
Abstract
Replication-selective oncolytic adenoviruses have proven safety records with promising clinical outcomes. However, strategies to improve efficacy are still required. Here we report greatly improved antitumor efficacy for both attenuated (dl1520) and highly potent (dl922–947) oncolytic mutants in combination with the current standard of care for late-stage hormone-independent prostate cancers, mitoxantrone or docetaxel. In agreement with previous reports, dl922–947 had superior potency compared with dl1520 both as a single agent and in combination with cytotoxic drugs. The dl922–947 mutant caused significant synergistic cell killing in both drug-insensitive and -sensitive prostate cancer cell lines, PC3 and DU145, respectively, when combined with docetaxel or mitoxantrone. The magnitude of the synergistic response was greatest for dl1520 whereas overall efficacy was greatest for dl922–947, and the latter was also more efficacious in vivo in prostate cancer models. In DU145 and PC3 cells increased viral uptake (up to 9- and 8-fold, respectively), E1A expression, and altered cell cycle progression contributed to the synergistic cell killing. A similar trend was also detected in LNCaP cells. Potent E1A expression was essential for the response. In murine xenograft models (DU145 and PC3) tumor growth inhibition was improved when suboptimal doses of docetaxel and viral mutants were combined. These findings demonstrate that the efficacy of highly potent oncolytic mutants such as dl922–947 that target the retinoblastoma protein (pRb) pathway could be further enhanced even with low drug doses, and support the deletion of the E1ACR2 region in future candidate adenoviruses for treatment of hormone-independent prostate cancers.
Introduction
A promising targeted anticancer treatment under development is virotherapy. Several engineered, replication-selective, oncolytic adenoviral mutants have been developed that efficiently kill cancer cells in culture and eliminate tumor xenografts in vivo (Parato et al., 2005; Waehler et al., 2007; Matthews et al., 2009). Mutants were designed to enable viral gene expression and replication only in tumors, with minimal toxicity to normal tissue. To date, clinical safety has been demonstrated in hundreds of patients with various oncolytic adenoviruses without cross-resistance to conventional clinical therapies (Aghi and Martuza, 2005; Liu et al., 2007; Pesonen et al., 2010). In the majority of trials, mutants with the E1B55K-gene deleted (e.g., dl1520) were evaluated for restricted replication in cancer cells with deregulated p53 and mRNA export pathways (Harada and Berk, 1999; O'Shea et al., 2004). Selectivity was confirmed for dl1520 mutants (e.g., Onyx-015, H101) in the majority of tumors whereas efficacy was reported only in combination with the cytotoxic drugs cisplatin, 5-fluorouracil (5-FU), or gemcitabine in phase II and III trials (Khuri et al., 2000; Reid et al., 2002; Aghi and Martuza, 2005; Garber, 2006). A modified dl1520 mutant expressing two suicide genes (Ad5-CD/TKrep) was evaluated in prostate cancer patients with localized disease in combination with prodrugs or radiation therapy and was reported to have potential long-term benefits in patients (Freytag et al., 2007a–c). Prostate-specific viruses were also evaluated in phase I–II trials, using prostate-specific antigen (PSA) and probasin enhancers to drive viral gene expression and replication after intratumoral or intravenous administration (DeWeese et al., 2001; Small et al., 2006). Although all mutants had low toxicity profiles in patients with prostate cancer, efficacy was not definitively confirmed. However, favorable PSA responses were demonstrated in combination with chemotherapy or radiation therapy. One of these prostate-selective mutants (CV787) was further demonstrated to act synergistically with taxanes in LNCaP preclinical models (Yu et al., 2001). In addition, a replication-defective mutant (Ad-OC-TK) targeting bone metastasis was also reported to decrease pain in combination with prodrug administration (Terao et al., 2009).
More potent oncolytic mutants have been developed, such as dl922–947 and several other Ad5Δ24 variants, which complement the frequently deregulated pRb pathway in cancer cells (Heise et al., 2000; Stolarek et al., 2004; Page et al., 2007; Öberg et al., 2010). A clinical trial with one Ad5Δ24 mutant for recurrent ovarian cancer and malignant gliomas was recently completed (NCT00562003 at
Here, we explored the potential to improve further the efficacy of the potent dl922–947 and attenuated dl1520 mutants in prostate cancer models. We hypothesized that activation of specific cellular programs in response to replicating complementation mutants and cytotoxic drugs might converge to amplify efficacy synergistically. Cell killing was greatly improved when either mutant was combined with mitoxantrone or docetaxel, the standard treatment of care for advanced prostate cancer. Increased uptake of virus in the presence of chemotherapeutics followed by higher expression levels of the chemosensitizing E1A gene was part of the response. These findings demonstrate that suboptimal doses of virus and drugs were highly efficacious when combined, implying that unwanted toxicity to normal cells could be minimized in future clinical applications.
Materials and Methods
Cancer cell lines and adenoviruses
The human prostate carcinoma cell lines LNCaP (lymph node metastasis) and DU145 (brain metastasis), human lung cancer cell line H460 (American Type Culture Collection [ATCC], Manassas, VA), and a subclone of human prostate PC3 cells (higher virus sensitivity than the parental cells; Clare Hall, Cancer Research UK, London, UK) were grown under standard conditions in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum. The following viral mutants were used: Ad5 (adenovirus type 5, wild-type), dl312 (ΔE1A, ΔE3B), AdGFP (ΔE1), dl922–947 (ΔCR2, ΔE3B), and dl1520 (ΔE1B55K, ΔE3B). All viruses had a particle-to-infectious unit ratio of 10–40 viral particles per plaque-forming unit (vp/pfu).
E1A-expressing DU145 cells were generated by retrovirus transduction according to standard protocols (E. Miranda, unpublished data). Briefly, using a pLPC plasmid (a kind gift from S. Ramón y Cajal, Universitat Autònoma de Barcelona, Barcelona, Spain), retroviruses were constructed for expression of E1A12S or green fluorescent protein (GFP). DU145 cells were infected with the corresponding retroviral vectors and selected with puromycin before cytotoxicity assays.
Cell-killing assay and synergistic effects
Evaluation of cell-killing efficacy with the combination treatments was performed as previously described (Cheong et al., 2008). In brief, cells were plated at 1 × 104 cells per well in 96-well plates and, 24 hr later, infected with viruses or treated with drugs. Cell viability was determined 6 days later (MTS assay) and dose–response curves were generated to determine the concentrations killing 50% of cells (i.e., 50% effective concentration, EC50). Drugs were used at the following dose ranges: mitoxantrone (Onkotrone; Baxter, Deerfield, IL), docetaxel (Taxotere; Sanofi-Aventis, Paris, France), and cisplatin (Sigma-Aldrich, St. Louis, MO), all at 8 × 10–5 to 2 × 102 μM; paclitaxel (Calbiochem) and doxorubicin and etoposide (Sigma-Aldrich), all at 3 × 10–5 to 30 μM; and viruses at 8.4 × 10–4 to 1 × 105 particles/cell (ppc). Four different dilution ratios were included in each experiment, keeping virus-to-drug ratios (ppc/nM) constant. Isobolograms were generated to determine combination indexes (CIs) and effects on cell death as previously described (Chou, 2006; Cheong et al., 2008). Each data point was determined from triplicate samples, and repeated three to five times. Synergy was defined as a greater effect on cell death than the theoretical additive effect according to the following: CI ≤ 0.9 = synergy (S); CI ≥ 1.1 = antagonism (A); and for additive (Add) effect, 0.9 < CI < 1.1 (Chou, 2006). The straight lines in the isobolograms represent the theoretical line of additivity.
Replication assay
Cells were seeded at 1 × 105 to 106 cells per well in 6-well plates and, 24 hr later, were infected at 10 or 100 ppc and/or treated with drugs at concentrations less than the EC15: mitoxantrone at 10 and 50 nM and docetaxel at 0.1 and 1.0 nM. Additions were either simultaneous or 24 hr apart. Cells and media were collected 24–96 hr postinfection and analyzed as the 50% tissue culture inhibitory dose (TCID50) as previously described (Wang et al., 2003). Each sample was investigated in triplicate and data from two or three separate studies were averaged and expressed as plaque-forming units per cell.
Immunoblot analysis
Cells were treated as in the replication assay, harvested, and lysed 24 and 48 hr postinfection (50 mM HEPES [pH 7.4], 250 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol [DTT], 1 mM NaF, and 1% Triton X-100 containing a protease inhibitor cocktail; Roche, Indianapolis, IN). Aliquots of 10–20 μg total protein were separated on reducing sodium dodecyl sulfate (SDS)–polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA). Viral and cellular proteins were detected with the following antisera: rabbit anti-Ad2 E1A diluted 1:2000 (SC-430; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-hexon diluted 1:2000 (Autogen Bioclear/Source BioScience LifeSciences, Nottingham, UK), mouse anti-topoisomerase IIα diluted 1:500 (SWT3D1) (SC-56805; Santa Cruz Biotechnology), and mouse anti-β-tubulin diluted 1:20,000 (Sigma-Aldrich). Detection was by application of horseradish peroxidase-conjugated secondary antibody, that is, goat anti-rabbit IgG or goat anti-mouse IgG (Dako, Carpinteria, CA), and chemiluminescence reagent (GE Healthcare Life Sciences, Piscataway, NJ) followed by autoradiography (Kodak BioMax film; Carestream Health, Rochester, NY).
Flow cytometry analysis
Cells were seeded at 2 × 104 cells per well in 24-well plates and 24 hr later treated with 0.1, 1.0, or 5.0 nM docetaxel or 10 or 50 nM mitoxantrone. Cells were infected with Ad5GFP at 10 or 100 PPC and harvested 24–96 hr later in fluorescence-activated cell-sorting (FACS) buffer (phosphate-buffered saline [PBS], 2% bovine serum albumin [BSA], 1 mM EDTA). Cellular receptors were detected with mouse monoclonal antibodies targeting human coxsackievirus and adenovirus receptor (CAR) diluted 1:500 (ATCC), αv-integrin at 2.5 μg/ml (Abcam, Cambridge, UK), αvβ3-integrin (MAB1976Z) and αvβ5-integrin (MAB1961Z) diluted 1:100 and 1:65, respectively (Millipore), followed by fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse antibody diluted 1:30 (F0479; Dako). For cell cycle analysis, cells were fixed in 70% ethanol for 30 min, treated with 5 μg of RNase A (Sigma-Aldrich), and analyzed with a FACSCalibur instrument (BD Biosciences, San Jose, CA) acquiring 10,000 events per sample from triplicate wells, using propidium iodide (PI; Sigma-Aldrich).
Quantitative PCR
Cells were infected with viral mutants at 100 ppc and/or treated with mitoxantrone at 10 and 50 nM or docetaxel at 0.1 and 1.0 nM for 3–48 hr followed by RNA extraction (Qiagen, Valencia, CA). First-strand cDNA was synthesized from 1 μg of total RNA, using mouse mammary leukemia virus (MMLV) reverse transcriptase (RT) and random hexamer primers (Applied Biosystems, Foster, City, CA). Expression levels of E1A and cellular 18S mRNAs were determined with the following primers: E1A-forward, 5′-TGCCAAACCTTGTACCGGA-3′; E1A-reverse, 5′-CGTCGTCACTGGGTGGAAA-3′; 18S-RNA forward, 5′-CGCCGCTAGAGGTGAAATTC-3′; and 18S-RNA reverse, 5′-CATTCTTGGCAAATGCTTTCG-3′. Power SYBR green master mix was added and quantitative PCR (qPCR) was performed (7500 real-time PCR system; Applied Biosystems). Results were analyzed with the system SDS software and expressed as the ratio of E1A cDNA to cellular 18S cDNA (g/g × 103) in each sample (n = 3). Data presented are from representative studies.
In vivo tumor growth
Tumors were grown in one flank of C57BL/6 athymic (ICRF nu/nu) mice by subcutaneous implantation of 1 × 107 or 1 × 106 PC3 or DU145 cells in 50% Matrigel (BD Biosciences) as previously described (Öberg et al., 2010). Dose responses to viral mutants and docetaxel were determined by intratumoral administration of 1 × 108 to 1 × 1010 vp/injection three times at 48-hr intervals and docetaxel at 1.0 to 15.0 mg/kg intraperitoneally two or three times from 2 to 8 days after the first virus injection. Suboptimal doses and treatment intervals for each agent alone were selected to enable detection of additive/synergistic effects on tumor growth inhibition. Tumor volumes were estimated twice weekly: volume = (length × width2 × π)/6. Treatments were initiated when tumors were 100 ± 20 μl and tumor growth and progression were monitored until tumors reached 1.44 cm2 (according to animal welfare regulations; U.K. Home Office). Treatment groups were balanced by tumor size at the time of treatment initiation in all cases, differences in tumor growth between treatment groups were analyzed by one-way analysis of variance, and p < 0.05 was considered significant. Survival analysis was expressed as time to progression (tumor volume ≥ 500 μl) performed according to the Kaplan–Meier method (log rank test for statistical significance).
Results
Wild-type adenovirus synergizes with a range of cytotoxic drugs to kill both drug-sensitive and -insensitive prostate cancer cells
Sensitivity to common chemotherapeutics was evaluated by determining EC50 values of six clinically used drugs in three metastatic prostate cancer cell lines. The microtubule-stabilizing drugs docetaxel and paclitaxel had similar efficacy in all cell lines tested (EC50 = 1.9–10.6 nM), with PC3 and LNCaP cells the least sensitive. PC3 cells were highly insensitive to the topoisomerase inhibitors, especially to mitoxantrone, at EC50 = 842 ± 183 nM, compared with DU145 and LNCaP cells at 361 ± 4.3 and 162 ± 59 nM, respectively. PC3 cells were also more resistant to the DNA-intercalating agent cisplatin, with LNCaP and DU145 cells 5-fold more sensitive. The EC50 value for wild-type virus (Ad5) was also 5- and 100-fold higher in PC3 cells compared with DU145 and LNCaP cells, respectively.
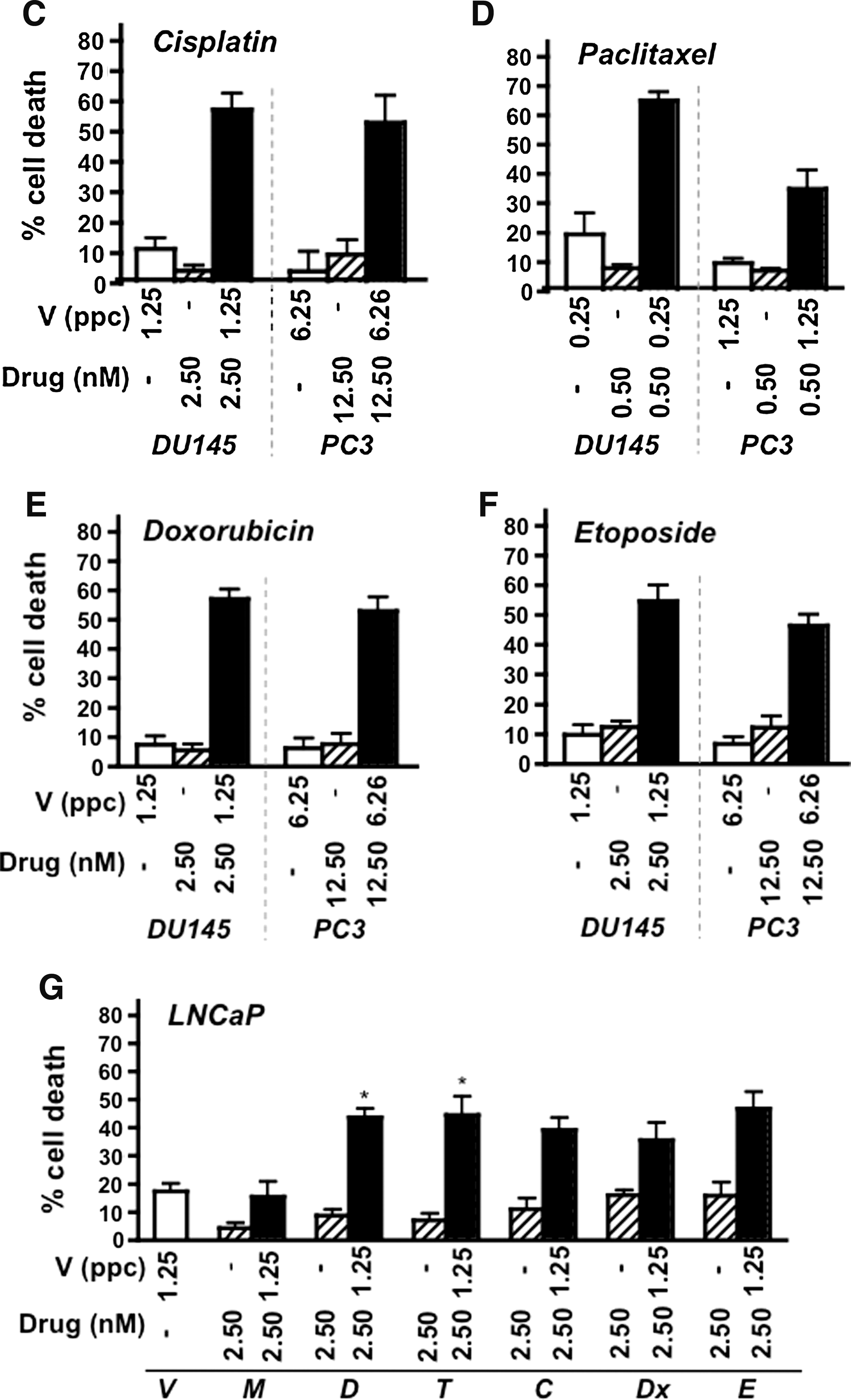
To test whether cell killing could be improved in the more resistant PC3 cells, virus and drugs were combined at fixed suboptimal concentrations (<EC20 doses). Combinations of Ad5 and mitoxantrone or docetaxel in PC3 cells were more efficacious than the predicted additive effects (Fig. 1A and B). For example, mitoxantrone at 2.5 or 12.5 nM and virus at 1.25 or 6.25 ppc resulted in 15–30% increases in cell death above the theoretical additive values. Interestingly, similar combinations also resulted in synergistic cell death in the relatively sensitive DU145 cells (Fig. 1A and B). To explore virus–drug interactions, additional DNA-damaging and cell cycle-interfering drugs were tested. Synergistic effects on cell death were obtained with all tested cytotoxic drugs in PC3 and DU145 cells at the selected doses (Fig. 1A–F). In the highly virus-sensitive LNCaP cells a similar trend of greater cell killing in combination with drugs was observed although the effect was synergistic only with docetaxel and paclitaxel at these concentrations (Fig. 1G).

Suboptimal concentrations of virus and cytotoxic drugs synergize to enhance prostate cancer cell killing in culture. Cells were treated for 3–6 days with fixed concentrations of Ad5 wild-type virus (V) and various drugs, at virus-to-drug ratios of 0.5 to 2.5 ppc/nM. Cell viability was determined for single agents and combination treatments.
Strong synergistic interactions with adenovirus and mitoxantrone or docetaxel
To explore further the enhancement of cell killing, combinations of virus and mitoxantrone or docetaxel, the standard of care for prostate cancer (dependent on local health policies), were tested, covering a range of concentrations and ratios to calculate combination indices (CIs). Potent synergistic killing of PC3 and DU145 cells resulted under all treatment conditions (see Supplementary Fig. 1 at
Abbreviations: CI, combination index; MV and DV, simultaneous addition of Ad5 (V) and mitoxantrone (M) or docetaxel (D); M–V and D–V, drug 24 hr before virus; V–M and V–D, virus 24 hr before drug.
ppc, particles per cell.
CI values were calculated from one representative experiment (n = 3).
CI ≤ 0.9, synergy; 0.9 < CI < 1.1, additive; CI ≥ 1.1, antagonistic. Entries in bold face indicate conditions with the highest degree of synergy.
Docetaxel and mitoxantrone enhance cellular uptake of virus
Next, we investigated whether increased uptake of virus in the presence of cytotoxic drugs could play a role in synergistic cell killing. In DU145 cells, viral uptake was significantly higher (p < 0.05 compared with virus alone) after pretreatment for 24 hr with drugs, up to 4-fold with mitoxantrone and 9-fold with docetaxel (Fig. 2A). In PC3 cells, the corresponding increases were 3- and 8-fold. The increases in infectivity in both cell lines were drug dose dependent. A trend toward increased viral uptake was also detected in LNCaP cells, but the changes were not statistically significant. The higher levels of cell infectability were greatest when cells were pretreated with drugs but were also higher (2- to 3-fold; data not shown) when treated simultaneously. No changes in GFP expression were observed when drugs were added after viral infection. These data are in agreement with our previous observations in H460, DU145, PC3, and LNCaP cells when the cells were treated with taxol or cisplatin (data not shown and Cheong et al., 2008).
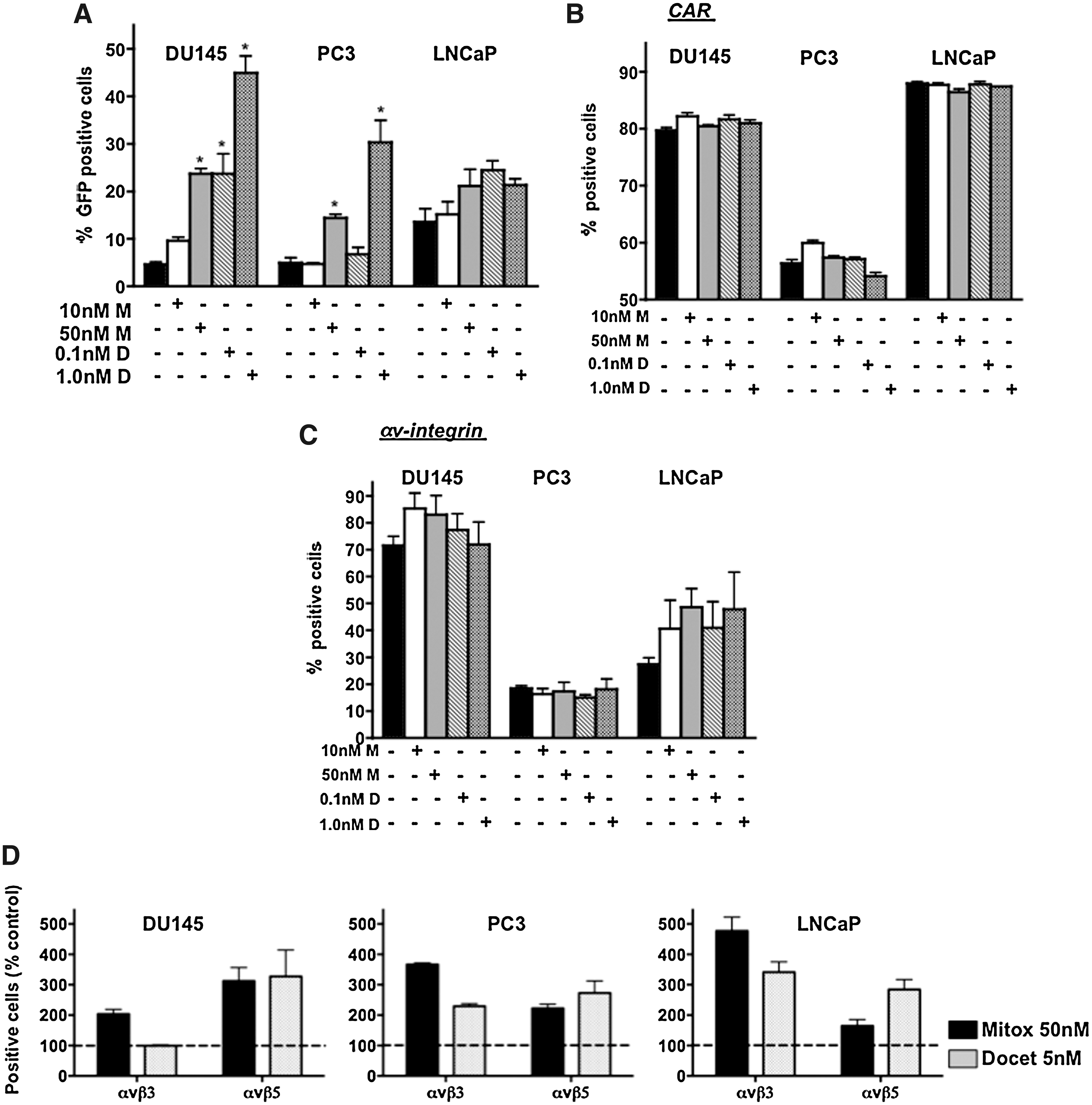
Cellular uptake of virus is stimulated by docetaxel and mitoxantrone in DU145 and PC3 cells.
Despite the potent increases in viral uptake, no significant changes in expression levels or cell membrane localization of the major viral attachment and internalization receptors, CAR and αv-integrins, were detected in response to either drug (Fig. 2B and C). However, trends were observed toward higher levels of αv-integrin in DU145 and LNCaP cells and of CAR in PC3 cells. The major αv-integrin subunits that are involved in viral internalization, β3 and β5, showed higher cell surface localization in response to the drugs in all three cell lines (Fig. 2D). The increases were greatest for the β3 subunit in PC3 and LNCaP cells and to a lesser degree for the β5 subunit. In agreement with the lower infectivity of PC3 cells, CAR and αv-integrins were also lowest in these cells.
Viral replication is reduced in the presence of mitoxantrone and docetaxel
Despite the drug-induced increases in viral uptake, replication was attenuated up to 48 hr postinfection (Fig. 3A and B). The reduction was dose dependent in DU145 and PC3 cells whereas in LNCaP cells only the highest dose of mitoxantrone decreased replication and docetaxel had no effect. Although total replication was significantly lower with drug exposure the replication rate paralleled that in cells infected with virus alone, as shown for DU145 (Fig. 3C). The decrease in virus production was independent of the order of addition in all cell lines (data not shown) and was not due to premature cell death; all treatment conditions caused <20% cell death after 24–72 hr in all cell lines tested whereas the decreases in replication were 10- to 100-fold.
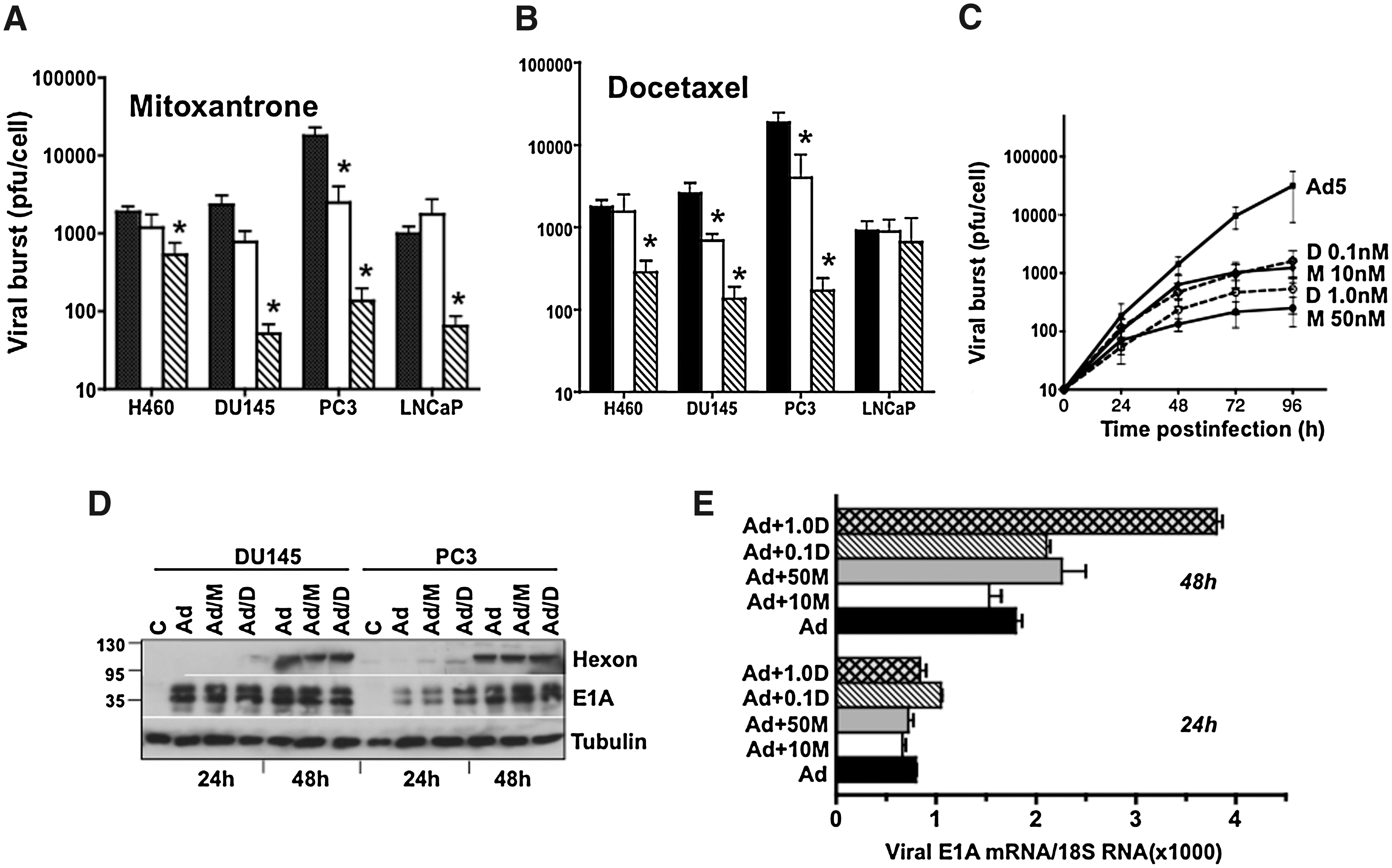
Mitoxantrone and docetaxel reduce viral replication but not gene expression in prostate cancer cell lines. DU145, PC3, and LNCaP cells were treated with cytotoxic drugs added 24 hr before infection with Ad5 at 100 ppc, and cell lysates were collected 48 hr postinfection.
Viral gene expression correlates with increased viral uptake in the presence of drugs
To investigate factors other than viral replication as the cause of synergistic cell killing we explored whether higher expression levels of early viral genes could contribute to the response. It was previously established that E1A expression sensitized cancer cells to cytotoxic factors even in the absence of viral replication (Ueno et al., 2000; Zhou et al., 2005; Cheong et al., 2008). In DU145 cells all conditions resulted in maximal E1A expression after 24 hr and no significant changes could be detected even though earlier expression of hexon was observed in cells treated with docetaxel (Fig. 3D). In PC3 cells, a slight induction of E1A after 24 hr was paralleled by earlier expression of hexon with both drugs and was verified by increased levels of E1A mRNA 48 hr after drug treatment (Fig. 3E). We conclude that E1A transcription was more potently activated in the presence of both mitoxantrone and docetaxel as a reflection of increased uptake of virus. In addition, neither drug inhibited late viral gene expression under these conditions, despite the decreases in replication as previously observed in murine cell lines not supporting viral replication (Cheong et al., 2008).
Efficacy of replication-selective dl1520 and dl922–947 mutants is strongly enhanced in combination with mitoxantrone or docetaxel
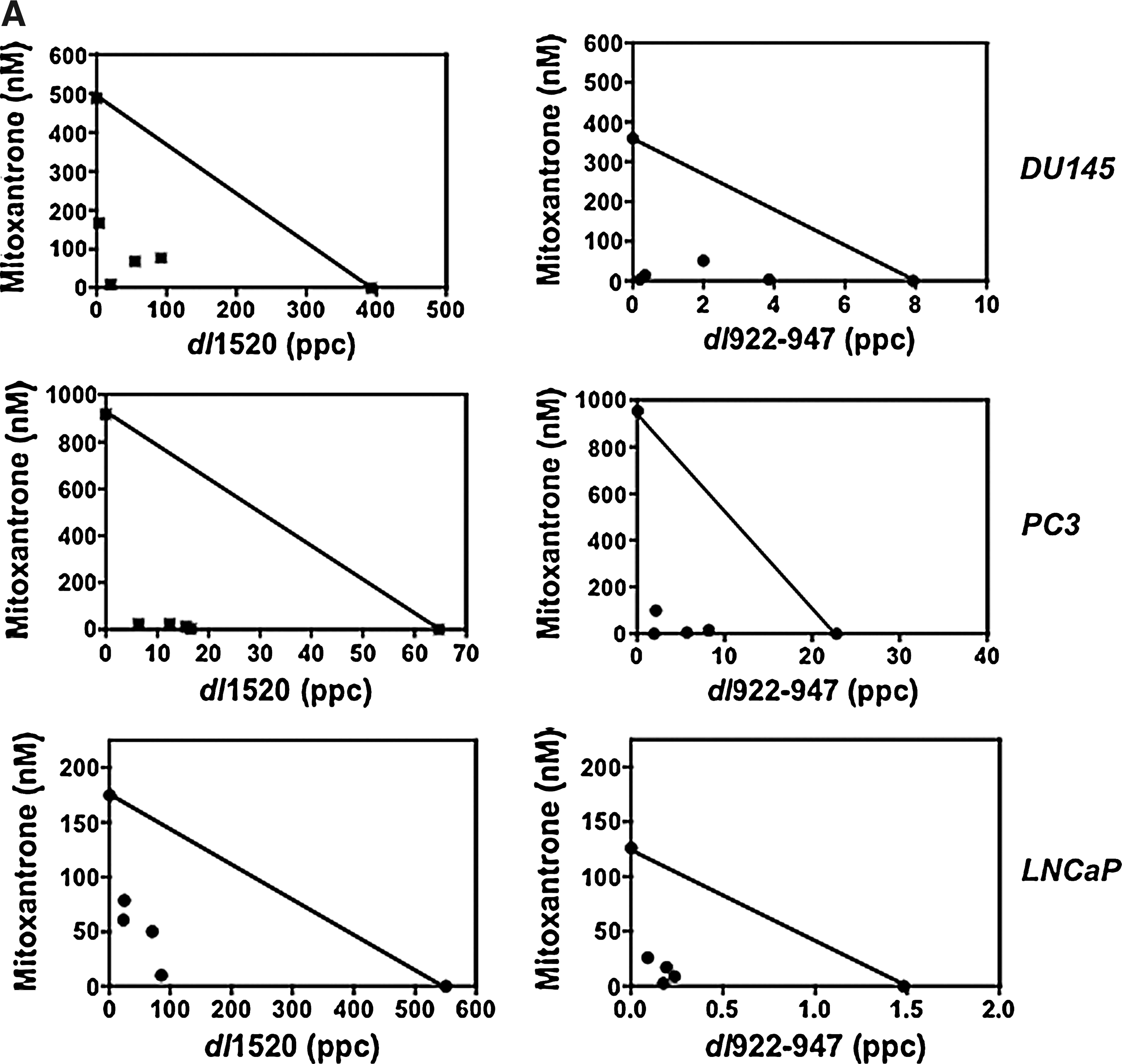
To explore whether efficacy could also be improved for the attenuated mutant dl1520 (ΔE1B55) or the more potent viral mutant dl922–947 (ΔCR2), CI values were determined for combinations with drugs (Table 2). In all cell lines dl922–947 was at least as potent as Ad5, and dl1520 was significantly less efficacious. LNCaP cells, expressing functional p53, were highly resistant to dl1520 with EC50 values (458 ± 52 ppc) more than 500-fold higher than for Ad5. In the p53-negative PC3 and DU145 cells, the corresponding values were 34.9 ± 7.1 and 180 ± 48 ppc, only 3- and 50-fold higher, respectively. Interestingly, in LNCaP cells both mutants acted synergistically with mitoxantrone or docetaxel, in contrast to the wild-type virus (Table 2 and Fig. 4A). Enhanced cell killing with dl1520 also occurred in PC3 cells, with both drugs resulting in CI values lower than those for Ad5 and dl922–947 (Table 2 and Fig. 4A). In DU145 cells, death was synergistic with both wild-type and mutants, with a trend toward less potent cell killing when dl922–947 was combined with docetaxel. The greatest potential for decreased drug concentrations was in the DU145 cells, where suboptimal concentrations caused the same degree of cell killing as 1000- to 3600-fold higher doses without virus, with trends toward better dose reductions for Ad5 or dl922–947 with mitoxantrone and dl1520 with docetaxel. Under these conditions viral mutants were also efficacious at lower doses than when given alone. Even in the more insensitive PC3 cells, drug and virus concentrations could be reduced up to 3500 and 600 times, respectively compared with each agent alone.
Strong synergy in DU145, PC3, and LNCaP cells over a range of concentrations for mitoxantrone in combination with dl922–947 or dl1520.

CI values were calculated from EC50 values determined for each agent alone and at four constant combination ratios for each cell line. Data are from one representative experiment (n = 3). CI ≤ 0.9, synergy; 0.9 < CI < 1.1, additive; CI ≥ 1.1, antagonistic.
Dose reductions for drug or virus were determined from conditions resulting in maximal synergy from three sets of studies. The maximal observed reductions are shown. Entries in bold face indicate a higher degree of synergy with mutant compared with Ad5 in each cell line.
Abbreviations: D, docetaxel; M, mitoxantrone; also see Table 1.
Virus-induced S and G2/M phases are altered in the presence of docetaxel and mitoxantrone, respectively
Mitoxantrone and docetaxel frequently arrest cells in G2 and G2/M phases, respectively, with variations dependent on cell type (Durr et al., 1983; Mackler and Pienta, 2005). Although the low doses used in our studies did not cause cell cycle arrest, the proportion of cells in G2/M (mitoxantrone) and S (docetaxel) phases were increased in all cell lines, shown for DU145 cells 48 hr after treatment (Fig. 4B and C). Ad5 and to a lesser degree the dl922–947 mutant caused potent S-phase induction 24–72 hr after infection, whereas dl1520 induced a significantly lower S-phase peak (Fig. 4B and C, left). Virus-induced S phase was sustained, and even increased, in the presence of docetaxel at all doses, but only with the lowest dose of mitoxantrone. The increase in S-phase population with virus and docetaxel and the decrease with mitoxantrone were paralleled by decreases (docetaxel) and increases (mitoxantrone) in G2/M phase (Fig. 4B, right). With the highest dose of docetaxel a significant increase in sub-G1, from 1.0 to 16–22%, was observed, indicating a higher degree of early DNA damage. No increase in the sub-G1 fraction was detected at 48–72 hr with mitoxantrone, whereas after 72 hr an increase in polyploidy and cell cycle aberrations dominated the analysis, indicating that aberrant mitosis might be involved in the response ultimately resulting in cell death (data not shown). The most significant difference between the viral mutants was the lower proportion of cells in S phase with dl1520, which was increased by docetaxel and decreased by mitoxantrone. Although clear differences in cell cycle progression and cell death mechanisms were observed for each mutant and drug combination at these early time points (48–72 hr), potent synergistic cell killing still resulted with all combinations at later time points (6 days; Fig. 4A and Table 2).
E1A expression sensitizes DU145 cells to mitoxantrone and docetaxel
To investigate the role of E1A expression in the synergistic cell killing in the absence of viral replication, cells were transduced with retrovirus expressing E1A or a control gene (GFP) for evaluation in viability assays (Fig. 4D). E1A-expressing DU145 cells showed greatly increased sensitivity to both docetaxel and mitoxantrone, with 70–75% lower EC50 values than the corresponding nontransduced or GFP-expressing cells. These findings are in agreement with previous reports using E1A-expressing plasmids in breast and ovarian cancers (Ueno et al., 2000). E1A has also been demonstrated to induce topoisomerase II promoter activation (Zhou et al., 2005). To this end we investigated whether increased enzyme expression in cells in response to viral infection might contribute to the synergistic cell killing with mitoxantrone (Fig. 4E). Although both wild-type virus and mitoxantrone increased topoisomerase IIα levels by as soon as 24–48 hr, we did not observe further increases with the combination treatments.
Combination treatments cause potent tumor regression in prostate cancer xenograft models
Treatment of DU145 and PC3 tumors with viral mutants or docetaxel resulted in dose-dependent inhibition of tumor growth and prolonged time to progression (Fig. 5A and B). In the DU145 model suboptimal doses of virus and docetaxel at 1 × 109 vp and 5 or 10 mg/kg, respectively, caused only slight increases in median survival from 44 days for control treated animals to 84 days with virus and to 52 or 68 days with docetaxel alone at 5 or 10 mg/kg. However, combination treatments significantly prolonged time to progression to 103 and 168 days when virus was administered with docetaxel at 5 and 10 mg/kg, respectively (p < 0.01) (Fig. 5A). In the PC3 xenograft model, higher doses of docetaxel and viral mutants were required to achieve antitumor efficacy when given alone (Fig. 5B). Although Ad5 at 1 × 109 vp or docetaxel at 10 mg/kg did not significantly prolong time to progression, virus at 1 × 1010 vp and docetaxel at 15 mg/kg resulted in a median survival of 51 and 40 days, respectively, compared with 21 days for the control group. However, when combining the lower doses of virus and drug a significant increase in median survival to 45 days was observed (p < 0.01). These results demonstrate that by lowering the dose of virus 10-fold and the drug dose by 30% the same degree of antitumor efficacy could be achieved as with the much higher doses of virus (1 × 1010 vp) and drug (docetaxel, 15 mg/kg) (Fig. 5B). Prolonged time to tumor progression and growth inhibition was also observed with the dl1520 or dl922–947 mutant in combination with docetaxel (Fig. 5C–F). In the DU145 model viral mutants alone had poor efficacy whereas dl922–947 and to a lesser degree dl1520 in combination with docetaxel (10 mg/kg) significantly inhibited tumor growth (Fig. 5E and F). In the PC3 model, similar to wild-type virus, no significant inhibition of tumor growth was detected with the mutants alone (1 × 109 vp) whereas in combination with docetaxel, growth regression was observed 24 days after treatment (Fig. 5C and D). In conclusion, all tested combination treatments had superior antitumor efficacy compared with single-agent treatments, including those with the attenuated dl1520 virus.
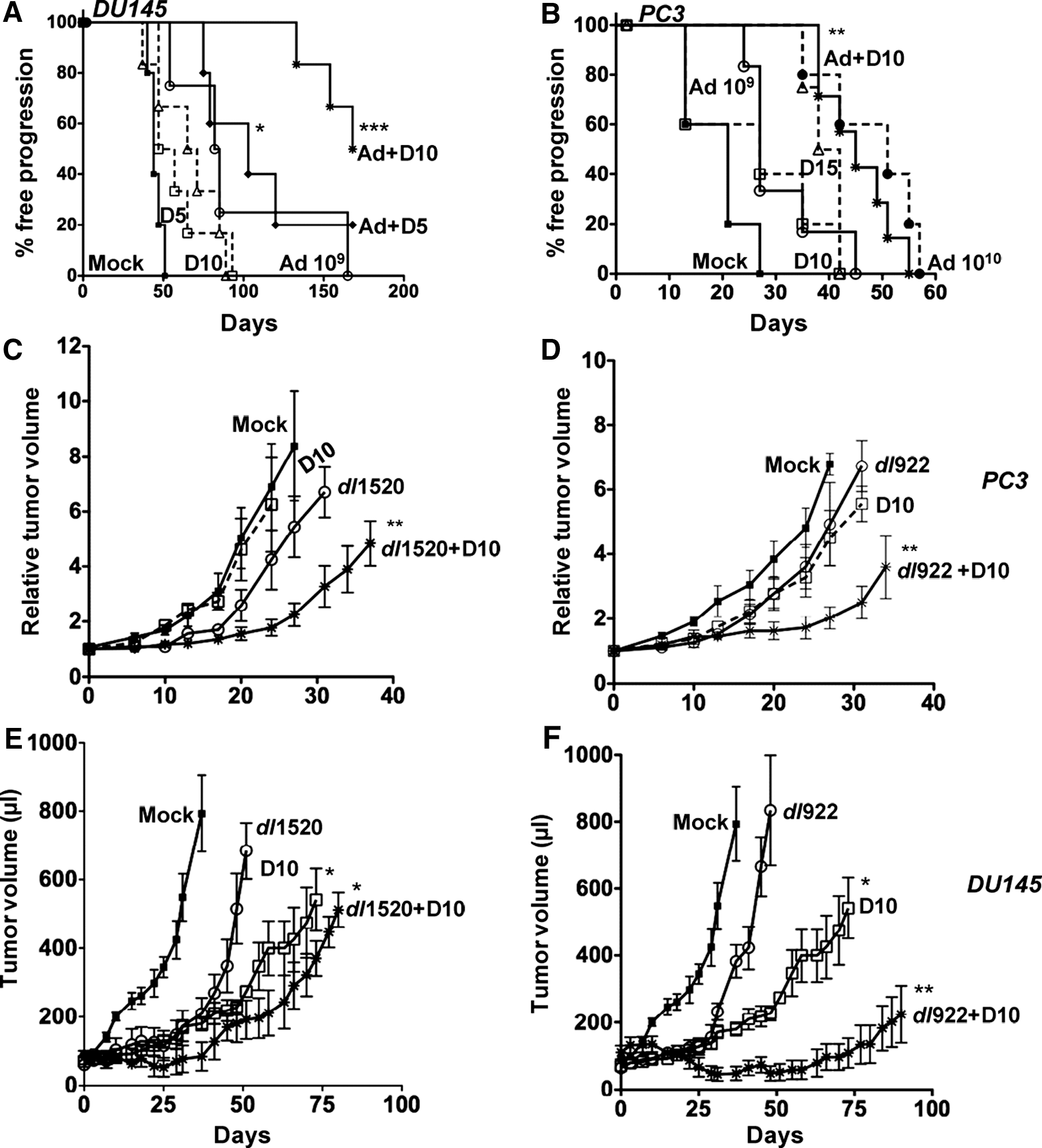
Enhanced antitumor activity in vivo by wild-type virus and the dl922–947 and dl1520 mutants in prostate tumor xenografts.
Discussion
We demonstrated that that antitumor efficacy of the attenuated E1B55K-deleted dl1520 virus, the most frequently clinically evaluated oncolytic mutant to date, could be potently enhanced when combined with chemotherapeutics in prostate cancer models. More interestingly, efficacy was also greatly improved with the more recently developed and highly potent E1ACR2-deleted mutant dl922–947 and with wild-type virus. The improved cell killing was caused by synergistic interactions of mutants targeting both deregulated pRb and p53 pathways in combination with the current standard of care (in the United States and the United Kingdom) for late-stage prostate cancers, mitoxantrone and docetaxel. The dl922–947 mutant was superior to dl1520 in all tested prostate cancer cells and in xenograft models in vivo, in agreement with numerous previous reports targeting various tumor types (Heise et al., 2000; Stolarek et al., 2004; Alonso et al., 2008; Bazan-Peregrino et al., 2008; Libertini et al., 2008). In fact, two CR2-deleted mutants (AdΔ24RGD and AdΔ24RGD-4C) are already undergoing testing in clinical trials for ovarian and brain cancers (
We found that in addition to the high levels of synergistic death in response to mitoxantrone or docetaxel, several other standard chemotherapeutics acting on different cellular factors and/or different cell death programs also synergized with adenovirus. The therapeutic toxicity of mitoxantrone occurs mainly through stabilization of cleavable complexes between topoisomerase II and DNA during replication, through direct enzyme inhibition and DNA intercalation (Tanaka et al., 2007; Zhao et al., 2008). Although the resulting inter/intrastrand cross-links and DNA breaks are not cell cycle phase specific, mitoxantrone was reported to more efficiently induce apoptosis in cells actively replicating DNA in S phase (Durr et al., 1983). Consequently, adenovirus is likely to promote mitoxantrone-induced cell death by stimulating S-phase entry. Like mitoxantrone, etoposide and doxorubicin are topoisomerase II inhibitors but with lower potency (Azarova et al., 2007). The therapeutic efficacy of docetaxel and paclitaxel occurs through stabilization of microtubules (MTs), causing mitotic aberrations and cell death (Mackler and Pienta, 2005; Hernandez-Vargas et al., 2007). Docetaxel is more potent than paclitaxel, with higher tubulin affinity; it binds to centromeres and causes cell damage in the S, G2, and M phases, depending on the dose. In addition to drug-specific mechanisms, all tested drugs induce alterations in cell cycle progression, oxidative stress, and DNA damage ultimately leading to apoptotic death. In contrast, adenoviruses prevent apoptosis during early stages of infection and induce cell lysis through nonapoptotic events once progeny virions are assembled (Abou El Hassan et al., 2004; Ito et al., 2006; Baird et al., 2008). We found that despite attenuated viral replication in the presence of drugs, virus-induced S phase was not prevented by suboptimal doses of mitoxantrone or docetaxel. Consequently, the increased S-phase activity could further promote DNA damage during synthesis in response to drugs. These findings suggest that expression of early viral genes (mainly E1A) that promote DNA synthesis proceeds even in the presence of cytotoxic drugs, thereby causing higher levels of drug-induced apoptosis. Although viral replication might also contribute to the synergistic responses, expression of the early apoptotic E1A proteins (Fig. 4D) appears to be an absolute requirement by acting on both cell cycle and cell death pathways as previously reported (e.g., Ueno et al., 2000; White, 2006; Cheong et al., 2008).
The reduced viral replication was likely due to drug-dependent inhibition of specific cell cycle-regulatory factors. For example, although cdc25A/cyclin E/cdk2 expression and activation are required for viral replication, these factors can be inhibited in response to cytotoxic stress (Leitner et al., 2009). As expected, we found that cyclin E expression was not significantly increased in cells treated with virus and drugs at early time points (<48 hr; data not shown). However, viral replication might resume at higher levels once the drugs have decayed >72 hr after treatment), similar to previous findings with gemcitabine (Raki et al., 2005; Leitner et al., 2009). More extensive studies would be necessary to determine the specific mechanisms such as additional effects on gene expression, translation, or viral assembly preventing efficient viral production. In this paper we conclude that viral replication did not appear to be proportional to the synergistic response.
On the other hand, viral uptake was increased by low concentrations of mitoxantrone and docetaxel. Increased viral uptake has been associated with increased expression of CAR, a cellular protein participating in the formation of tight junction complexes, also reported to be upregulated by inhibition of key cellular pathways (Anders et al., 2003; Lacher et al., 2006). We did not detect significant changes in cell surface expression levels of either CAR or αv-integrin for viral attachment and internalization, respectively. However, expression of the αv-integrin subunits β3 and β5, which are essential for viral uptake, were increased in response to both drugs in all three cell lines. In addition, a trend toward higher expression levels and membrane localization of at least one receptor type in each cell line was observed. Interestingly, no clear correlation between receptor levels and viral uptake was detected; LNCaP cells, with high levels of CAR and average integrin expression, were more highly infectable than were DU145 cells, which had higher expression levels of both receptors. However, the poorly infectable PC3 cells did express low levels of both CAR and all integrin subunits. Although CAR plays a role in facilitating virus binding to nonpolarized cells in culture, it is well documented that the receptor is not required for adenoviral infection in vivo whereas the presence of αvβ5- and/or αvβ3-integrin complexes appears to be essential for viral internalization (Arnberg, 2009). It is possible that additional cellular factors not evaluated in our study, such as unidentified viral receptors or cofactors, could also play a role as previously suggested (Johansson et al., 2007; Arnberg, 2009). Other causes for the increased viral uptake include drug-induced changes in cytoskeletal proteins facilitating viral internalization and/or transport and gene expression. It was previously reported that low concentrations of drugs that suppress microtubule dynamics, such as taxanes, greatly enhanced microtubule-dependent adenovirus transport to the nucleus (Giannakakou et al., 2002). Cellular uptake of adenovirus has also been reported to prevent microtubule catastrophe and to promote its own nuclear trafficking through integrin-related activation of phosphatidylinositol 3-kinase (PI3K) and Rac1, which initiates membrane ruffling and microtubule net growth (Warren et al., 2006). Also, inhibition of the cellular microtubule-destabilizing protein stathmin through ribosome delivery in LNCaP cells synergistically inhibited cell proliferation in combination with taxanes (Mistry and Atweh, 2006). These reports, together with our findings, indicate that taxane-induced microtubule stabilization might be further enhanced by the virus-induced uptake by acting on the same structural cellular networks.
In agreement with the increased infectivity we observed higher levels of early viral gene expression (E1A) in cells pretreated with chemotherapeutic drugs. A smaller increase was observed with simultaneous treatments whereas addition of drugs after infection had no significant effects (data not shown). These data indicate that enhanced viral cell attachment/entry and/or nuclear transport rather than drug-induced promoter effects caused the increased infectivity followed by higher E1A expression.
E1A proteins can act directly on death signaling pathways and increase cell sensitivity to cytotoxic drugs such as paclitaxel, doxorubicin (Adriamycin), etoposide, and gemcitabine in carcinoma cells as previously demonstrated (Lee et al., 2003; Liao et al., 2004; Zhou et al., 2005). Wild-type E1A was also reported to increase the expression of topoisomerase II (Zhou et al., 2005). Here we demonstrate that in addition to mitoxantrone-induced stabilization of topoisomerase IIα, wild-type Ad5 could also increase the enzyme levels (Fig. 4E). It is possible that E1A promoter activation could sensitize the cells to drug-induced DNA cleavage, although we could not detect further changes in enzyme expression levels with the combination treatment. Even though the dl1520 virus did not induce S phase as potently as Ad5 under our test conditions, a high degree of synergy in combination with either drug was observed, supporting the possibility that E1A expression alone could sensitize the cells. Absence of the antiapoptotic E1B55K in dl1520 likely contributed to the greater synergistic effects with this mutant through enhancement of drug-induced apoptosis, as previously reported for viruses deleted in the antiapoptotic E1B19K gene (Ueno et al., 2000; White, 2006; Leitner et al., 2009). In addition, docetaxel increased the S-phase population and likely facilitated the expression of early viral genes. Although no significant differences in cell death with the viral mutants were observed at these early time points (24–72 hr), specific cell cycle aberrations were obvious for each mutant and each drug, likely causing the differences in synergistic cell death after longer treatment times (5–6 days). The higher degree of cell death in response to both dl922–947 alone and in combination with drugs makes this potent virus a good candidate for clinical evaluation. In addition, in LNCaP cells, in contrast to other tested prostate cancer cell lines, the magnitude of the synergistic response was also greater with dl922–947 than with dl1520 or wild type.
In conclusion, all tested combination treatments had superior antitumor efficacy compared with single-agent treatments, both in cultured cells and in xenografts in vivo. Tumor growth was greatly reduced and time to progression prolonged even in the highly drug- and virus-resistant PC3 model, although this PC3 clone had higher sensitivity to virus than the parental cells and showed a higher degree of synergy (Öberg et al., 2010). At doses of virus and docetaxel that did not result in significant efficacy alone, median survival was increased by the combinations to levels similar to those of drugs administered at a higher dose. These results indicate that chemotherapeutics could be given at lower doses in the clinic if coadministered with replication-selective mutants (e.g., CR2 deleted) to reduce unwanted side effects and toxicity. Additional studies to optimize doses, effects in normal tissue, scheduling, and delivery could further improve the antitumor efficacy of ΔCR2 mutants as well as determine the effects of an intact immune system that will be important in the clinical setting.
Footnotes
Acknowledgments
This work was supported by grants from the Barts and the London Charity and Cancer Research UK (C633-A6253/A6251 programme grant). The authors thank Gary Martin and colleagues at Clare Hall (Cancer Research UK) for excellent experimental assistance and Virginie Adam for insightful discussions.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.