
Abstract
Adoptive T cell therapy is aimed at overcoming constraints of the endogenous immune response. In patients with malignancies, this approach is based on the possibility of administering sufficient numbers of tumor-reactive lymphocytes under conditions in which they will promote a therapeutic response. Although this strategy is potentially applicable to a vast number of malignancies, its efficacy, to date, has been limited. This is likely related to several factors including an insufficient persistence and reactivation of infused cells, insufficient tumor infiltration, and the presence of an immunosuppressive environment. Here, we review the importance of pretransplantation host conditioning and posttransplantation strategies that have been shown to contribute to the therapeutic efficacy of infused T lymphocytes.
Introduction
Tumor-infiltrating lymphocytes, tumor-primed lymph node cells, and in vitro-sensitized peripheral blood lymphocytes have all been shown to harbor in vivo antitumor efficacy (Gattinoni et al., 2006). More recently, the possibility to redirect T lymphocyte specificity by the transfer of TCRs and chimeric antigen receptors (CARs) has created the opportunity to identify high-affinity/avidity receptors and expand the number of clinical targets (Berry et al., 2009; Dotti et al., 2009; Schmitt et al., 2009). Current limitations of ACT still remain the source and nature of the lymphocytes to be transferred, their long-term persistence on in vivo infusion, their homing to appropriate anatomical locations (i.e., the site of the tumor), their effector function within the tumor, and their long-term persistence for immune surveillance against metastatic disease. Human trials have generally reported only short-term persistence of infused tumor-reactive T lymphocytes, possibly accounting for the limited clinical success of this approach. New strategies are aimed at obtaining sufficient numbers of tumor-reactive lymphocytes for infusion, as well as creating a favorable environment in the patient at the time of transfer. Here, we discuss the importance of lymphocyte selection, conditioning regimens before infusion, and the provision of posttransplantation supportive strategies in the success of ACT (see Fig. 1).
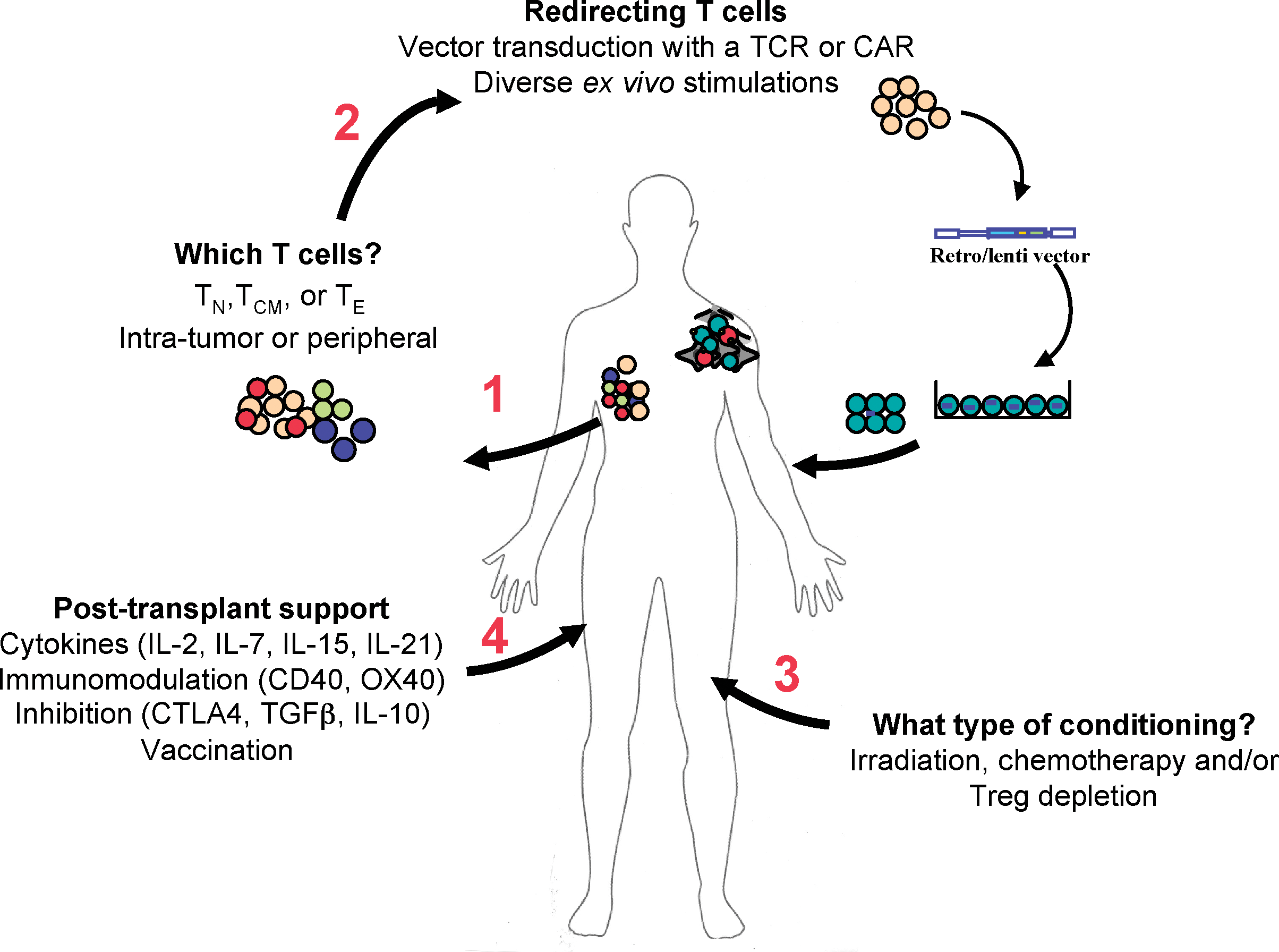
Factors implicated in the success of adoptive T cell therapy (ACT) against tumors. Shown are some of the important steps and critical questions in ACT. It is initially important to determine whether naive (TN), central memory (TCM), or effector (TE) T cells are optimal for transfer and whether it is best to isolate them from peripheral blood or from the tumor itself (1). To obtain the most “fit” tumor-reactive T cells, a tumor-specific chimeric antigen receptor (CAR) or T cell receptor (TCR) can be introduced by retrovirus- or lentivirus-mediated technology after ex vivo stimulation and/or expansion (2). These tumor-specific lymphocytes are then infused into tumor-bearing individuals preconditioned to favor the survival/persistence and activity of the transferred cells (3 and 4). Posttransplantation treatment of the patients with cytokines, stimulatory or antagonist antibodies, and vaccination further favors the in vivo expansion of tumor-specific lymphocytes. CTLA, cytotoxic T lymphocyte antigen; IL, interleukin; TGFβ, transforming growth factor β; Treg, regulatory T cells. Color images available online at
Pre-transplantation Lymphocyte Selection: Choosing the Right Cells for the Job
Adoptive cellular immunotherapy aims to eradicate malignancies by the transfer of reactive T cells. These T cells can be derived from the tumor-bearing host and all of the following have been tested; tumor-infiltrating lymphocytes, tumor-primed lymph node cells, in vitro-sensitized peripheral blood lymphocytes, and tumor-specific T cells generated in vitro by TCR/CAR gene transfer (Berry et al., 2009; Dotti et al., 2009; Schmitt et al., 2009). Ex vivo gene transfer strategies provide new opportunities to choose appropriate targets, optimize TCR structures and composition, and define T cell culture conditions promoting the differentiation of lymphocytes with high in vivo persistence and effector function.
There is some debate as to the relative persistence and efficacies of naive, central memory, and effector memory T cells for ACT. Central memory cells, with high proliferative and reconstituting capacity, maintain the ability to relocate to secondary lymphoid organs and are involved in recall responses. In contrast, terminally differentiated effectors, present primarily in peripheral tissues, are endowed with immediate effector function on antigen reencounter but show poor proliferative and reconstituting abilities (Sallusto and Lanzavecchia, 2009). This is of importance as terminally differentiated effector cells might exert potent, but transient antitumor activity, whereas central memory T cells may confer a more durable T cell immunity required for long-lasting immunosurveillance.
The ex vivo expansion of tumor-reactive lymphocytes and gene transduction of T cells (by retroviral vectors) requires that the cells be activated and undergo cell division. In general, antigen-specific or polyclonal stimuli (CD3 or CD3/CD28 monoclonal antibodies [mAbs]) used to activate T cells have been coupled to interleukin (IL)-2 stimulation. More recently, T cells have been stimulated ex vivo, using artificial antigen-presenting cells that can enhance their efficacy and function (Powell and Levine, 2008). These activation protocols are associated with the acquisition of memory markers and differentiation. However, increasing evidence indicates that although antitumor effector T cells, obtained after multiple rounds of ex vivo stimulation, possess highly effective in vitro cytotoxic activity, they are less effective than naive or memory-like T cells in vivo (Klebanoff et al., 2005; Berger et al., 2008; Hinrichs et al., 2009).
Notably, it has been found, in mice as well as in macaques, that antigen-specific CD8+ T cells derived from central memory T cells show significantly higher long-term persistence/survival than effector memory T cells (Klebanoff et al., 2005; Berger et al., 2008). Moreover, these central memory T cells mediate superior antitumor immunity than effector memory T cells on adoptive transfer into tumor-bearing mice (Klebanoff et al., 2005; Berger et al., 2008). It is also important to note that under conditions in which a tumor-specific TCR is introduced ex vivo into either naive or central memory T cells (by retrovirus-mediated gene transfer), infusion of the former results in a significantly more robust antitumor response (Hinrichs et al., 2009).
Considerable research efforts have thus been devoted to the development of ex vivo activation strategies that preserve a naive- or central memory-like phenotype. Whereas prolonged IL-2 signaling promotes the terminal effector differentiation of CD8+ T cells (Kalia et al., 2010; Pipkin et al., 2010), homeostatic γ-chain cytokines (IL-7, IL-15, and IL-21) have proven efficacious in sustaining T cell proliferation and in vivo antitumor function without favoring terminal differentiation (Huarte et al., 2009; Kaneko et al., 2009). Of note, it has been shown that IL-7, IL-15, and IL-21 support the generation of CD8+ cells with superior therapeutic activity as compared with IL-2 alone (Liu et al., 2006; Daudt et al., 2008; Hinrichs et al., 2008; Caserta et al., 2010; Pouw et al., 2010). The combined use of these cytokines needs to be carefully studied as high-dose IL-2 may sensitize IL-7-stimulated cells to Fas-mediated apoptosis (Jaleco et al., 2003). IL-7 and IL-15 have also been exploited for ex vivo gene transfer; these cytokines sustain sufficient activation of lymphocytes in the absence of TCR stimulation, rendering them susceptible to lentiviral infection without favoring cell differentiation (Dardalhon et al., 2001; Cavalieri et al., 2003; Verhoeyen et al., 2003; Circosta et al., 2009; Gilham et al., 2010). As an alternative to the use of recombinant cytokines, genetic modification of T cells with a vector expressing a homeostatic cytokine such as IL-15 or IL-21 may favor the generation of lymphocytes with a central memory phenotype (Kaka et al., 2009).
Although it is clear that CD8+ T cells play a critical role in antitumor immunity, we and others have documented a function for CD4+ T cells (Pardoll and Topalian, 1998; Antony et al., 2005; Benigni et al., 2005). Moreover, two publications have demonstrated a direct role for these lymphocytes in adoptive immunotherapy (Pellegrini et al., 2009; Quezada et al., 2010). On retroviral transfer of a tumor-specific receptor into T lymphocytes, it has been shown that gene-modified CD8+ as well as CD4+ T cells are required for an efficient immune response. The most potent antitumor responses were observed when the ratio of gene-modified CD4+:CD8+ T cells was 1:1 (Moeller et al., 2005). Moreover, we and others have found that adoptively transferred memory-like CD8+ T cells are subject to peripheral cross-tolerance; breakdown of this tolerance and differentiation of CD8+ T cells into effectors requires CD4+ T cell help (Hamilton and Jameson, 2008; Le Saout et al., 2008). Thus, the cooperativity between CD4+ and CD8+ T cells needs to be taken into account in ACT protocols.
The efficacy of tumor-reactive lymphocytes is also likely to be modulated by the numbers of infused cells. It has been shown that when precursor frequencies of tumor-specific T cells are below a competitive threshold, the lymphocytes undergo increased proliferation, are polyfunctional, and eradicate tumors more effectively (Rizzuto et al., 2009). When tumor-specific lymphocytes are primed at frequencies above this threshold, the functional benefit is impaired, possibly because of intraclonal competition (Rizzuto et al., 2009). Overcoming intraclonal competition, while still transferring sufficient numbers of lymphocytes able to target large tumor masses, may require that the transferred T cells target different antigens on the tumor or on tumor-associated stroma via receptors selected for defined MHC–peptide combinations (Xue and Stauss, 2007). Indeed, the rejection of spontaneous tumors by tumor-specific T cells is improved by harnessing responsiveness against minor histocompatibility antigens expressed on the tumor (Hess Michelini et al., 2010). Thus, future developments in ACT will benefit from taking these parameters into consideration: optimizing the source and phenotype of T cells to be used for transfer, both before and after ex vivo expansion; defining the most favorable ratios of CD4+:CD8+ T cells; using optimal numbers of T cells; and taking advantage of alloreactivity.
Although it will clearly be advantageous to retarget T cells to tumor antigens by the introduction of TCR/CAR molecules and select those lymphocytes with regenerative capacities and long-term survival potential, this may not be sufficient to achieve a clinical response (Walker et al., 2000). The efficacy of adoptively transferred T cells also relies on their capacity to migrate to the sites of tumor development. T cell migration is dependent on the expression of a combination of selectins, chemokine receptors, and integrins that regulate extravasation and trafficking in different microenvironments (Butcher and Picker, 1996). Thus, differential expression of adhesion and chemokine receptors on T cells (and the corresponding expression of ligands by tumor cells/stroma) will modulate their ability to provide immune surveillance and migration to the tumor. Accordingly, forced expression of defined chemokine receptors (e.g., CXCR2 and CCR4) has been shown to improve migration of redirected T cells to tumor sites (Kershaw et al., 2002; Di Stasi et al., 2009).
Host Conditioning: Creating a Friendly Environment
Persistence and long-term functionality of adoptively transferred tumor-reactive T cells within the tumor-bearing host are critical for therapeutic efficacy. Modulation of the immunologic environment in the patient plays a major contribution in the engraftment, differentiation, clonal expansion, anergy, and/or death of transferred T cells. This is generally referred to as “host conditioning”, and is attained with myeloablative drugs (i.e., combinations including busulfan, cyclophosphamide, fludarabine, melphalan, and/or treosulfan) or irradiation. Although it is still not completely clear how host conditioning influences the development of antitumor T cell responses, the following parameters have been shown to play important roles: (1) Chemotherapy can render solid tumors accessible to transferred T cells by changing the vascular tumor endothelium (Ganss et al., 2002), sensitizing tumor stroma for killing (Zhang et al., 2007), and inducing tumor cell death (Nowak et al., 2003); (2) by depleting endogenous T cells, there is an increased availability of endogenous prosurvival homeostatic cytokines for the remaining endogenous T cells and/or adoptively transferred T cells, resulting in their improved functionality (Gattinoni et al., 2005; Muranski et al., 2006); and (3) total body irradiation and/or chemotherapy damage the integrity of mucosa barriers in the intestinal tract, thereby resulting in the translocation of microbial products (Nakayama et al., 1997; Abad et al., 2008). These serve as immunological adjuvants as proven by their ability to increase production of inflammatory cytokines, activate dendritic cells (DCs), and augment T cell responses (Kieper et al., 2005; Hamilton and Jameson, 2008).
The role of host conditioning in the elimination of suppressive regulatory T cell and myeloid populations is also a major factor in the success of subsequent adoptive T cell therapy (Polak and Turk, 1974; Ghiringhelli et al., 2004). Regulatory T cells (Tregs) clearly inhibit T cell responses against tumors and their abundance within tumors has been shown to be inversely correlated with survival (Curiel et al., 2004). Furthermore, in several mouse models, ablation of CD25+ T cells before tumor implantation results in T cell-mediated tumor eradication (reviewed in Frumento et al., 2006; Petrausch et al., 2009). It is notable that the recognition of tumor cells by self-specific memory Tregs has been shown to be extremely rapid, preceding the activation of naive antitumor T cells and thereby “protecting” the tumor from an immune response (Darrasse-Jeze et al., 2009). Regarding myeloid-derived suppressor cells (MDSCs), these cells are induced by proinflammatory factors and their accumulation downregulates immune surveillance. Nevertheless, MDSCs are heterogeneous and the mechanisms regulating their expansion and suppressor activities have still not been clearly elucidated (reviewed in Ostrand-Rosenberg and Sinha, 2009). Thus, although clinical protocols promoting the selective depletion of Tregs in ACT are well advanced (Thistlethwaite et al., 2008), the implementation of protocols for eliminating/antagonizing MDSCs will require further basic and translational research.
Chemotherapeutic agents commonly used to treat human malignancies, such as cyclophosphamide, exert both direct and indirect antitumor activities. In addition to targeting actively proliferating transformed cells, chemotherapy ameliorates the antitumor efficacy of adoptively transferred lymphocytes in preclinical models (Vierboom et al., 2000; Bracci et al., 2007; Cheadle et al., 2008) and, most importantly, in clinical trials (Dudley et al., 2005; Nistico et al., 2009). This effect has been ascribed, at least in part, to the drug-induced recruitment, expansion, maturation, and activation of endogenous antigen-presenting cells (APCs), and especially DCs (Paulos et al., 2007; Saini et al., 2009; Salem et al., 2009, 2010). Chemotherapeutic effects on DCs are observed both immediately after lymphopenia induction and during the recovery phase (reviewed in Salem et al., 2007).
The establishment of a lymphopenic environment at the time of ACT also provides a competitive advantage for infused tumor-reactive T cells (reviewed in Gattinoni et al., 2006). After lymphopenia induction, the lymphocyte compartment is regenerated mainly by the thymic-independent homeostatic proliferation of peripheral T cells. Murine studies have shown that homeostatic proliferation of naive T cells requires TCR signals delivered by peptides presented by MHC molecules (Goldrath and Bevan, 1999; Surh and Sprent, 2000; Seddon and Zamoyska, 2002b) and by IL-7 (Schluns et al., 2000; Seddon and Zamoyska, 2002a). Lymphodepleting regimens result in increased circulating levels of IL-7, favoring the proliferation of the remaining T cells (Bolotin et al., 1999; Fry et al., 2001). Moreover, it has been shown that IL-7 signaling on DC subsets can modulate the responsiveness of mature CD4+ and CD8+ T cells (Guimond et al., 2009; Saini et al., 2009; Vogt et al., 2009). Finally, the peripheral cross-tolerance regulating autoreactive naive CD8+ T cells is overcome under conditions of lymphopenia, as long as CD4+ T cell help is provided (Calzascia et al., 2008; Hamilton and Jameson, 2008; Le Saout et al., 2008). The ensemble of these data demonstrates the importance of immune depletion in the recipient host before adoptive transfer of tumor-reactive lymphocytes. Conditioning increases long-term engraftment, efficacy, and function of the transferred cells.
Postinfusion Supportive Strategies: Helping from the Outside
Posttransplantation survival and function of transferred tumor-reactive T cells can be enhanced and supported by active immunotherapy. Vaccination using tumor cells, tumor lysates, and tumor antigen-derived peptides, either emulsified in adjuvant or loaded onto dendritic cells (Pilla et al., 2009), can augment the efficacy of infused lymphocytes. Thus far, however, therapeutic vaccines have not been successful in the clinic. This is possibly due to the clinical state of enrolled cancer patients (who have generally failed to respond to previous treatments), and to their profound state of tolerance and immunosuppression (Rosenberg, 2001). However, in the context of ACT, vaccination has proven efficacious (Koike et al., 2008) and able to favor the expansion of TCR/CAR-redirected T cell after infusion (Jiang et al., 2006; de Witte et al., 2008), at least in the context of animal models. This may be because under conditions of ACT, “fresh” lymphocytes, capable of responding to the vaccination strategy in a nontolerizing environment, are provided. The finding that chemotherapy does not hinder the immunogenic potential of various vaccine formulations (Casati et al., 2005) and can favor an antitumor response (Apetoh et al., 2007) has prompted the implementation of combined chemoimmunotherapies. For example, gemcitabine and a nonspecific immunotherapy such as anti-CD40 treatment act synergistically in the treatment of animals with large tumors (Broomfield et al., 2005). Likewise, concomitant cyclophosphamide administration and OX40 costimulatory receptor engagement mediated improved CD8+ T cell priming and an improved intratumoral T effector/T regulatory cell ratio (Hirschhorn-Cymerman et al., 2009).
Similar results have been obtained in ACT settings. Chemotherapy preconditioning, ACT, and vaccination followed by adjuvant peritumoral injections of immunostimulatory nucleic acids elicit potent cytotoxic inflammatory responses (Kohlmeyer et al., 2009). Cyclophosphamide preconditioning markedly increases the expansion and function of adoptively transferred T cells in response to vaccination while delaying the contraction phase. These effects can be further enhanced by the use of the TLR3 ligand poly(I:C), likely by favoring DC maturation (Salem et al., 2007). In addition to chemotherapeutic drugs, local tumor irradiation combined with intratumoral dendritic cell administration can significantly enhance the therapeutic efficacy of adoptively transferred tumor-reactive T cells (Teitz-Tennenbaum et al., 2009). As discussed previously, immunomodulating agents such as Toll-like receptor agonists, CD40 agonists, as well as combinations of adjuvants targeting components of the adaptive (IL-2/anti-IL-2 mAb complexes) and innate [poly(I:C)] immune responses (Verdeil et al., 2008) as well as IL-7 administration (Pellegrini et al., 2009) support the reactivation, expansion, and survival of infused cells. Similarly, the administration of antibodies able to neutralize immunosuppressive cytokines such as transforming growth factor (TGF)-β and IL-10, inhibitors of indoleamine 2,3-dioxygenase (IDO)-positive dendritic cells, and antagonistic antibodies against the inhibitory receptor cytotoxic T lymphocyte antigen (CTLA)-4, improve T cell responsiveness and enhance vaccine potential (Hodi et al., 2008; Ribas et al., 2009).
The promise of recombinant homeostatic cytokines has been demonstrated in animal models and, more recently, clinical trials in patients have been initiated. Notably, short-term administration of recombinant IL-7 has been shown to increase the homeostatic proliferation of endogenous CD4+ and CD8+ lymphocytes in patients with metastatic diseases (Capitini et al., 2009; Sportes et al., 2009, 2010). IL-15α can similarly promote T cell function in vitro (King et al., 2009) and in vivo (Miyagawa et al., 2008; Berger et al., 2009) and patients are currently being recruited for a phase I clinical trial of intravenously administered rIL-15 (NCI, NCT01021059). Moreover, encouraging results have been observed in patients with malignant melanoma and renal cell carcinoma treated with rIL-21 (Thompson et al., 2008; Davis et al., 2009). Thus, the use of recombinant IL-7, IL-15, and IL-21, either alone or in combination, may greatly enlarge our arsenal of antitumor immune adjuvants, significantly enhancing adoptive T cell therapies (Fig. 1).
Conclusions and Future Considerations
The availability of realistic mouse models and innovative strategies to identify T cell function at the single-cell level (Hadrup et al., 2009) has provided a growing understanding of the biological processes underlying the development of antitumor protective immunity. Of note, they have also contributed to the design of improved and safer therapeutic strategies. These studies, together with the optimization of cell expansion methodologies and TCR/CAR gene transfer technologies, have boosted interest in ACT. Combining ACT with lymphodepleting conditioning regimens, posttransplantation vaccination, and immunomodulatory agents will enhance therapeutic efficacy.
Translation of complex protocols involving several combinatorial approaches might, however, compromise feasibility. Good Manufacturing Practice and Good Laboratory Practice principles are needed to ensure quality, safety, purity, and potency of the product and are required for clinical trials (Laurencot and Ruppel, 2009). However, compliance with these practices might prove financially overwhelming and inapplicable for nonspecialized clinical centers. It will therefore be important to define minimal ACT conditions and optimize resources between manufacturing and clinical facilities. The future experience of multicenter clinical trials will contribute to the definition of safe and affordable strategies tailored to individual needs.
Footnotes
Acknowledgments
This work was supported by a grant from the European Community (contract LSHC-CT-2005-018914 ATTACK) (to A.M. and N.T.) and grant R01AI059349 from the National Institute of Allergy and Infectious Diseases (to N.T.). S.L.-M. and V.D. were supported by ANRS and INSERM-ARC, respectively. The authors thank ATTACK members for constructive discussions and open collaborative efforts. The authors are grateful to Valérie Zimmermann for constructive input and critical reading of the manuscript.
Author Disclosure Statement
No competing financial interests exist.