
Abstract
Hyperlipidemia is a common feature of type 2 diabetes and is related to cardiovascular disease. The very low-density lipoprotein receptor (VLDLR) binds to and internalizes triglyceride-rich lipoproteins with high specificity. In this study, we evaluated the role of VLDLR in hyperlipidemia in type 2 diabetic rats. Type 2 diabetes was induced in male Wistar rats by injection of low-dose streptozotocin coupled with a high-fat diet. Recombinant adeno-associated viral (rAAV) vectors encoding the human VLDLR gene (rAAV·VLDLR) were intravenously administered to diabetic rats. Results showed that in vivo, total VLDLR mRNA and protein levels were significantly decreased in skeletal muscle (type I VLDLR mainly reduced) and adipose tissue (type II VLDLR mainly reduced) but not in heart in hypercholesterolemic, hypertriglyceridemic diabetic rats compared with normal rats. And in vitro, in 3T3-L1 adipocytes, insulin-induced (1.0 mmol/liter) insulin resistance significantly decreased VLDLR mRNA expression. In rats, rAAV·VLDLR delivery resulted in a reduction in serum cholesterol and triglyceride that lasted for the duration of the study (12 weeks). Fasting blood insulin was significantly lower in the rAAV·VLDLR group versus untreated diabetic rats although fasting blood glucose levels were not significantly different in both groups at the end of the study. rAAV·VLDLR-treated animals had significantly increased lipoprotein lipase activity and reduced aortic atherosclerosis. Taken together, our data suggest that type 2 diabetes and related insulin resistance manifest decreased VLDLR with elevated serum cholesterol and triglyceride levels, and overexpression of VLDLR through a single injection of rAAV·VLDLR reversed these effects and consequentially attenuated aorta atherosclerotic plaque progression.
Introduction
The VLDL receptor (VLDLR) is a member of the LDL receptor (LDLR) family and is uniquely expressed in the capillary endothelium of skeletal muscle, heart, and adipose tissue and only in trace amounts in the liver (Oka et al., 1994b; Sakai et al., 1994). Initial in vitro studies have demonstrated that the VLDLR is a receptor for several apolipoprotein E (apoE)-containing lipoprotein ligands, such as VLDL, intermediate density lipoproteins, and chylomicrons (Webb et al., 1994; Kim et al., 1997). The binding of these lipoprotein particles to the VLDLR is stimulated by apoE and lipoprotein lipase (LPL) and inhibited by the 39-kDa receptor-associated protein (Gafvels et al., 1993, 1994). It is now believed that the VLDLR may function primarily by facilitating the binding of TG-rich lipoproteins in the capillary bed in concert with LPL, leading to the subsequent delivery of TG-derived fatty acids to the underlying tissues active in fatty acid metabolism (Takahashi et al., 1992).
It has been reported that VLDLR mRNA in skeletal muscle is reduced in experimental hypothyroidism and increased in hyperthyroidism (Jokinen et al., 1994). Reductions of VLDLR expression have been described in rats with chronic renal insufficiency and nephrotic syndrome and in rats with spontaneous focal glomerulosclerosis (Liang and Vaziri, 1997; Vaziri and Liang, 1997; Sato et al., 2002). Rats with these conditions showed marked hypertriglyceridemia and impaired VLDL clearance.
Several reports have demonstrated that VLDLR gene therapy in LDLR-deficient mice produced a substantially sustained hypocholesterolemic response. MacDougall and colleagues (2006) reported that aggressive VLDL/LDL lowering achieved by hepatic overexpression of VLDLR, combined with a low-fat diet regimen, induces regression of advanced plaques in the brachiocephalic artery of LDL receptor-deficient mice. Therefore, the VLDLR appears to be an appropriate target gene for the treatment of dyslipidemia. In the present study, we investigated the therapeutic role of the VLDLR in the dyslipidemia of type 2 diabetes. We studied VLDLR expression in induced type 2 diabetic rats and mouse adipocytes (3T3-L1 cells), and also evaluated the potency of overexpression of the VLDLR gene via recombinant adeno-associated virus (rAAV) as a sole therapy to correct the dyslipidemia in type 2 diabetic rats.
Materials and Methods
Materials
Horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse IgGs were obtained from Jackson ImmunoResearch Laboratories (Soham, Cambridgeshire, UK). Enhanced chemiluminescence reagent was purchased from Pierce Chemical (Rockford, IL), prestained molecular weight standards were from Bio-Rad (Hercules, CA), and polyvinylidene difluoride (PVDF) membranes were from Schleicher & Schuell (Dassel, Germany). VLDLR monoclonal antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). All other chemicals and reagents were from Sigma-Aldrich (St. Louis, MO) unless otherwise specified.
Construction and preparation of rAAV
The recombinant adeno-associated viral vector (rAAV) plasmid pXXUF1, and an rAAV plasmid carrying the report gene, pdxII-lacZ, were from X. Xiao (Division of Molecular Therapeutics, University of North Carolina, Chapel Hill, NC). pXXUF1 contains two inverted terminal repeats, the cytomegalovirus (CMV) promoter, and a poly(A) tail. The rAAV packaging plasmids were constructed on the basis of plasmid pAAV/Ad as we reported previously (Monahan and Samulski, 2000; Yuan et al., 2007). Packaging plasmid pXX2 (for serotype 2 of adeno-associated virus) was constructed from pACG2 by inserting a promoter p5 element downstream of the capsid gene. The adenovirus helper plasmid pXX6 was constructed by inserting the large ClaI–SalI fragment of pXX5 into plasmid pBluescript KS. A 2600-bp human VLDLR cDNA fragment (NotI–NotI) was subcloned into pXXUF1 downstream of the CMV promoter to produce the plasmid pUF1·VLDLR. rAAVs carrying the LacZ and VLDLR genes were prepared and purified, respectively, as described previously (Pollare, 1990; Jaffa et al., 2003) and the rAAV titers were determined by dot-blot hybridization. The resultant rAAVs were assigned the nomenclature rAAV·LacZ and rAAV·VLDLR, respectively.
Experimental animals and cultured 3T3-L1 preadipocytes
The animal model has been described in detail previously (Douillet et al., 2000). Briefly, 2-month-old male Wistar rats weighing 180 to 200 g were supplied by the Experimental Animal Center of China in Shanghai. Experimental protocols complied with National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and were approved by the Academy of Sciences of China. All animals were housed (four per cage) in a room with controlled temperature (25 ± 3°C) and humidity (50 ± 20%) with 12-hr light/dark cycles and allowed free access to normal rat chow plus water for 1 week to obtain baseline data. The animals were then distributed randomly into two groups. Normal rats (n = 10) were kept untreated and were studied for 16 weeks. Experimental rats (n = 20) were fed diets enriched in fat (25% fat, 15% protein, 51% starch, and 5% fiber) (Greene et al., 2000). After exposure to the individual diets for 1 month, the experimental rats were injected intravenously with streptozotocin (STZ, 25 mg/kg; Sigma-Aldrich) and the normal rats were injected with vehicle (citric acid, 0.05 mol/liter, pH 4.5) and fed the same diet. Rats showing a glucose level >200 mg/dl were considered diabetic.
Preadipocyte 3T3-L1 cells were cultured and induced to differentiate as previously described (Liu et al., 2001). In brief, cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The cells were grown in 6-well plates until 2 days postconfluence. Differentiation was induced by the addition of various concentrations of insulin (0, 10, 100, and 1000 nmol/liter), 3-isobutyl-1-methylxanthine (IBMX, 0.5 mmol/liter), and dexamethasone (DEX, 0.25 μmol/liter) (IS-IBMX-DEX) and 2 days after induction, the medium containing IS-IBMX-DEX was replaced with medium containing insulin (1000 nmol/liter) alone. The medium was subsequently replaced again with fresh culture medium (DMEM supplemented with 10% FBS) after 2 days and then every other day thereafter. Oil red O staining was performed to observe the morphological change in cell differentiation. After 3T3-L1 preadipocytes were induced to differentiate into adipocytes with insulin (1000 nmol/liter), various concentrations of insulin (0, 10, 100, and 1000 nmol/liter) were added to the medium for a further 4 days. Semiquantitative RT-PCR was used to evaluate the effect of insulin on expression of the VLDLR during 3T3-L1 cell differentiation and in mature adipocytes. The GOD-PAP (GOD, glucose oxidase; PAP, peroxidase–aminophenazone–phenol) method was used to measure glucose remaining in the medium.
Gene delivery
Diabetic animals were randomly distributed into two groups, receiving intravenous rAAV·VLDLR or rAAV·LacZ injection (1 × 1011 virion particles in 1 ml of saline solution) (n = 10 for each group) via the tail vein, respectively, after intraperitoneal anesthetization with pentobarbital at a dose of 50 mg/kg body weight. Animals were kept warm with an infrared lamp until return of consciousness. All animals were killed 16 weeks after rAAV delivery under pentobarbital anesthesia (50 mg/kg) and heart, skeletal muscle, liver, aorta, and fat were collected, flash frozen in liquid nitrogen, and then stored at −80°C until further analysis were done.
Serum measurements
Fasting blood samples (overnight fast) were taken from the tail vein before injection of rAAV and every 2 weeks afterward. Plasma was prepared and stored at −80°C. Plasma glucose, cholesterol, and triglyceride were measured in duplicate with an AEROSET clinical chemistry system (Abbott Laboratories, Abbott Park, IL). Plasma insulin was measured with the use of a magnetic solid-phase enzyme immunoassay kit from Biochem ImmunoSystem (Singapore). Insulin resistance was calculated by the homeostasis model assessment of insulin resistance (HOMA-IR) method (Srinivasan et al., 2003).
To obtain postheparin plasma, fasted rats were bled for 5 min after a tail vein injection of 100 U of heparin per kilogram body weight. Postheparin LPL activity was measured by the method described by Hocquette and colleagues (1998). To distinguish lipolysis mediated by hepatic lipase, the plasma samples were assayed under high-salt conditions (1 M NaCl, final concentration).
Histological analysis of the aortic root
Animals were killed 16 weeks after rAAV delivery for evaluation of aortic root atherosclerosis. The arterial tree was perfused in situ with phosphate-buffered saline (PBS) (100 mmHg) for 20 min via a cannula in the left ventricular apex. The heart plus aortic root and descending aorta were excised and preserved in 4% PBS-buffered formaldehyde solution and embedded in paraffin. Serial sections were cut and stained with hematoxylin–eosin (H&E) reagent, starting at the appearance of the tricuspid valves Atherosclerotic lesion areas were evaluated in a blinded manner by microscopy inspection.
Isolation of membrane fractions from tissues and Western blot analysis
Membrane fractions were prepared according to a standard methods (Bollag and Edelstein, 1991). Protein concentrations were estimated by the Bradford method (Bradford, 1976). Equal amounts of protein sample were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). The proteins were visualized by enhanced chemiluminescence using horseradish peroxidase–labeled anti-mouse IgG (GE Healthcare, Piscataway, NJ) and quantified by densitometric analysis, using a bioimaging system (Syngene, a division of Synoptic, Cambridge, UK).
RNA extraction and RT-PCR
Total RNA was extracted from frozen tissues of hearts, skeletal muscle, livers, aorta, and fat with TRIzol reagent (Invitrogen, Carlsbad, CA). For RT-PCR analysis, the following primers were used: total VLDLR mRNA (5′ primer, 5′-AGG CAG TAG GCA AAG AG-3′; 3′ primer, 5′-CCA GTA AAC AAA GCC AGA C-3′); spanning the O-linked sugar region, distinguishing the two VLDLR subtypes (5′ primer, 5′-CAA TAA ATT CAC TGG GTC A-3′; 3′ primer, 5′-TTC CTC CAC ATC AAG TAG CC-3′). For β-actin, the primers used were as follows: 5′ primer, 5′-GGAGAAGATGACCCAGATC-3′; 3′ primer, 5′-GATCTTCATGAGGTAGTCAG-3′. The RT-PCR method was performed according to manufacturer's instructions (Takara Bio, Shiga, Otsu, Japan). Conditions were as follows: denaturing phase (1 min, 94°C), annealing phase (30 sec, 52.8°C), and extension phase (30 sec, 72°C) for 30 cycles. PCR products were electrophoresed on 2% agarose gels. The quantities of specific VLDLR transcripts were normalized to the expression of β-actin to control for RNA quality and amount.
Statistical analysis
Results are expressed as means ± SEM. Data were analyzed by unpaired Student t test or analysis of variance (ANOVA), with the use of Systat software (Systat, San Jose, CA). Values were considered statistically significant at p < 0.05.
Results
Metabolic characteristics of experimental animals
The general characteristics of the rats analyzed in this study are shown in Table 1. Body weight, serum glucose, insulin, triglycerides, cholesterol and HOMA-IR showed no significant differences between the two groups at baseline. After STZ injection and high-fat feeding, compared with normal rats, experimental rats were characterized by markedly higher body weight (p < 0.05), serum fasting glucose (p < 0.01), insulin levels (p < 0.01), and significantly increased HOMA-IR. These data indicate that the metabolic abnormalities of experimental rats closely resemble those seen in type 2 diabetes.
HOMA-IR, homeostasis model assessment of insulin resistance.
Values represent means ± SEM. Normal rats were fed with normal rat chow (normal). Diabetic rats were administered an STZ injection and high-fat feeding (diabetes).
p < 0.05 versus normal.
p < 0.01 versus normal.
Expression of VLDLR in diabetic rats
mRNA levels of VLDLR were analyzed by RT-PCR. VLDLR mRNA was detected mainly in the hearts, skeletal muscle, aorta, and adipose tissue, but not in the liver, in both normal and diabetic rats. In skeletal muscle, aorta, and adipose tissue, VLDLR mRNA levels were significantly lower in diabetic rats than that in control rats (Fig. 1A and B), but there was no change in heart tissue. We also examined the splice isoform of the VLDLR mRNA by RT-PCR and results showed that the type I VLDLR mRNA (Fig. 1C) level was decreased by 44 and 46% in diabetic rats in skeletal muscle and aorta, respectively, whereas it was not changed in heart or adipose tissue. A significant 32% decrease was detected in type II VLDLR mRNA (Fig. 1D) in adipose tissue in diabetic rats, but there were no differences in skeletal muscle or heart in both groups. The expression of VLDLR protein was consistent with the VLDLR mRNA level in diabetic rats. As shown in Fig. 2A–C, type I VLDLR protein levels were decreased by 71 and 25% in skeletal muscle and aorta, respectively, and the type II VLDLR protein level was decreased by 44% in adipose. Levels of both splice isoforms of the VLDLR protein were not different in heart tissue between normal and diabetic rats.

Representative blot of expression of total VLDLR, and VLDLR subtype mRNAs and β-actin in muscle, heart, aorta, liver, and adipose tissue. The quantities of specific VLDLR transcripts were normalized to the expression of β-actin. Normal, normal rats (lanes 3, 6, 9, 12, and 15); rAAV-VLDLR, diabetic rats treated with the VLDLR gene (lanes 1, 4, 7, 10, and 13); rAAV-0, diabetic rats treated with the LacZ gene (lanes 2, 5, 8, 11, and 14). Values represent means and SEM (n = 6). *p < 0.05, **p < 0.01, ***p < 0.001 versus the rAAV-0 group.
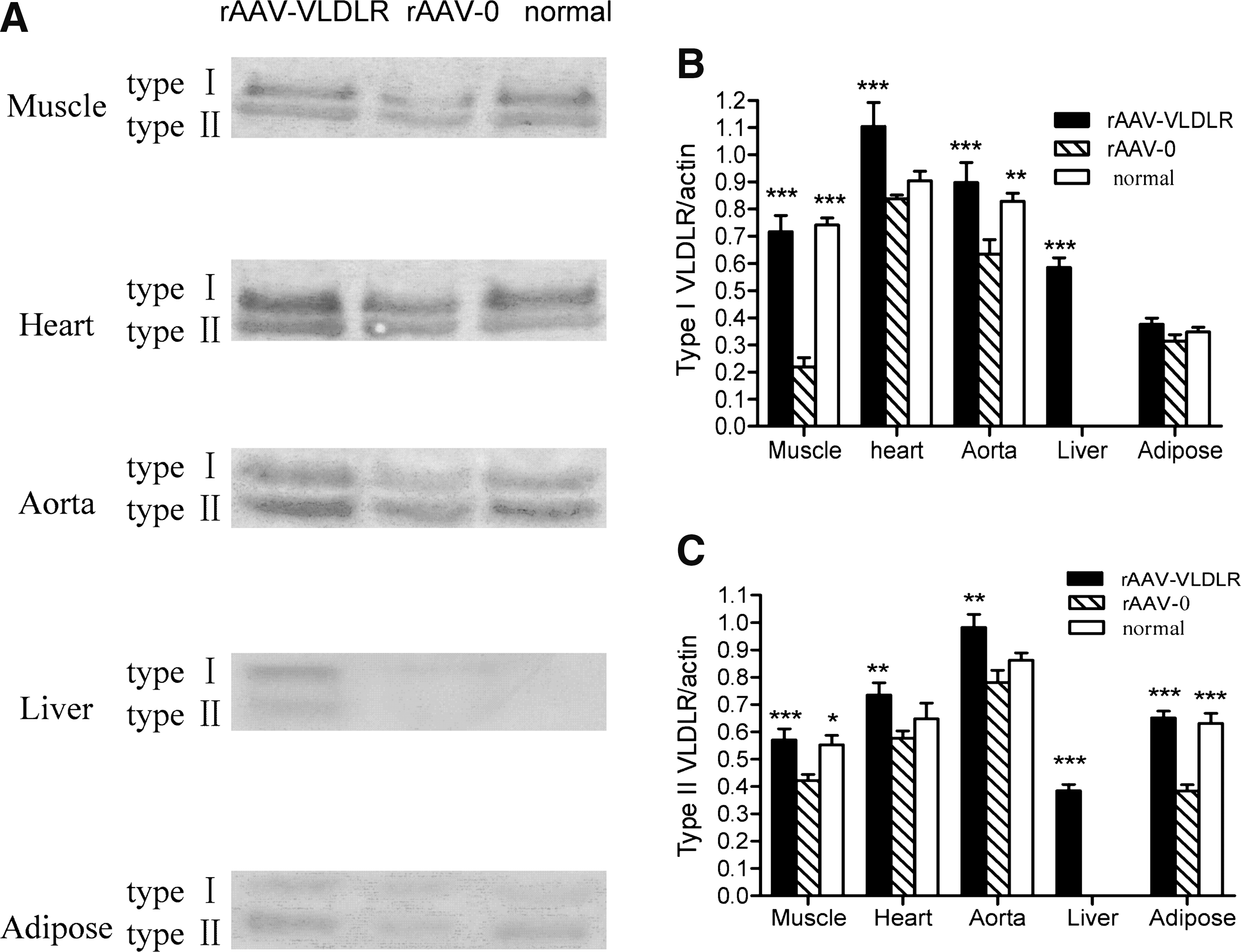
Expression of VLDLR protein in muscle, heart, aorta, liver, and adipose tissue of normal rats (normal), diabetic rats treated with the VLDLR gene (rAAV-VLDLR), and diabetic rats treated with the LacZ gene (rAAV-0). Values represent means and SEM (n = 6). *p < 0.05, **p < 0.01, ***p < 0.001 versus the rAAV-0 group.
VLDLR mRNA and protein in rats after rAAV delivery was also analyzed by RT-PCR and Western blot. As seen in Fig. 1, in skeletal muscle, aorta, and adipose tissue, total VLDLR mRNA and both VLDLR isoform types were significantly higher in rAAV·VLDLR rats than in rAAV·LacZ rats. There was no VLDLR mRNA expression in the liver in normal and rAAV·LacZ rats. After rAAV·VLDLR injection, VLDLR mRNA was clearly detected in rAAV·VLDLR rats. The VLDLR protein levels were similar to VLDLR mRNA expression after rAAV·VLDLR treatment (Fig. 2). The results indicate that VLDLR mRNA and protein were stably expressed in the tissues after rAAV·VLDLR administration in rats.
Effect of insulin on expression of VLDLR during 3T3-L1 cell differentiation and in mature adipocytes
To explore the effect of insulin on expression of the VLDLR, 48-hr confluent cells were exposed to differentiation medium containing IBMX, DEX, and various concentrations of insulin (up to 1000 nmol/liter), and VLDLR mRNA was measured on days 0, 3, 5, 7, and 9. As seen in Fig. 3A, the expression of VLDLR mRNA was increased by insulin in a concentration- and time-dependent manner. This result suggests that insulin stimulates the expression of VLDLR mRNA during 3T3-L1 cell differentiation.
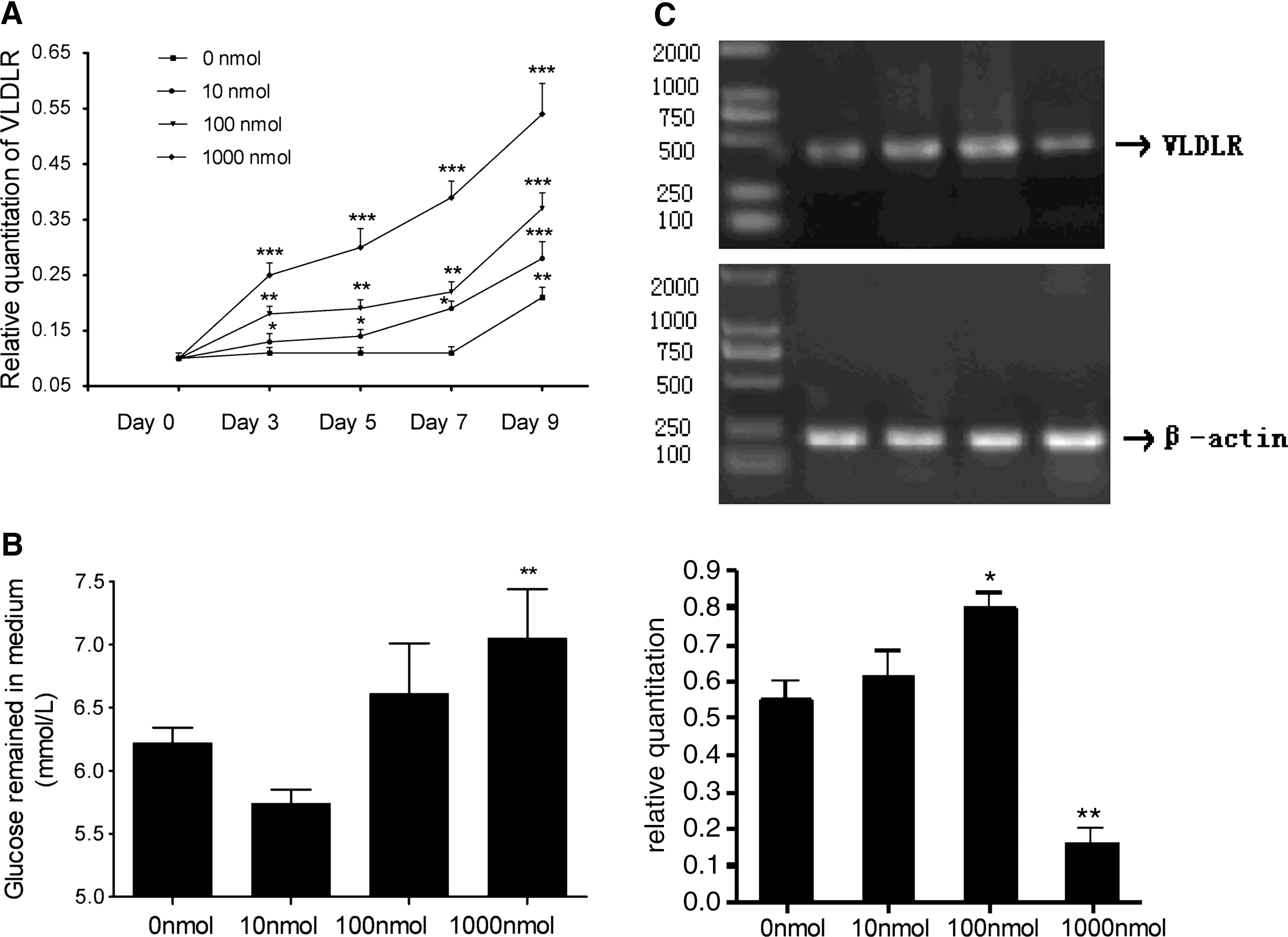
Effect of insulin on the expression of VLDLR during 3T3-L1 cell differentiation and in mature adipocytes. (
To investigate the relationship between insulin resistance and VLDLR expression, we prolonged insulin treatment of 3T3-L1 adipocytes to induce an insulin-resistant state (Ricort et al., 1995). Various concentrations of insulin (0, 10, 100, and 1000 nmol/liter) were added directly to mature adipocytes. Four days later, the amount of glucose remaining and the expression of VLDLR mRNA were tested. As seen in Fig. 3B, the medium concentration of glucose remaining was decreased by insulin at 10 nmol/liter, but significantly increased by insulin at 1000 nmol/liter, which suggested that long-term high concentrations of insulin induced 3T3-L1 adipocytes to become insulin resistant for glucose transport. The expression of total VLDLR mRNA was quantified by RT-PCR. As shown in Fig. 3C, in cells treated with insulin at 100 nmol/liter, the expression of VLDLR mRNA was significantly increased. By contrast, in cells treated with insulin at 1000 nmol/liter, VLDLR mRNA level was significantly lower than in control cells, suggesting that the decrease in VLDLR mRNA level was associated with insulin resistance.
Treatment of diabetic rats by intravenous injection of rAAV·VLDLR
To evaluate the effect of the VLDLR gene on lipid metabolism, an equivalent dose of rAAV·VLDLR or rAAV·LacZ (1 × 1011 particles per rat) was injected intravenously via the tail vein into diabetic rats. Results showed that basal plasma cholesterol and triglyceride levels were the same in the two groups, but that rAAV·VLDLR treatment induced a marked hypolipidemic effect 2 weeks after gene delivery compared with control rAAV·LacZ treatment (Fig. 4A and B).
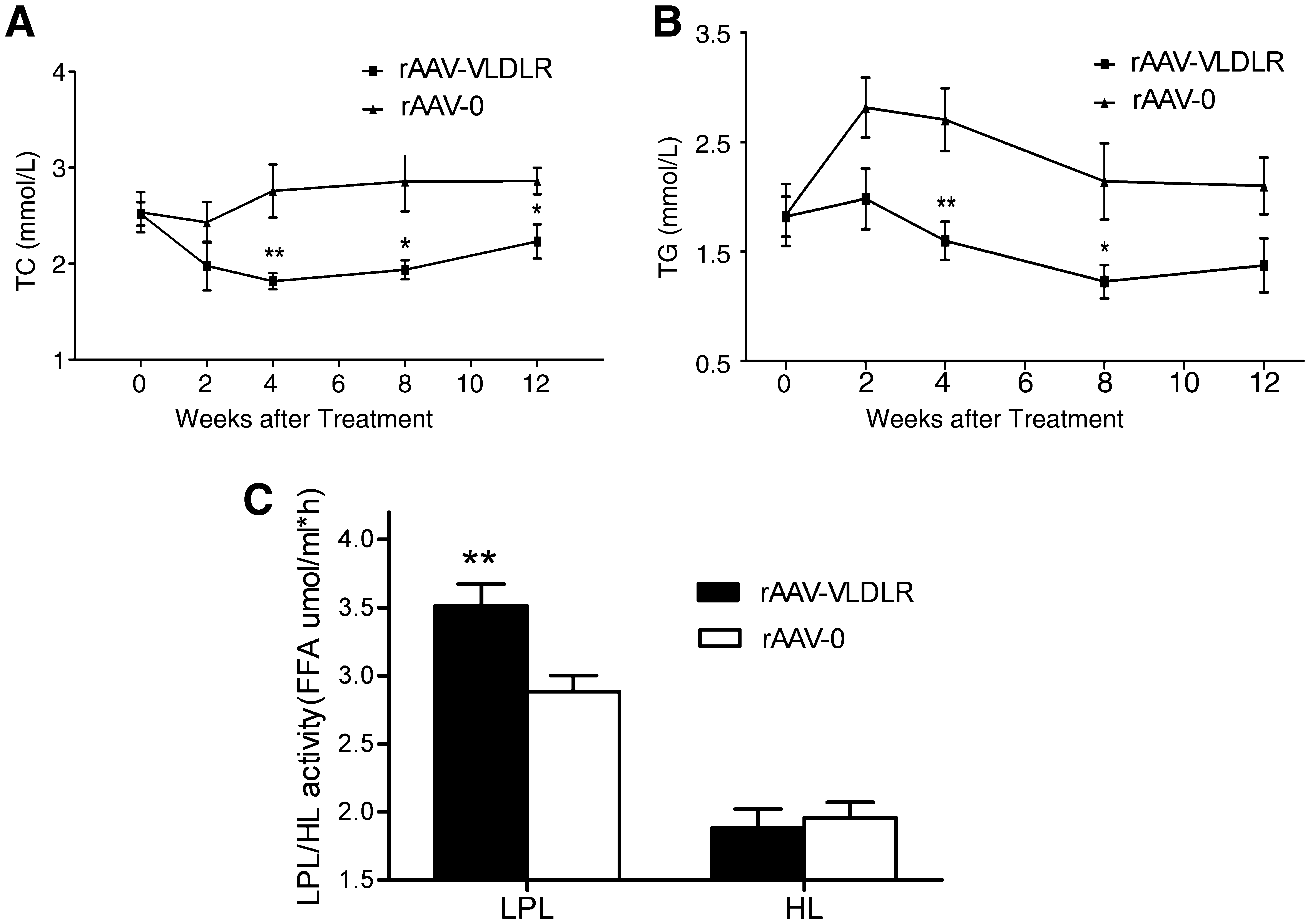
Altered lipid metabolism due to rAAV-VLDLR injection. (
Yagyu and colleagues (2002) reported that the VLDLR is required for normal LPL regulation in vivo, and the that disruption of the VLDLR results in hypertriglyceridemia associated with decreased LPL activity. Accordingly, we tested postheparin plasma LPL activity. As seen in Fig. 4C, infection with rAAV·VLDLR led to a significant increase in LPL activity compared with control rAAV·LacZ treatment. There was no difference in hepatic lipase activity between the two groups. These results suggest that increased plasma lipids in diabetic rats are due both to the changes in LPL activity and VLDLR expression. Overexpression of VLDLR may attenuate hyperlipidemia via increasing LPL activity.
Weight and fasting plasma glucose and insulin levels were also measured before rAAV injection and every 2 weeks thereafter. Insulin resistance was calculated by means of HOMA-IR. Results showed that fasting plasma glucose was higher in both the rAAV·VLDLR and rAAV·LacZ groups than in the normal group by the end of the study. Although the levels of fasting blood insulin were significantly lower in the rAAV·VLDLR group than in the rAAV·LacZ group, the levels of fasting blood glucose were not significantly different between the two groups, suggesting that less insulin is required in the VLDLR group than in the LacZ group to maintain the same glucose level. Likewise, insulin resistance was significantly attenuated in the rAAV·VLDLR group compared with the rAAV·LacZ group (Fig. 5A–D).
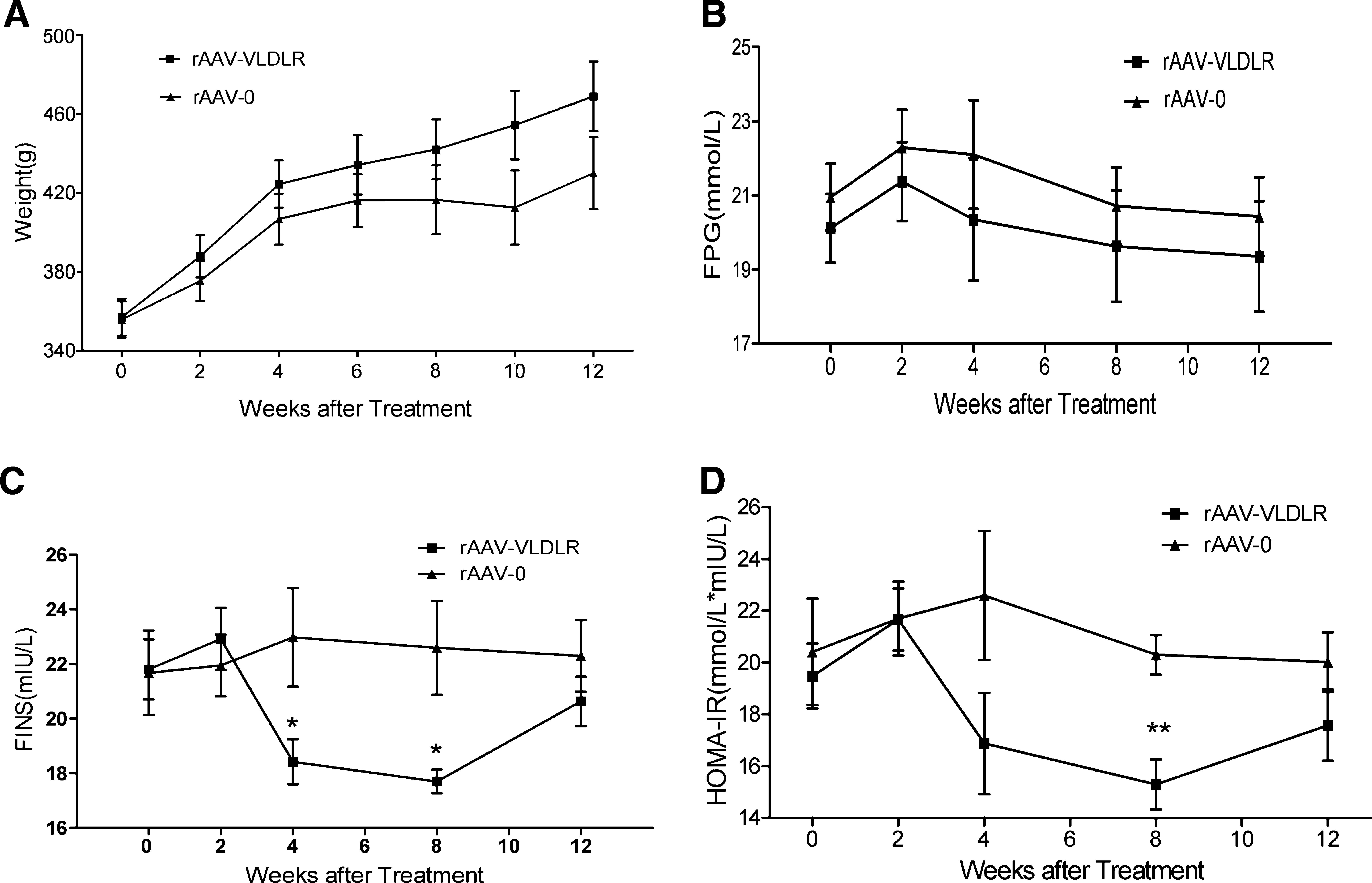
Weight, fasting plasma glucose, insulin, and HOMA-IR in diabetic rats treated with the VLDLR gene (rAAV-VLDLR) or LacZ gene (rAAV-0) at a starting time point before treatment (0 weeks) and every 2 weeks after treatment. FPG, fasting plasma glucose; FINS, fasting insulin; HOMA-IR, homeostasis model assessment of insulin resistance. Values represent means ± SEM (n = 8). *p < 0.05, **p < 0.01 versus the rAAV-0 group.
Atherosclerotic lesion development was analyzed in the aortic root 12 weeks after rAAV delivery (Fig. 6). All rats in all three groups have no visible macroscopic dorsal xanthoma. When observed by microscopy, fatty foam cells and atherosclerotic plaque could be seen in the diabetic rAAV·LacZ rats. By contrast, aortas of rAAV·VLDLR diabetic rats were nearly normal.
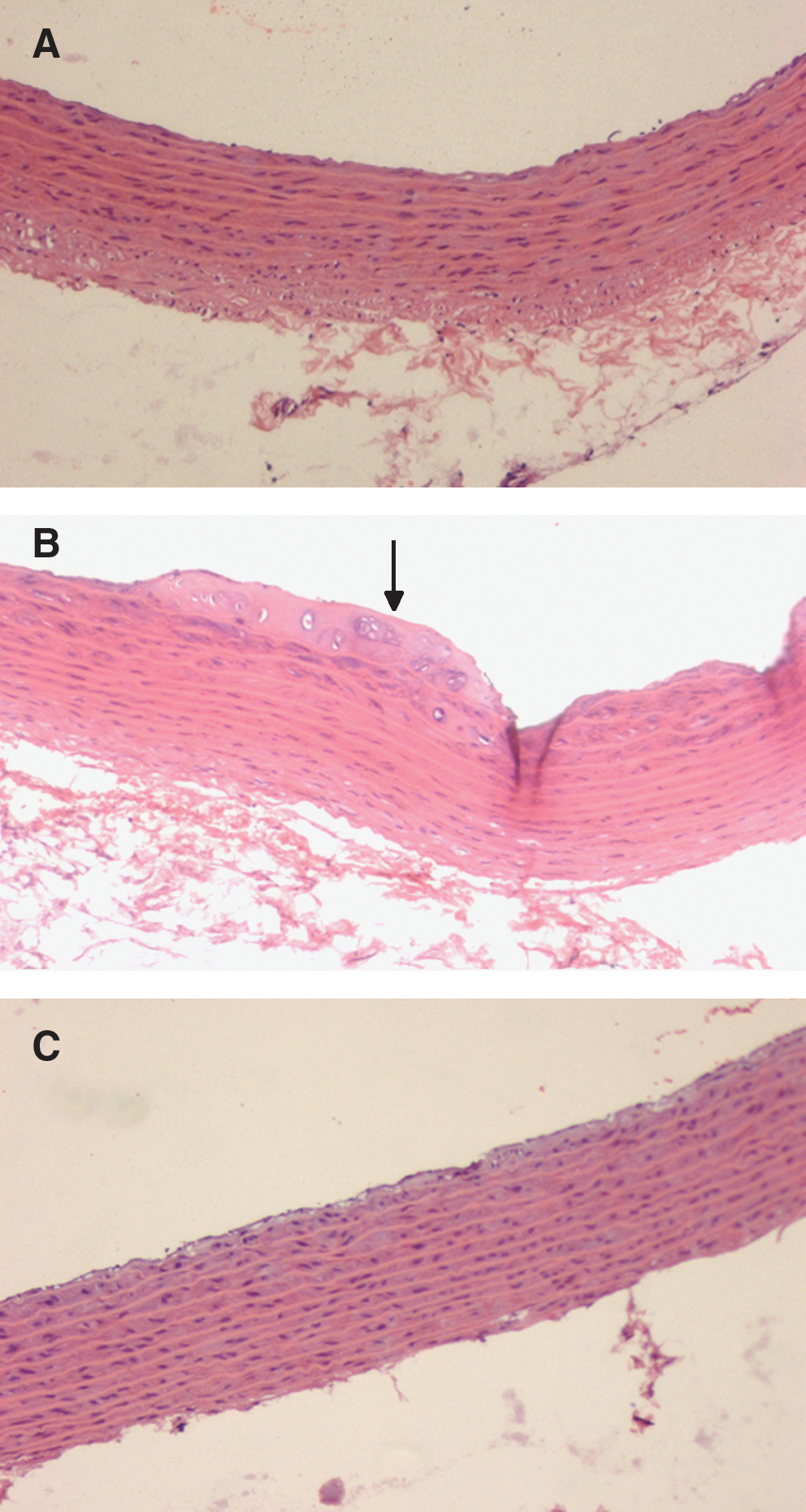
Histological analysis of the aortic root. (
Discussion
The purpose of this study was to gain insight into the role of the VLDL receptor in type 2 diabetes. The results of this study raise the possibility that hyperlipidemia in type 2 diabetic rats may be caused in part by reduced VLDLR expression in skeletal muscle and adipose tissues, in addition to decreased LPL activity. The mechanism of reduced VLDLR expression may be explained in part by insulin resistance. Overexpression of VLDLR by a single injection of rAAV·VLDLR caused significant reductions in plasma cholesterol and triglyceride in diabetic rats, and consequently the regression of aortic atherosclerotic plaque.
The VLDLR is most abundantly expressed in heart, skeletal muscle, and adipose tissue, but not in liver (Oka et al., 1994a). VLDLR expression is also localized in endothelial and smooth muscle cells of arteries and veins (Multhaupt et al., 1996). Because heart and skeletal muscle use fatty acids as an energy source, and adipose tissue uses fatty acids for energy storage, the VLDLR was hypothesized to play a role in the delivery of fatty acids derived from VLDL-triglycerides to these peripheral tissues. To our knowledge, the role of VLDLR has not been reported previously in type 2 diabetes. We found that total VLDLR mRNA was reduced in skeletal muscle and adipose tissue in type 2 diabetic rats. In skeletal muscle, type I isoform VLDLR mRNA was decreased, whereas type II VLDLR mRNA was significantly decreased in adipose tissue. Total VLDLR mRNA and both types of VLDLR isoform were not changed in heart tissue. Reduction of VLDLR protein was associated with a reduction of VLDLR mRNA. Further, type II VLDLR protein was slightly decreased in skeletal muscle in diabetic rats, but type II VLDLR mRNA was not significantly changed. Last, postheparin plasma LPL activity was also reduced in diabetic rats.
Our data indicated that hyperlipidemia in type 2 diabetic rats might be in part due to decreased VLDLR protein in skeletal muscle and adipose tissue. Although the initial studies on VLDLR knockout mice (Frykman et al., 1995) revealed no alterations in lipoprotein profiles or in total plasma cholesterol, triglyceride, and free fatty acid levels as compared with wild-type mice, later studies (Tacken et al., 2000) showed that the VLDLR does affect serum triglyceride levels when specific conditions are applied (such as cross-breeding of VLDLR-deficient and VLDLR-overexpressing mice onto an LDLR-deficient background). Absence of the VLDLR was associated with elevated serum triglyceride levels under conditions of dietary stress. In contrast, overexpression of the VLDLR under similar conditions was associated with lowering of serum triglyceride levels. Goudriaan and colleagues (2004) also demonstrated a major role of the VLDLR among postprandial lipoproteins by enhancing LPL-mediated TG hydrolysis, rather than by mediating fatty acid uptake. These results indicate that the VLDLR plays an important role in VLDL-TG metabolism in heart, skeletal muscle, and adipose tissue under conditions of severe stress (fasting or a high-fat diet) or on an LDLR-deficient or ob/ob background. Iwasaki and colleagues (2005) also reported that the severe elevations of serum cholesterol and TG concentrations were accompanied by a deficiency of VLDLR protein in heart, skeletal muscle, and adipose tissue in conjunction with reduced postheparin plasma LPL activity in STZ rats. The VLDLR levels were dependent on insulin levels in a rat muscle cell line as well as in skeletal muscle in insulin-deficient STZ rats. Because insulin resistance also played a crucial role in addition to decreased insulin secretion in type 2 diabetes, we studied the effect of insulin resistance on VLDLR expression in vitro. Our data indicated that the expression of VLDLR mRNA was increased by insulin in a concentration- and time-dependent manner during 3T3-L1 cell differentiation and that insulin resistance decreased VLDLR mRNA expression in mature adipocytes.
The possibility of VLDLR gene treatment in familial hypercholesterolemia (FH) has been investigated in several studies. Kazuhiro and colleagues reported that intravenous delivery of helper-dependent adenoviral vector carrying the VLDLR gene produces long-term lowering of plasma cholesterol and prevents atherosclerosis development in ldlr-deficient mice. MacDougall and colleagues also reported that aggressive VLDL/LDL lowering achieved by hepatic overexpression of VLDLR combined with a low-fat diet regimen induces regression of advanced plaques in the brachiocephalic artery of LDL receptor-deficient mice. However, a low-fat diet alone also decreased plasma cholesterol levels but cannot regress atherosclerotic lesions. Another study (Chen et al., 2000) delivering the VLDLR gene by adeno-associated viral vectors to the liver of a murine model of FH obtained the same result. With respect to the decreased VLDLR mRNA and protein levels in type 2 diabetic rats, we hypothesize that overexpression of VLDLR might result in reduction of serum cholesterol and triglyceride. In the present study, we used rAAV vectors to obtain stable gene transduction and long-term transgene expression. At the end of the experiments, RT-PCR and Western blotting showed that the expression of VLDLR mRNA and protein was increased in skeletal muscle and fat in rAAV-VLDLR-treated diabetic rats. After VLDLR treatment, high levels of VLDLR mRNA and protein were also detected in liver, where it is not endogenously expressed. Studies using adenoviral vectors (Kobayashi et al., 1996; van Dijk et al., 1998) have shown that ectopic expression of the VLDLR in mouse liver results in enhanced internalization of lipoproteins. Thus, when expressed in liver, the VLDLR appears to act as a clearance receptor for lipoproteins, similar to the action of the LDLR and LRP. However, studies that used VLDLR transgenic and knockout mice (Tacken et al., 2000) revealed that the VLDLR in the periphery affects primarily VLDL triglyceride content. This suggests that, at its natural site of expression, the VLDLR facilitates hydrolysis of triglycerides, rather than clearing entire lipoprotein particles. Our data show that both plasma cholesterol and triglyceride were significantly decreased by rAAV·VLDLR, which drives the VLDLR gene to express both in liver and peripheral tissues compared with untreated diabetic rats. In the present study, insulin resistance was also attenuated in rats treated with the VLDLR gene, as evaluated by HOMA-IR. Although the levels of fasting blood insulin were significantly reduced in the VLDLR-treated group versus the untreated group, the fasting glucose levels were not significantly different; this suggests that less insulin is required in the VLDLR group to maintain the same glucose level. This might be explained in part by decreased plasma lipid levels.
Our data also indicated that VLDLR gene delivery increased VLDLR expression as well as LPL levels. The VLDLR and LPL are present on the vascular wall in the same tissues, can bind to each other, and both appear to be involved in hydrolysis of triglycerides. The VLDLR might regulate LPL levels on the vascular wall by mediating transport of LPL derived from adipocytes and muscle cells from the basal to the luminal side of endothelial cells. LPL is reported to bind to LRP (Williams et al., 1994), the LDLR (Salinelli et al., 1996), megalin (Kounnas et al., 1993), and the VLDLR (Takahashi et al., 1995), thereby enhancing binding and internalization of lipoproteins. Deficiency of adipose tissue LPL in ob/ob mice results in a similar reduction in weight gain and adipose tissue mass as has been detected in VLDLR-deficient ob/ob mice. Goudriaan and colleagues reported that adipocyte triglyceride storage is reduced when the VLDLR is absent. In the present study, we find a slight increase in body weight after VLDLR overexpression, although not significantly different compared with the untreated group, possibly because the untreated high blood glucose level affects weight gain.
In summary, the elevation of serum cholesterol and TG concentration in type 2 diabetic rats was accompanied by reduced expression of VLDLR mRNA and protein in skeletal muscle and adipose tissues in conjunction with reduced postheparin plasma LPL activity. rAAV-mediated VLDLR gene delivery can efficiently lower elevated plasma cholesterol and TG concentrations as well as reduce the formation of aortic atherosclerotic plaque via increasing VLDLR expression in skeletal muscle, liver, heart, and adipose tissues and enhancing plasma LPL activity. These data suggest that the VLDLR might play a important role in diabetic hyperlipidemia.
Footnotes
Acknowledgment
This work was supported by a National Nature Science Foundation Committee grant (30400220, to G.Y.).
Author Disclosure Statement
No competing financial interests exist.