors, into established murine melanomas inhibited tumor growth and neovascularization, without evidence of cytotoxic T lymphocytes or NK cell-mediated cytotoxicity, suggesting that the therapeutic effect was a result of antiangiogenic properties of the therapy (Asselin-Paturel et al., 1999).
Oncolytic herpes simplex virus armed with tissue inhibitor of matrix metalloproteinase (TIMP)-3 gene reduced tumor vascularity and circulating endothelial progenitor cells while delaying tumor growth in neuroblastoma and malignant peripheral nerve sheath tumor models, suggesting the possibility of antivasculogenesis (Mahller et al., 2008).
Other gene therapy strategies with antiangiogenic properties
Some other gene therapy strategies are known to possess antiangiogenic properties, apart from their main mode of action. Adenovirus-mediated antisense urokinase plasminogen activator receptor (uPAR) and cathepsin B ex vivo gene therapy applied to glioma cells inhibited tumor growth, invasiveness, and angiogenesis (Gondi et al., 2004b). Similar results were observed with an siRNA sequence targeting uPAR as a plasmid DNA (Gondi et al., 2004a, 2003) and with antisense uPAR and MMP-9 gene therapies mediated by adenoviral vector in murine models of lung cancer and glioma (Lakka et al., 2003; Rao et al., 2005).
Dominant-negative epidermal growth factor receptor (EGFR) gene therapy (Lammering et al., 2003) and HSV-tk/GCV suicide gene therapy, with its ability to transduce and kill dividing endothelial cells, have antiangiogenic properties apart from their main mode of action (Ram et al., 1994).
Genetic delivery of an anti-p65 intracellular antibody (intrabody) suppressed invasion and angiogenesis in glioma cells by inhibiting nuclear factor (NF)-κB, which led to decreased expression of MMP-9, uPAR, uPA, and VEGF (Li et al., 2007).
PTEN (phosphatase and tensin homolog on chromosome 10) and p53 correction gene therapies have shown bystander effects due to their antiangiogenic properties (Abe et al., 2003). Ovarian cancer cells transduced ex vivo with the PTEN gene had a slower growth rate and a lower number of blood vessels in murine tumor models, suggesting antiangiogenic properties of this gene therapy modality (Takei et al., 2008).
Discussion
Major concerns about antiangiogenic gene therapies
Gene therapy is not without adverse effects. Several authors have previously suggested the possibility of systemic toxicity with certain antiangiogenic gene therapies (Kong and Crystal, 1998; Crystal, 1999). Severe hemorrhage, massive hepatic necrosis, and residual hepatocyte apoptosis, leading to early death, was reported in a murine ovarian cancer model after systemic administration of sVEGFR-1 (Mahasreshti et al., 2003). Another study revealed that intravenous administration of sVEGFR-1 in normal mice resulted in ascites and early death (Kuo et al., 2001). Preeclampsia patients with high serum sVEGFR-1 levels are known to have hypertension, proteinuria, and renal damage. Likewise, the same was observed with systemic administration of sVEGFR-1 in an animal model (Sugimoto et al., 2003), suggesting that systemic sVEGFR-1 gene therapy may have similar consequences. Weight loss, bleeding, and increased incidence of death were seen in mice treated with systemic endostatin gene therapy (Wen et al., 2001). However, in most preclinical studies gene therapy was directly administered into the tumor, thereby reducing the possibility of systemic toxicity. In clinical settings this is still achievable if the tumors are localized. Unfortunately, most of the current antiangiogenic therapies have been approved for metastatic tumors, where gene therapy would have to be administered systemically. Transductional and transcriptional targeting of gene delivery vectors may hold promise to solve this problem.
High intratumoral and interstitial pressures within solid tumors, caused by the presence of multiple aberrant, leaky blood vessels, may limit the vector volume that can be injected into solid tumors (Boucher et al., 1997). High interstitial pressure may hinder the dissemination of vectors within solid tumors, thereby having a negative effect on gene transduction efficacy. Technological advances in high-titer vector production and biotechnological modifications of vectors may increase transduction efficacy. However, the greatest challenge to gene therapy is still the limited transduction efficiency of the current vectors.
Some studies have shown that the long-term presence of antiangiogenic agents in high concentrations is needed for therapeutic success (Goldman et al., 1998; Harding et al., 2006). However, others have demonstrated therapeutic efficacy with adenoviral vectors that have a relatively short duration of transgene expression (Takayama et al., 2000; Pulkkanen et al., 2002; Liu et al., 2007). One must be cautious when extrapolating results from preclinical studies in aggressive tumor models. One typical caveat is that in animal models survival is usually short compared with the duration of gene expression, as opposed to human cancers, in which patient survival and the need for treatment effect can be much longer. For example, adenoviruses, being nonintegrating vectors, have a limited duration of transgene expression (Kremer and Perricaudet, 1995; Uchida et al., 2001). Studies have shown this to be as low as a few weeks in tissues such as liver (Yang et al., 1994), lung (Simon et al., 1993), and muscle (Dai et al., 1995), but may last as long as 60 days in the brain (Byrnes et al., 1995; Kajiwara et al., 1997), because of its immune-privileged state. Host immune responses may be responsible for the short duration of adenovirus-mediated gene expression (Driesse et al., 1998). The risk would be even greater with repeated vector administrations, making the efficacy of repeated gene transfer limited (Yang et al., 1994). Prior exposure to the adenoviral vector is believed to reduce transduction efficacy and the duration of transgene expression (Ohmoto et al., 1999). However, results suggest that the effect of preexisting antivector immunity will depend on many other factors in the microenvironment and on the vector dose (Tsai et al., 2008). AAV and lentiviral vectors, on the other hand, have a long-term gene expression pattern. However, compared with adenoviral vectors the titers of these vectors are still much lower.
Why do antiangiogenic therapies fail?
The exact antitumor mechanisms of anti-VEGF therapies are yet to be determined. The original notion of inhibiting blood vessel growth and starving tumors to death has evolved into a more complex picture involving multiple antitumor mechanisms. Many studies have demonstrated that antiangiogenic therapy in cancer causes tumor necrosis/apoptosis, destruction of tumor cells, and reduction in tumor size (Goldman et al., 1998; Pulkkanen et al., 2002; Heidenreich et al., 2004) whereas others suggest that this treatment method is cytostatic, needing the aid of other treatment modalities, such as chemotherapy, for the eradication and destruction of tumor cells (Kirsch et al., 2000). In the clinical setting anti-VEGF therapies have shown their efficacy as single agents in some cancers whereas in others their effect was produced in combination with chemotherapy (Ellis and Hicklin, 2008). The main mechanism behind most anti-VEGF therapies is the inhibition of growth of new blood vessels in tumors. However, the use of vessel count/microvessel density as a marker of treatment effect has been questioned (Hlatky et al., 2002). Studies have shown that VEGF is needed for the survival of tumor endothelial cells (Benjamin and Keshet, 1997) and lack of it leads to their apoptosis even though regression of the tumors has not been a frequent finding (Ellis and Hicklin, 2008). It is believed that lack of pericyte coverage makes tumor endothelium vulnerable to the effects of antiangiogenic agents, suggesting the advantage of therapies acting on both of these cell types.
Inhibition of the incorporation of bone marrow-derived hematopoietic cells and endothelial progenitor cells into the tumor vasculature is another possible mechanism of anti-VEGF therapies. However, the percentage of the tumor vasculature composed of these cells is variable, and is probably less in human tumors compared with preclinical animal models. Their contribution to tumor vasculature has even been challenged (Purhonen et al., 2008).
It is widely accepted that antiangiogenic therapy normalizes intratumoral blood vessels, decreases their permeability, and thereby reduces the interstitial pressure within the tumor. The resulting restoration of the hydrostatic pressure gradient across the vascular wall facilitates deeper penetration of chemotherapeutic agents into the tumor, explaining the possible synergism between antiangiogenic therapy and chemotherapy (Jain et al., 2007). There is evidence suggesting that this will create a therapeutic window, where tissue oxygenation and thereby the response to radiotherapy is increased (Winkler et al., 2004). Another possibility is that transient vessel normalization will reduce the VEGF-induced intratumoral hemorrhages and edema, relieving the effects of tumor burden. Antiangiogenic therapies targeting both endothelial cells and pericytes may be counterproductive in this regard, because disruption of pericytes may hamper the vessel normalization that increases the delivery of chemotherapeutic agents into tumors (Bergers and Hanahan, 2008). On the other hand, there is evidence to suggest that concomitant administration of antiangiogenic agents reduced the entry of chemotherapeutic agents into tumors, leading to an antagonistic effect (Ma et al., 2001; Claes et al., 2008). Chronic inhibition of angiogenesis is known to cause similar consequences. Some authors believe that synergism is due to sensitization of the tumor vasculature to chemotherapy by antiangiogenic agents (Kerbel and Kamen, 2004). The effect of antiangiogenic therapy on the blood–brain barrier may have an impact on drug delivery into CNS tumors. One study revealed a reduction in the permeability of the blood–brain barrier after adenovirus-mediated sVEGFR-1 gene therapy into the ventricular system in an ischemic rat model (Kumai et al., 2007). Similar mechanisms can impair drug delivery into brain tumors with systemic antiangiogenic therapy. Antiangiogenic gene therapy, if targeted into the tumors, may circumvent this disadvantage.
It has been reported that VEGF secretion from tumors undergoes feedback upregulation during VEGF blockade via overexpression of other VEGF receptors such as neuropilin-1. Apart from VEGF, other angiogenic pathways such as the FGF and PDGF pathways are upregulated whereas the antiangiogenic pathways, such as Erb-2, angiopoeitin-2, and TIMP-2, are downregulated in response to VEGF inhibition therapy, suggesting the presence of compensatory mechanisms (Harding et al., 2006; Bergers and Hanahan, 2008). Redundancy of angiogenic pathways is further suggested by the upregulation of PlGF during bevacizumab and sunitinib therapy (Kerbel, 2008). One study has implicated VEGF-D as a factor that can maintain tumor blood flow in the absence of VEGF-A (Moffat et al., 2006). VEGF-A expression is known to be induced by genotoxic stress caused by chemotherapy and radiotherapy. These phenomena may have to be taken into account when evaluating the therapeutic usefulness of concomitant antiangiogenic therapies (Ellis and Hicklin, 2008).
Tumors in highly vascularized organs such as liver, lung, and brain may employ vessel cooption as a mode of blood supply (Ellis and Hicklin, 2008). Thus, antiangiogenic therapies targeting VEGF may not be as effective against tumors in these organs. On the other hand, some tumor cell lines are known to poses intrinsic resistance to anti-VEGF therapies and some metastatic tumors may be devoid of VEGF/VEGFRs, making them resistant to anti-VEGF therapies. Acquired resistance can be due to activation of alternative angiogenic pathways, selection of resistant tumor subclones, or rapid vascular remodeling after therapy, which can make the vasculature unresponsive to antiangiogenic therapies (Kerbel, 2008).
Preclinical studies with antiangiogenic gene therapy have not demonstrated increased tumor invasiveness so far. In fact, some preclinical endostatin gene therapy studies have demonstrated reduced tumor invasiveness and dissemination (Hajitou et al., 2002; Yokoyama et al., 2007). One explanation could be that in most preclinical studies gene therapy was directed to the tumor itself, thereby avoiding counterproductive host responses. On the other hand, genetic deletion of angiogenic factors such as VEGF, HIF1α, and MMP-9 has demonstrated increased invasive properties in some tumor cell lines, suggesting that this serious adverse effect is not limited to conventional antiangiogenic agents. Moreover, antiangiogenic gene therapy still lacks clinical evidence to support this claim.
Duration of the gene expression and the concentration of the therapeutic agent within the tumor microenvironment must be optimal for therapeutic effect and to avoid the adverse effects and escape mechanisms. In our opinion, this will be one of the biggest challenges for gene therapy in the clinical setting.
Future Prospects
As with current antiangiogenic agents, antiangiogenic gene therapies will most probably find their place in oncological practice in combination treatment with conventional chemotherapy and radiotherapy, or perhaps even in combination with other gene therapy strategies that have synergistic effects. Identifying treatment combinations and schedules that will minimize the emergence of resistance and engage tumors at different fronts by being cytotoxic or immunomodulatory, or by inhibiting invasion and metastasis, may provide vital keys to success in future antiangiogenic gene therapies and may also broaden the use of this therapy.
Adenovirus-mediated HSV-tk/GCV suicide gene therapy has been combined successfully with adenovirus-mediated endostatin gene therapy in an orthotopic human renal cell cancer model in nude mice (Pulkkanen et al., 2002). Suicide gene therapy in this regard is a potential partner for antiangiogenic gene therapy to kill tumor cells with far less toxic side effects, compared with conventional chemotherapeutic agents. The combination of antiangiogenic gene therapy with other gene therapy strategies, such as oncolytic virotherapy, immune gene therapy, and gene therapies targeting tumor invasion and metastasis, may hold promise for future treatments.
Another possible target for antiangiogenic therapy would be the tumor stem-like cell fraction, which is believed to maintain tumor growth and make them resistant to most available therapies. Emerging data suggest that it might be possible to target these cells with antiangiogenic therapies. There is considerable evidence that therapies targeting multiple antiangiogenic pathways have better therapeutic efficacy compared with those targeting only single targets (Sallinen et al., 2009). However, these “broad spectrum” therapies will probably come with the cost of additional side effects.
Implantation of genetically modified cells that secrete antiangiogenic agents would be another way to deliver antiangiogenic therapy. Cell encapsulation systems can be used to protect these cells from the immune system and regulatable systems can be incorporated to control gene expression in these systems (Samaranayake et al., 2009). However, systemic side effects would be a disadvantage in this method.
Lack of reliable biomarkers to evaluate the efficacy of antiangiogenic therapies and to identify patients who either respond well or may develop resistance to these therapies represents an important area for future research. Most clinical trials do not permit direct evaluation of antiangiogenic effects and imaging studies are usually not done frequently enough to confirm efficacy. The development of such markers and imaging methods, and the designing of clinical trials with these challenges in mind, will help to broaden our understanding of these therapies and mechanisms of action and resistance. The development of biomarkers that will predict which patients will or will not respond to a particular type of antiangiogenic therapy is equally important, when it comes to designing cost-effective, rational, and ethical antiangiogenic gene therapies.
Footnotes
Acknowledgments
This work was supported by the Finnish Academy, a Clinigene EU grant, and Ark Therapeutics, Kuopio, Finland.
Author Disclosure Statement
Haritha Samaranayak, Ann-Marie Määttä, and Jere Pikkarainen are employees of Ark Therapeutics, Kuopio, Finland.