
Abstract
Duchenne muscular dystrophy (DMD) is an X-linked genetic disease characterized by the absence of dystrophin (427 kDa). An approach to eventually restore this protein in patients with DMD is to introduce into their muscles a plasmid encoding dystrophin cDNA. Because the phenotype of the dystrophic dog is closer to the human phenotype than is the mdx mouse phenotype, we have studied the electrotransfer of a plasmid carrying the full-length dog dystrophin (FLDYSdog) in dystrophic dog muscle. To achieve this nonviral delivery, the FLDYSdog cDNA was cloned in two plasmids containing either a cytomegalovirus or a muscle creatine kinase promoter. In both cases, our results showed that the electrotransfer of these large plasmids (∼17 kb) into mouse muscle allowed FLDYSdog expression in the treated muscle. The electrotransfer of pCMV.FLDYSdog in a dystrophic dog muscle also led to the expression of dystrophin. In conclusion, introduction of the full-length dog dystrophin cDNA by electrotransfer into dystrophic dog muscle is a potential approach to restore dystrophin in patients with DMD. However, the electrotransfer procedure should be improved before applying it to humans.
Introduction
There are two genes that could be used to stop the progression of DMD. The first encodes the full-length dystrophin and the second encodes a truncated version called micro- or minidystrophin. The possibility of using a truncated gene to treat DMD originated from the observation of patients with mild Becker muscular dystrophy (Beggs et al., 1991). Indeed, in Becker patients there are different mutations in the dystrophin gene leading to partially functional dystrophin. However, because full-length dystrophin expression would provide better stability to myofibers than minidystrophin or microdystrophin (Harper et al., 2002; Banks et al., 2009), we have studied the introduction of the full-length dystrophin cDNA into muscle. Effective results have previously been obtained with the delivery of full-length dystrophin by an adenoviral vector into neonatal mdx mouse muscle (Gilbert et al., 2001). However, the same injection of this virus into adult mdx mouse muscle is less effective. Moreover, adenovirus injection has adverse effects, such as an immune response against the viral capsid and toxicity after the dissemination of the virus throughout the body (Douglas, 2007).
An approach to avoid this problem is to directly inject a cDNA encoding the gene of interest into muscle (Wolff et al., 1990). A clinical trial performed on nine patients with DMD has shown the expression of human dystrophin in up to 6% of the myofibers after intramuscular injection of a plasmid encoding human full-length dystrophin (Romero et al., 2004). This technique is safe, fast, and easy to use, but needs to be substantially improved. This could be done by using an electrotransfer technique to increase the incorporation of the plasmid into more myofibers (Aihara and Miyazaki, 1998; Mir et al., 1999). Indeed, it is difficult to introduce into muscle fibers large plasmids such as those carrying dystrophin cDNA. Since the first clinical trial in 1991 (Mir et al., 1991), this technique involving gene therapy and electrotransfer has proven its safety and efficiency by introducing plasmids into several tissues such as skin, melanoma, and others (Favard et al., 2007; Mir, 2008; Bodles-Brakhop et al., 2009). With this approach, human and mouse full-length dystrophin cDNAs were introduced into mdx mouse muscle (Chapdelaine et al., 2000; Vilquin et al., 2001; Wells et al., 2008) but not yet into large animals.
Because there is no information about the use of the dog full-length dystrophin and because the dystrophic dog is the best model in which to study DMD pathology, a dog full-length dystrophin (FLDYSdog) cDNA was cloned into a plasmid to allow its electrotransfer into muscle.
Materials and Methods
Animals
Rag –/– mdx mice came from our own colony and C57BL/10SnJ mice were from Jackson Laboratory (Bar Harbor, ME). The normal beagle dog was provided by Marshall BioResources (North Rose, NY). The dystrophic dog was born and housed at the Muscular Dystrophy Research Center (Associação dos Amigos Portadores de Distrofia Muscular [AADM]/Universidade da Associação de Ensino de Ribeirão Preto [UNAERP], Ribeirão Preto, Brazil) in accordance with the guidelines set by the Institutional Laboratory Animal Care Committee of the University of Ribeirão Preto; it arrived in our laboratory when it was 2 years old and was housed at Université Laval (Quebec, QC, Canada) animal service. The Brazilian colony was founded from one carrier female donated by J.N. Kornegay (Université Laval). Genotype analyses of the wild-type dogs and of the dystrophic dog in pedigrees segregating golden retriever muscular dystrophy were previously described (Bartlett et al., 1996). All the experiments made on these animals were approved by the Animal Protection Committee of Université Laval.
Construction of full-length dog dystrophin plasmid
All the primers (Table 1) designed for the cloning of full-length dystrophin were deduced from the Canis lupus familiaris dystrophin mRNA (accession number AF070485.1) submitted by the laboratory of S.D. Wilton in 1998. Dog RNA was extracted with TRIzol (Invitrogen, Carlsbad, CA) from a biopsy of the brachialis anterior muscle of a beagle and afterward reverse transcribed with SuperScript III (Invitrogen), using reverse-specific primers named GS1, GS2, and GS3 (see Fig. 1A). The cDNA strands were transcribed with these specific primers covering the dystrophin mRNA (11 kb) entirely. The cloning procedure for full-length dystrophin is summarized in Fig. 1A, showing the amplification of four amplicons named A, B, C, and D resulting from a nested PCR strategy.
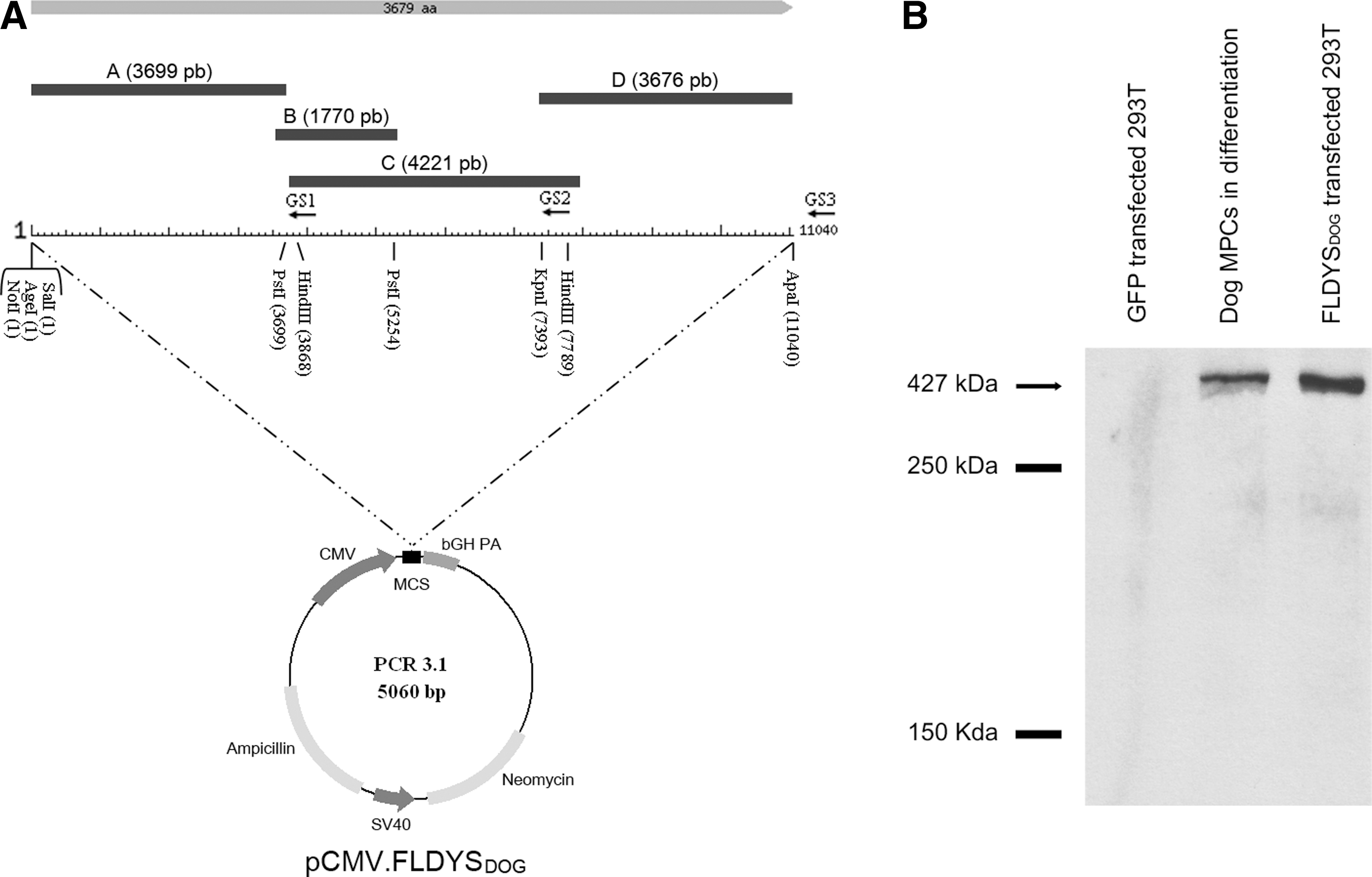
Full-length dog dystrophin plasmid.
These primers were designed from the predicted sequence of dog dystrophin.
The first round of PCRs was done with 2 μl of reverse transcription (RT) reaction for each amplicon, different sets of primers (noted 1s and 1r; see Table 1) used at 0.5 μM each, and Phusion DNA polymerase (New England BioLabs, Pickering, ON, Canada) in Phusion HF buffer as recommended by the manufacturer. The thermal cycle program was at 98°C for 30 sec for the initial denaturation step following by 25 cycles (for fragment B) or 30 cycles (for fragments A, C, and D) of denaturation (98°C, 10 sec), an annealing step (55°C, 30 sec), and an extension at 72°C for 1 min (B) or for 1 min 30 sec (A, C, and D). The final extension was at 72°C for 10 min and kept at 4°C. The second round of PCR was performed with 2 μl of the first round of PCR with primers annotated 2s and 2r (Table 1), and 35 cycles of PCR were performed as described for the first-round PCR except for the last 30 annealing steps, which were done at 60°C. At the end of the extension step, Taq DNA polymerase (New England BioLabs) was added to the PCR medium for a final extension for 10 min at 72°C.
The resulting amplicons A, B, C, and D were cloned in TA Cloning vector (pDrive; Qiagen, Mississauga, ON, Canada) after gel extraction of the agarose bands as suggested by the manufacturer. The sequential cloning of different amplicons was done in pCR3.1 EGFP vector (Chapdelaine et al., 2000), from which the gene encoding enhanced green fluorescent protein (EGFP) was removed by cutting the vector with AgeI and SalI followed by Klenow treatment and religation, regenerating a single SalI site in the cloning site. First, fragment A (coming from pDrive containing this fragment; pDrive-A) was digested with SalI and PstI and ligated with T4 DNA ligase (New England BioLabs) in pCR3.1, cut with the same restriction enzymes, and transformed chemically in competent DH5α bacteria. The resulting pCR3.1-A was then cut with PstI and ligated to fragment B obtained from the digestion of pDrive-B with PstI, the result of the ligation making pCR3.1-AB. To find the clones with the correct orientation of PstI-fragment B, an analysis with various restriction enzymes and sequencing was necessary to confirm its desired orientation within the vector. In a third cloning step, pCR3.1-AB was digested with HindIII (present only in PstI-fragment B and not in the vector) and the fragment C present in pDrive-C was also digested with HindIII. The ligation between the pCR3.1-AB vector and the HindIII-fragment C was performed and transformed in DH5α bacteria, making pCR3.1-ABC bearing two HindIII sites. As previously described, the orientation of the obtained HindIII-fragment C was necessary to establish its desired orientation in the vector construct. The last step consisted of the ligation of pCR3.1-ABC digested with KpnI and pDrive-D digested with KpnI and ApaI, making the final construct pCR3.1-ABCD (FLDYSdog). Afterward, it was transformed in competent SURE cells (Stratagene/Agilent, La Jolla, CA). FLDYSdog was driven by the cytomegalovirus (CMV) promoter present initially in the pCR3.1 vector containing also the bovine growth hormone (bGH) polyadenylation site at the 3′ end as shown in Fig. 1A. This vector was named pCMV.FLDYSdog.
A second FLDYSdog construct was made simply by replacing the CMV promoter with the muscle creatine kinase (MCK) promoter (Jaynes et al., 1988) to obtain expression only in myotubes or myofibers. This vector was named pMCK.FLDYSdog.
Sequence analysis
Twenty primers, described in Table 2, were necessary to entirely sequence our 11-kb transgene. The obtained sequence is available at GenBank (accession number GU137540). The Lalign program was used to compare two sequence alignments, using the algorithm of Myers and Miller (1988).
CMV, cytomegalovirus; FLDYSdog, full-length dog dystrophin; poly (A), polyadenylation signal.
These primers have been used directly in the sequencing of FLDYSdog plasmid.
Cell culture
293T cells were cultured in Dulbecco's modified Eagle's medium with high glucose (DMEM-HG; GIBCO, Burlington, ON, Canada) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin–streptomycin (GIBCO). Normal dog muscle precursor cells (MPCs) came from muscle biopsies of a 3-month-old dog. These cells were proliferated in MCBD120 medium (HyClone, Mississauga, ON, Canada) supplemented with 15% fetal bovine serum (GIBCO), 1% penicillin–streptomycin (GIBCO), basic fibroblast growth factor (bFGF, 10 μg/liter; Feldan, Saint-Laurent, QC, Canada), dexamethasone (0.4 mg/liter; Sigma-Aldrich, Oakville, ON, Canada), and insulin (5 mg/liter; Sigma-Aldrich). To cause their differentiation into myotubes, cells were cultured in differentiation medium (DMEM [GIBCO] supplemented with 2% fetal bovine serum [Invitrogen]). All the cells were maintained at 37°C under 5% CO2.
Plasmid transfection
Lipofectamine 2000 (Invitrogen) was used as a transfection reagent to obtain the expression of pCMV.FLDYSdog in 293T cells. The transfection protocol was that recommended by the manufacturer. Briefly, 10 μg of plasmid in DMEM-HG was mixed with 10 μl of Lipofectamine 2000 in 40 μl of Opti-MEM and added to 293T cells. For the transfection control, the same amount of pCMV.GFP was transfected into other 293T cells.
Western blot
Proteins from differentiated normal dog MPCs (3 days in differentiation medium) and from transfected 293T cells (2 days after the transfection) were extracted by a methanol–chloroform technique to perform Western blots. Twenty-five micrograms of protein was loaded per well. Dog dystrophin was detected with a mouse anti-dystrophin antibody (MANHINGE1A, diluted 1:100; Centre for Inherited Neuromuscular Diseases [CIND], Oswestry, UK).
Electrotransfer
A single transcutaneous longitudinal injection of 40 μg of pCMV.GFP or FLDYSdog plasmid in a final volume of 40 μl of Tyrode's buffer 1× (Sigma-Aldrich) was done in the tibialis anterior (TA) of Rag –/– mdx mice and electrode electrolyte cream (Teca, Pleasantville, NY) was applied on the skin to favor the passage of the electric field between the two electrode plates. These plates were fitted on the skin of the mouse TA (4 to 5 mm of separation between the plates) and the voltage was delivered. The generator used in this experiment was a Grass S48 stimulator (Astro-Med, West Warwick, RI) and delivered square-wave pulses. The parameters were 12 pulses of 180 V/cm (approximately 80 to 90 V) of 25 msec duration spaced by 300 msec. The electrotransferred muscles were harvested 2 weeks after the experiment and were snap-frozen in liquid nitrogen. Serial 12-μm cryostat sections were obtained throughout the entire muscle.
For the dystrophic dog, 200 μg of pCMV.FLDYSdog was injected, in a final volume of 200 μl of Tyrode's buffer 1× (Sigma-Aldrich), into the brachialis anterior. Two close injections of 100 μl were made directly into the muscle previously exposed by cutting the skin. Afterward, two needles (27 gauge; Hamilton, Reno, NV) separated by 4 mm were implanted in the muscle 1 cm deep around the plasmid injection area and 8 electric pulses of 100 V were applied without moving the needles. The pulsations lasted 25 msec and were spaced by 300 msec. For the negative control, 200 μl of Tyrode's buffer 1× (Sigma-Aldrich) was injected into the muscle. Suture points were placed in the muscle to distinguish the areas of electrotransfer. The regions that were electrotransferred were biopsied 2 weeks later and they were prepared as done for mouse muscle, except that the cryostat sections were 18 μm thick.
Histological analysis
Hematoxylin–eosin staining was used for morphological analysis of electrotransferred muscle. Immunohistofluorescence to detect dystrophin was performed with a mouse anti-dystrophin antibody, MANDYS104 (diluted 1:10; CIND), which reacts to dog but not mouse dystrophin. To detect the presence of dog CD8+ cells, a rat anti-CD8 antibody (diluted 1:80; AbD Serotec, Oxford, UK) was used. α-Sarcoglycan was revealed with a mouse anti-α-sarcoglycan antibody (NCL-a-SARC, 1:100; Novocastra, Newcastle, UK). Administration of these primary antibodies was followed by incubation with a biotinylated anti-mouse or anti-rat antibody (diluted 1:300; Dako, Mississauga, ON, Canada) and with streptavidin–Cy3 (diluted 1:300; Sigma-Aldrich). Afterward, the sections were mounted in phosphate-buffered saline (PBS)–glycerol (1:1). GFP and histological analyses were performed under fluorescence with an Axiophot microscope (Zeiss, Oberkochen, Germany).
Results
Full-length dog dystrophin plasmid
RNA was extracted from a muscle biopsy of a beagle dog and three cDNA strands covering the full-length dystrophin transcript were transcribed, using three specific reverse primers (GS1, GS2, and GS3) as shown in Fig. 1A. The use of a nested PCR strategy (Table 1 and Fig. 1A), as described in Materials and Methods, produced four amplicons (A, B, C, and D) containing at their extremities different restriction enzyme sites. These amplicons were cloned consecutively in four steps into the pCR3.1 vector according to their restriction sites, resulting in dog FLDYSdog. This transgene was driven by a CMV promoter present in the pCR3.1 vector. The resulting plasmid was named pCMV.FLDYSdog (Fig. 1A). The complete sequencing of the FLDYSdog plasmid was done with 20 primers (Table 2), which were necessary to cover the sequence of the transgene.
The obtained nucleotide and amino acid sequences (accession number GU137540) were then compared with the sequence submitted previously by the Wilton team (accession number AF070485.1). Our sequence had three nucleotides less than the Wilton sequence—11,040 versus 11,043 bp—and there were also 5 gaps and 28 nucleotide substitutions between the two sequences (Supplementary Table S1; supplementary data are available online at
In vitro expression of dog dystrophin
To verify the expression of FLDYSdog carried by the pCR3.1 vector, the plasmid was transfected into 293T cells using Lipofectamine 2000. Two days later, proteins were extracted and Western blotting was done. Detection of dog dystrophin was performed with the use of a monoclonal antibody (MANHINGE1A), confirming that our plasmid FLDYSdog was able to express the full-length dystrophin protein at the expected size (∼427 kDa) (Fig. 1B).
Electrotransfer into Rag–/– mdx mouse muscle
After this positive in vitro result, our plasmid was also transduced into mouse muscle by the electrotransfer technique to allow better penetration of plasmids previously injected into muscle fibers via an electric field. To set the best effective parameters for the electrotransfer, 40 μg of the plasmid encoding green fluorescent protein (pCMV.GFP) was electrotransferred into the TA of immunodeficient (Rag –/– ) mdx mice (n = 6). Two weeks later, the TAs were harvested and GFP was detected in cryostat sections (Fig. 2a). This method was thus efficient in introducing plasmid into muscle. Forty micrograms of pCMV.FLDYSdog was thus electrotransferred into six TAs of Rag –/– mdx mice (n = 6) and these TAs were analyzed 2 weeks later by immunohistofluorescence, using an antibody recognizing dystrophin (MANDYS104). Muscle electrotransferred with GFP plasmid was used as a negative control for dystrophin expression. As expected, no myofiber expressing dystrophin was detected in GFP-electrotransferred muscle (Fig. 2b), whereas 50 to 70 dystrophin-positive myofibers were detected per cryosection in muscle electrotransferred with pCMV.FLDYSdog (Fig. 2c and d).
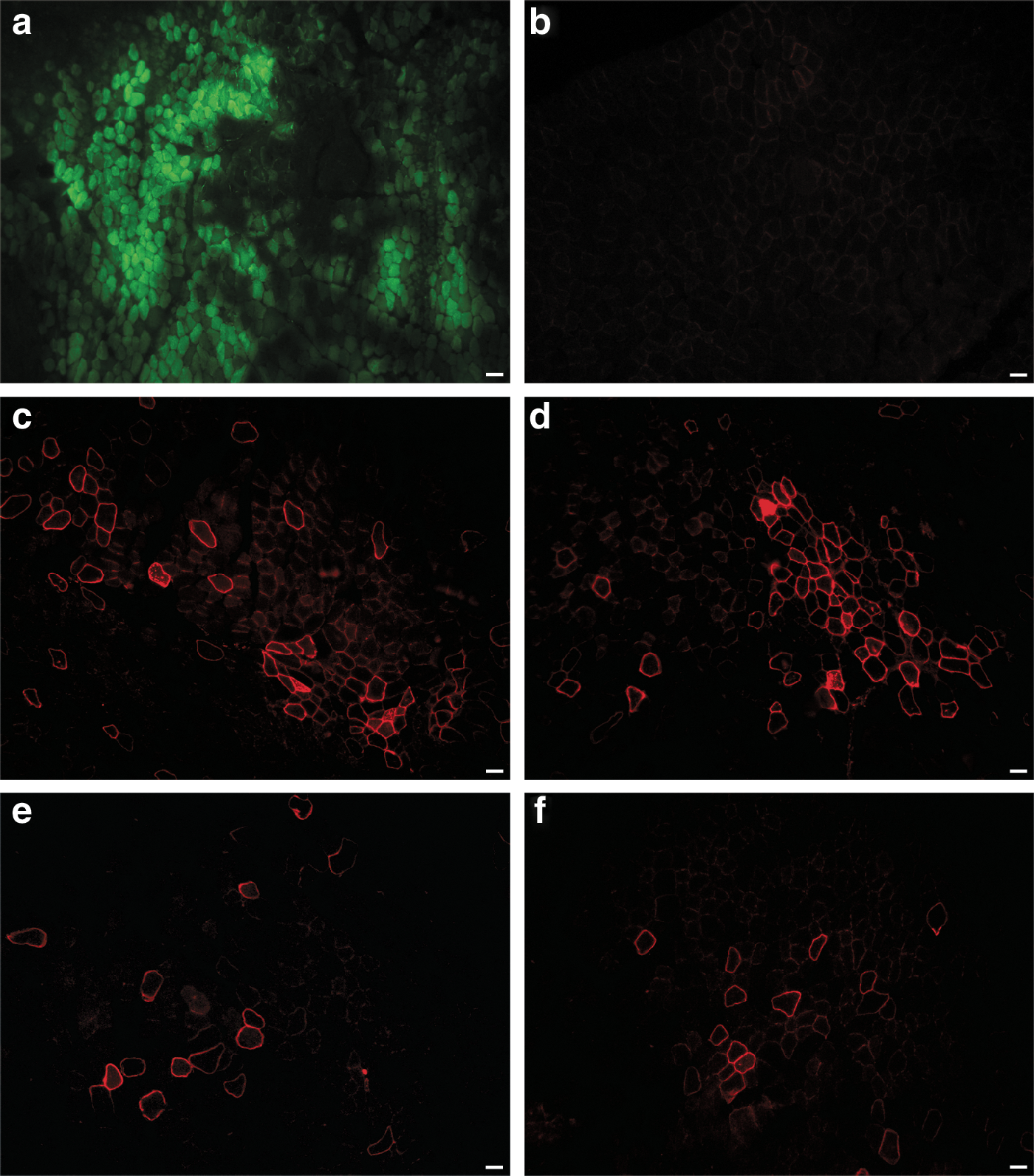
Electrotransfer in Rag
–/–
mdx mouse muscles.
FLDYSdog expression was also tested with the muscle-specific MCK promoter. The plasmid was obtained by replacing the CMV promoter with the MCK promoter, giving pMCK.FLDYSdog. Forty micrograms of this new plasmid was electrotransferred into the TAs of Rag –/– mdx mice (n = 4) and, 2 weeks later, the TAs were harvested. Immunohistofluorescence with the MANDYS104 antibody was used to detect myofibers expressing dystrophin (Fig. 2e and f). Between 25 and 40 myofibers expressing this transgene per cryosection were observed after this electrotransfer. The number of dystrophin-positive myofibers was thus lower with the MCK promoter than with the CMV promoter.
Restoration of α-sarcoglycan in electrotransferred mouse muscle
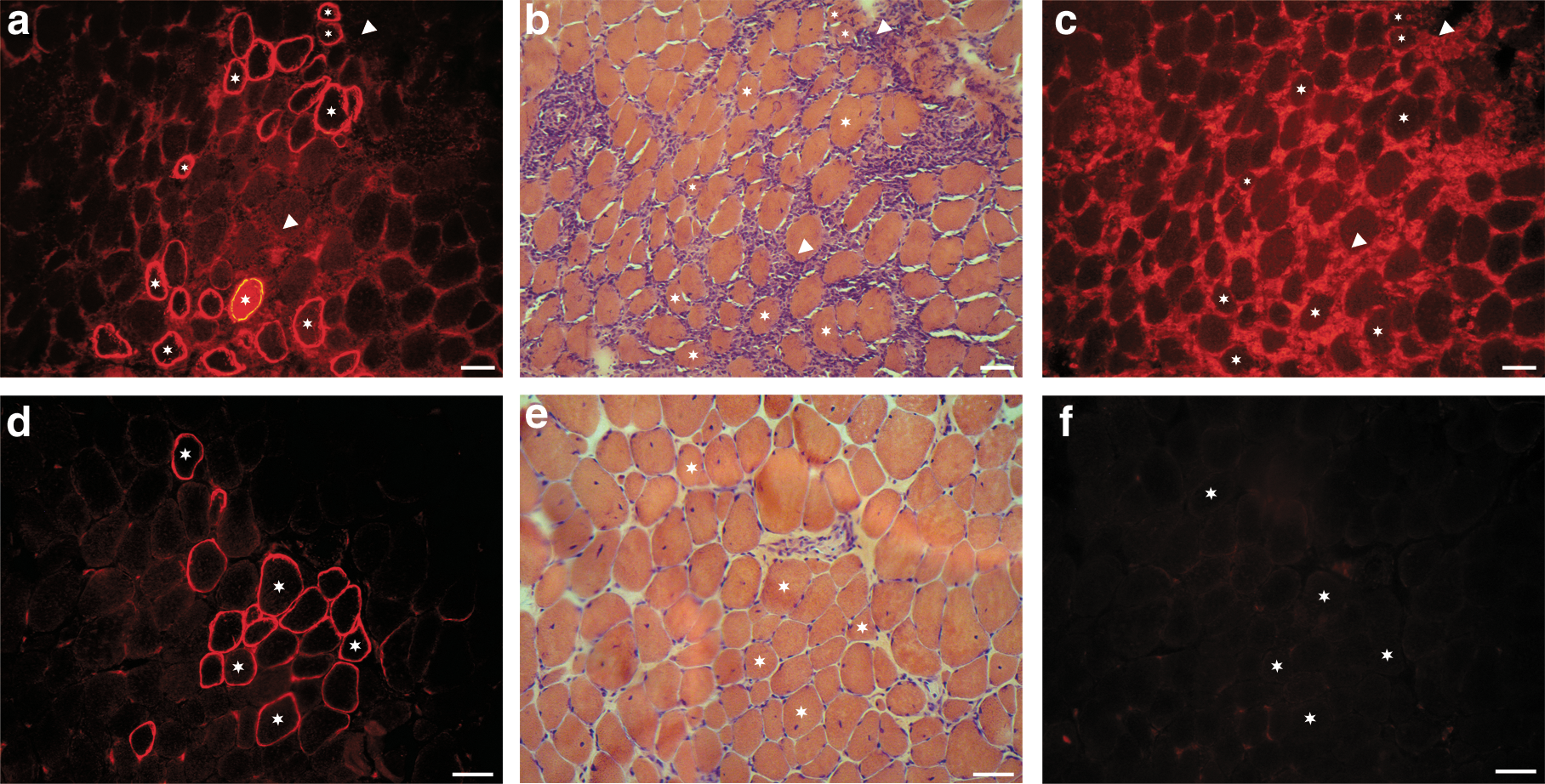
Dystrophin is normally associated with other proteins to form a complex, the dystrophin-associated glycoprotein complex (DGC), and the absence of dystrophin leads to instability of this complex (Ervasti et al., 1990). To confirm whether the expression of FLDYSdog was associated with restoration of the DGC, immunohistofluorescence analysis of α-sarcoglycan protein, one of its components, was performed on mouse muscle electrotransferred with pCMV.FLDYSdog. Rag –/– mdx mouse muscle and nondystrophic mouse (C57BL/10SnJ) muscle were, respectively, used as negative (Fig. 3a) and positive (Fig. 3b) controls for α-sarcoglycan expression. As shown in Fig. 3 (bottom), FLDYSdog (Fig. 3c) was colocalized with α-sarcoglycan (Fig. 3d) in electrotransferred mouse muscle, proving that the introduction of FLDYSdog leads to the expression of α-sarcoglycan.

Colocalization of FLDYSdog and α-sarcoglycan in electrotransferred mouse muscles.
Electrotransfer in dystrophic dog muscle
After these positive results in mice, an electrotransfer experiment was done in the dystrophic dog because it is the best model in which to study DMD pathology. Two hundred micrograms of pCMV.FLDYSdog was injected into the brachialis anterior of a dystrophic dog (n = 1). The electrotransferred region was biopsied 2 weeks later and myofibers expressing FLDYSdog were observed by immunohistofluorescence in cryosections (Fig. 4a and d). Up to 35 dystrophin-positive fibers were counted per cryosection of this muscle whereas muscle electrotransferred only with saline solution, that is, used as negative control for the experiment, did not present more than three dystrophin-positive fibers per cryosection (Fig. 5a). To verify whether plasmid expression can induce an immune response in the dystrophic dog, muscle sections were stained with hematoxylin–eosin. No cell accumulation was detected in muscle electrotransferred with saline solution (Fig. 5b) or in nonelectrotransferred muscle (data not shown). However, accumulations of mononuclear cells with the morphology of lymphocytes were observed in muscles electrotransferred with FLDYSdog. Indeed, lymphocyte infiltration was colocalized (Fig. 4b and c) or not (Fig. 4e and f) with myofibers expressing FLDYSdog (Fig. 4a and d). Immunohistochemical staining for CD8 confirmed that many cells in these cellular accumulations were cytotoxic T lymphocytes (Fig. 4c).
Electrotransfer of pCMV.FLDYSdog into dystrophic dog muscle. Two regions of electrotransferred muscle are shown as serial sections, respectively, in
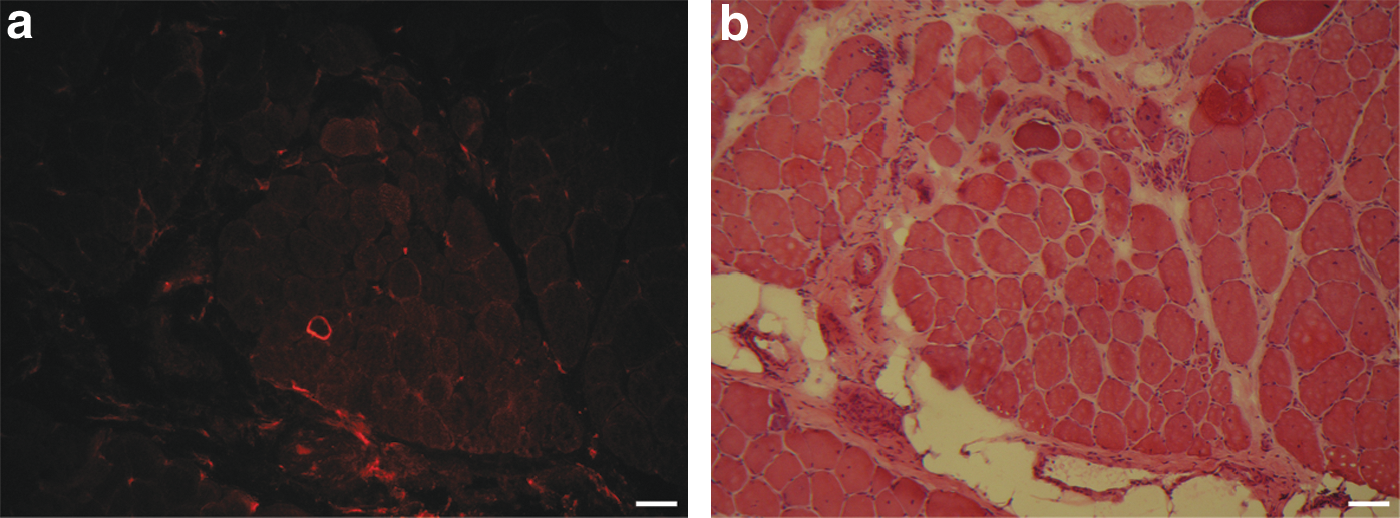
Dystrophic dog muscle electrotransferred with saline solution. Two electrotransferred muscle regions are shown in serial sections
Discussion
When we looked for the sequence of dog dystrophin in databases, we first found a sequence submitted in 1998 by the Wilton laboratory (accession number AF070485.1), another one accessible in 2008 (NM_001003343.1), and one predicted by an automated computational analysis (XM_850502.1). The first two sequences were identical, and thus these two sequences could be put together in GenBank. Concerning the last sequence, it lacks some nucleotides in the N terminus, which brings the first codon (ATG) to position 279 instead of being further up in the sequence, compiling a sequence of only 10,674 nucleotides. The fact that our sequence of 11,040 bp is different from that of Wilton's group (i.e., 5 gaps and 28 nucleotide substitutions) may be due to two main reasons: first of all, the dog used to obtain our dog dystrophin was a beagle, whereas they used a golden retriever. The second reason may be due to the progress in technology between the time of their sequencing and ours (more than 10 years). Nevertheless, there is more than 99% homology between the previously published sequences and our sequence, without the presence of significant variations.
The plasmid encoding our cloned full-length dog dystrophin cDNA was shown to be functional after dystrophin detection by Western blot in cells transfected with this plasmid. Expression was also obtained in vivo after its introduction into mouse and dystrophic dog muscles by a nonviral gene therapy method, that is, DNA electrotransfer. This technique allows the transfection of all myofiber types (Fewell et al., 2001; Bertrand et al., 2003) and allows long-term expression of a plasmid in skeletal muscle because myofibers are postmitotic. Compared with viral gene therapies, this is a faster method because no virus production is required and may also be less immunogenic and safer because no virus or viral capsid is introduced into the body (Wang et al., 2007; Trollet et al., 2008; Ohshima et al., 2009). Even if some plasmids injected during electrotransfer may enter the bloodstream, the use of a muscle-specific promoter will ensure that transgene expression is restricted to myofibers (Miranda et al., 1988); this can also be the case for virus injections. However, the incorporation of naked plasmids by cells is weak without the use of electrotransfer, whereas a virus circulating in the blood can spread throughout the body, reaching tissues other than muscle (Douglas, 2007; Zaiss and Muruve, 2008). This method combining electrotransfer and gene therapy has been used in muscles, tumors, and skin in large animal such as dogs, pigs, nonhuman primates, and so on, and is currently in clinical trials to treat various diseases (Bodles-Brakhop et al., 2009).
Other efficient nonviral gene therapies exist to introduce a plasmid into muscles. The first one involves the delivery of a plasmid by hydrodynamic limb vein injection, which was initiated by Wolff and colleagues (Hagstrom et al., 2004). Their most recent study using this technique showed up to 20% dystrophin expression throughout the injected mdx muscles (Zhang et al., 2009), but there is a lack of information about this procedure because it is still recent as of this writing. The use of minicircle plasmids seems also to be an interesting venue (Mayrhofer et al., 2009). This approach allows removal of the bacterial backbone sequence consisting of an antibiotic resistance gene, an origin of replication, and inflammatory sequences intrinsic to bacterial DNA genes, for an improved safety profile. In addition, the size of the plasmid is reduced to increase gene transfer efficiency. Another method allowing better plasmid dispersion is to fuse the transgene of interest with a peptide containing a protein transduction domain (PTD) and a nuclear localization signal (NLS), such as VP22 (Xiong et al., 2007) or TATκ (Flinterman et al., 2009).
GFP expression in muscle electrotransferred with pCMV.GFP was more important than that of dystrophin after electrotransfer of pCMV.FLDYSdog. This was expected because the efficiency of plasmid transduction decreases with increasing size of the plasmid (Wells et al., 2008). Indeed, the size of pCMV.FLDYSdog is almost three times superior to that of pCMV.GFP (17 vs. 5.8 kb). In mouse muscle electrotransferred with pCMV.FLDYSdog, dystrophin expression was double that in mouse muscle electrotransferred with pMCK.FLDYSdog. Even if the MCK promoter ensures that transgene expression is restricted to myofibers, it is less strong than the CMV promoter (Fabre et al., 2006); this could explain the difference in efficiency. For the dystrophic dog, dystrophin expression was less important in its electrotransferred muscle than that observed in mouse muscle electrotransferred with pCMV.FLDYSdog, but its level of expression corresponds to that obtained in mouse muscle electrotransferred when the transgene was under the control of the MCK promoter. This difference in efficiency is probably simply due to an easier and more effective electrotransfer technique in smaller muscles, such as those of mice, than in bigger muscles, such as those of dogs. In addition, the electrical parameters for electrotransfer in smaller muscles are already well established, which is not the case for larger dog muscles (Jiao et al., 1992; Fewell et al., 2001; Fattori et al., 2005).
The restoration of α-sarcoglycan, a protein of the DGC, was also verified in mouse muscle electrotransferred with pCMV.FLDYSdog. Indeed, dystrophin expression was correlated with α-sarcoglycan expression. This shows that FLDYSdog can bind to the DGC, restore this complex, and thus be functional.
The electrotransfer of pCMV.FLDYSdog in dog muscle led to the expression of FLDYSdog but also to mononuclear cell infiltration. Dystrophin expression was either (1) observed alone or (2) colocalized with cytotoxic T lymphocytes; (3) the presence of cytotoxic T lymphocytes without the expression of dystrophin was also observed. The observed infiltration could be due to either a nonspecific immune response or a specific immune response. Concerning a nonspecific immune response, some signs of inflammatory response may still be present in a muscle that has been electrotransferred 2 weeks previously. However, none of these signs was noticed in muscle electrotransferred only with saline solution. Therefore, the observed infiltration is related to an adaptive immune response. Indeed, it is as if we have managed to fix the sequence of events corresponding to an immune response: (1) the expression of an antigen, (2) the infiltration of cytotoxic cells near the muscle fibers expressing the antigen, and (3) the elimination of the muscle fibers expressing this antigen. The cause(s) of this specific immune response remain(s) to be determined, but it seems to be due to one or more components of the plasmid. The first component is FLDYSdog. There are contradictory results in dystrophic mouse experiments receiving transplantation of syngeneic MPCs expressing normal dystrophin. One study reported the production of antibodies reacting with dystrophin, but without inducing rejection long term (Vilquin et al., 1995). Another study reported both antibody production and specific T lymphocyte reaction against dystrophin, leading to rejection (Ohtsuka et al., 1998). In patients with DMD, anti-dystrophin antibodies were also observed after normal MPC grafts (Roy et al., 1993). However, we observed the same type of immune response in normal dog muscle electrotransferred with a dog microdystrophin plasmid (Pichavant et al., 2010). For this reason, it does not seem that dystrophin expression is the major cause of rejection in our case even if its expression were part of it. Therefore, it seems that other components of the plasmid are more involved in the observed immune response, such as the antibiotic resistance gene or CpG sequence motifs (Mayrhofer et al., 2009; Tolmachov, 2009).
In the dog experiment, a longer term study should thus be made to verify whether there will still be expression of dystrophin and to verify the absence or presence of cellular infiltrations. Therefore, in the future, plasmid modifications should be taken into consideration when checking the immunogenicity of plasmid components.
The first time that a restoration of full-length dog dystrophin was observed in dystrophic dogs was in 2006 by the Cossu laboratory, with an intraarterial delivery of wild-type dog mesoangioblasts. However, the treated dystrophic dogs needed immunosuppression because allogeneic mesoangioblasts were used (Sampaolesi et al., 2006). Thus, we are the first to demonstrate local restoration of full-length dog dystrophin in dystrophic dog muscle by gene therapy, and more precisely by electrotransfer, a method requiring neither cell nor virus and a possible alternative to viral approaches for in vivo gene transfer. Because the phenotype of dystrophic dogs is the closest to that of patients with DMD, plasmid electrotransfer could be a possible avenue to study the function of full-length dystrophin in the dystrophic dog and a potential treatment for muscular dystrophies such as DMD.
Although it seems that full-length dystrophin provides better stability to myofibers than minidystrophin or microdystrophin (Harper et al., 2002; Banks et al., 2009), the number of fibers expressing FLDYSdog, compared with the number of fibers expressing microdystrophin, was lower in dog muscle after electrotransfer (Pichavant et al., 2010). However, it remains to be determined whether it is better to have a higher number of microdystrophin-positive fibers or a lower number of full-length dystrophin-positive fibers, regarding the stability provided by these two transgenes to myofibers.
To restore muscle strength and thus to correct the phenotype of the dystrophic dog, the next step will be to improve the efficiency of the delivery method, such as by the use of minicircles or a protein transduction domain coupled with a nuclear localization signal (PTD-NLS). This will also require a more detailed study of the electrotransfer parameters to obtain wider expression of FLDYSdog, such as the use of a combination of low-voltage and high-voltage pulses (Andre et al., 2008; Hojman et al., 2008). These parameters should be considered to study the introduction of dystrophin into dog muscles.
Footnotes
Acknowledgments
The authors thank Dr. Glenn E. Morris and Dr. Le Thanh Lam (Wolfson Centre for Inherited Neuromuscular Disease, Oswestry, UK) for providing the MANHINGE1A and MANDYS104 antibodies, and Guylaine Jalbert and Dr. Brigitte Dubé for technical assistance during the experiments performed in dogs. Dr. Tremblay is the president and hold shares in CellGene, Inc., a biotech company involved in the development of cell and gene therapies.
Author Disclosure Statement
No disclosures.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.