
Abstract
Both gene replacement therapy and alteration of host gene expression are playing increasingly important roles in the treatment of ocular diseases. Ocular gene therapy may provide alternatives to current treatments for eye diseases that are either greatly invasive and thus run the risk of complications, that offer only short-term relief from disease symptoms, or that are unable to directly treat vision loss. The success of three separate phase I clinical trials investigating a gene therapy intervention for the treatment of the retinal degenerative disorder Leber's congenital amaurosis (LCA) has unveiled the therapeutic potential of gene therapy. Preliminary results have demonstrated ocular gene transfer, using nonpathogenic recombinant adeno-associated viral (rAAV) vectors specifically, to be a safe, effective, and long-term treatment for LCA, a previously untreatable disorder. Nonpathogenic rAAV vectors offer the potential for long-term treatment. Many of the genes implicated in human ocular diseases have been identified, and animal models for such diseases have been developed, which have greatly facilitated the application of experimental rAAV-mediated gene therapy. This review highlights the key features of rAAV-mediated gene therapy that make it the most suitable gene therapy treatment approach for ocular diseases. Furthermore, it summarizes the current progress of rAAV-mediated gene therapy interventions/applications for a wide variety of ophthalmologic disorders.
Ocular Gene Therapy: An Introduction
The rAAV vector, a nonpathogenic, human parvovirus with a linear, single-stranded DNA genome, consists of two reading frames, rep and cap, enclosed between inverted terminal repeats (ITRs)—two symmetric T-shaped palindromic terminal sequences. rAAV vectors have helped overcome the challenges of efficiency and prolonged gene transduction, although they do present some limitations, given the slow onset of transgene expression after transduction and their small packaging capacity (Smith et al., 2009). However, the development of hybrid rAAV vectors has expanded the genomic size limitation. The limitations of the rAAV vector are far outweighed by its efficacy, and features intrinsic to the eye make it well suited as a target organ for rAAV-mediated gene therapy (Surace and Auricchio, 2008). The discovery of novel rAAV vector serotypes has given way to the aforementioned hybrid vectors, which demonstrate varied kinetics of transgene expression and improved tropism for a broad range of ocular cell types (Martin and Quigley, 2004; Smith et al., 2009) (Fig. 1). The efficiency of ocular vector transduction depends on a number of factors, including the well-established specificity of ocular rAAV-mediated gene therapy, the various routes of vector administration (Figs. 2 and 3), and the promoter sequence of the vector (Martin and Quigley, 2004). Enhancement of rAAV vector transduction efficiency at low doses is also possible (Zhong et al., 2008). Table 1 summarizes rAAV-mediated gene therapies for ocular diseases.
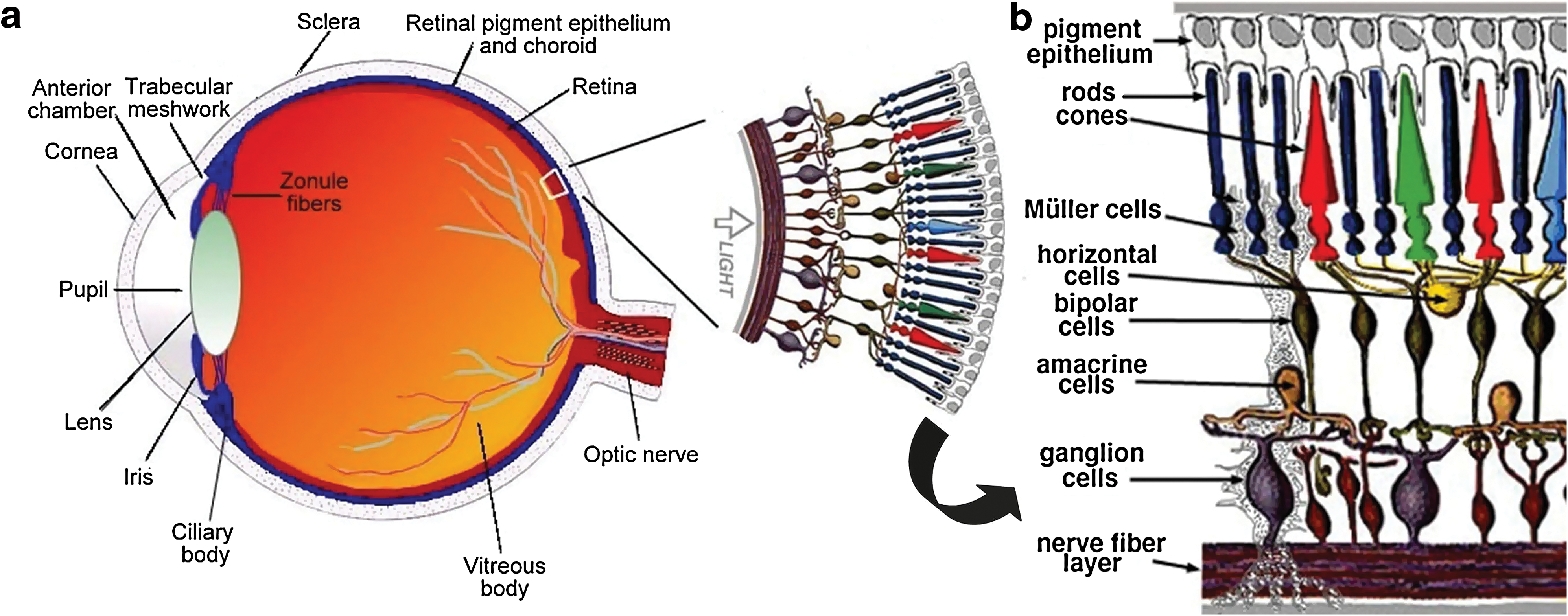
Structure and function of the eye. (
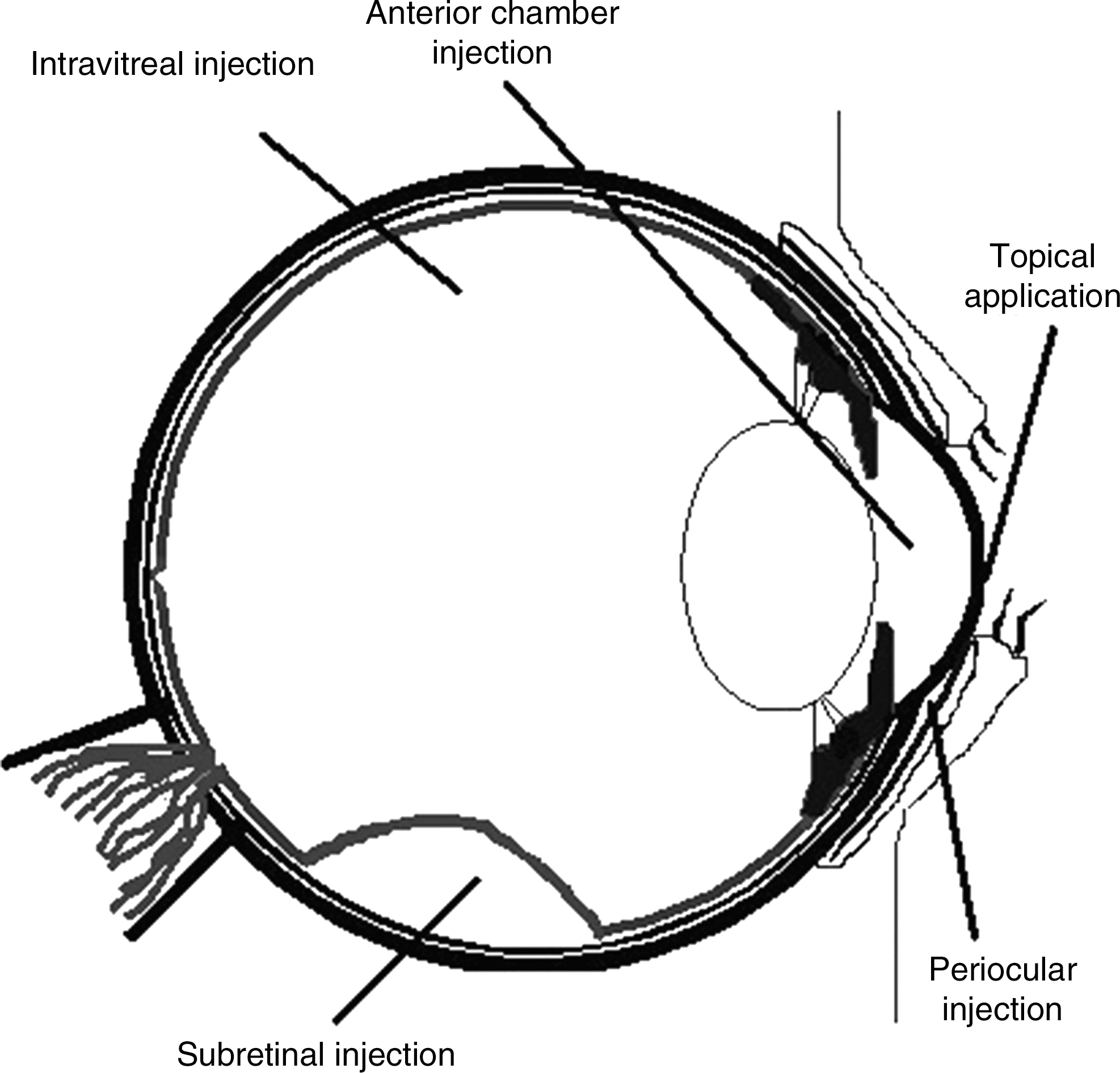
Administration routes for rAAV vector delivery. The route through which rAAV vector will be delivered depends on which of the numerous ocular cell types it will transduce. rAAV vector used to transduce photoreceptors and retinal pigment epithelial cells is administered via subretinal injection; cells of the inner retina, including retinal ganglion, intravitreal injection; cells of the conjunctiva and corneal epithelium, topical application; cells involved in neovascular and corneal diseases, periocular injection; and cells of the corneal endothelium, anterior chamber injection (Hauswirth and Beaufrere, 2000; Jun and Larkin, 2003; Bainbridge et al., 2006; Cheng et al., 2007; Surace and Auricchio, 2008; Colella et al., 2009).
Administration of rAAV2-CB-hRPE65 by subretinal injection to a clinical trial patient with Leber's congenital amaurosis (LCA). Color images available online at
rAAV-Mediated Gene Therapy for Corneal Diseases
The cornea is a transparent tissue chiefly involved in protecting the structure and function of the eye. As the outermost ocular structure, it protects the eye from any physical and pathogenic injury due to the external environment. It also plays an immunoprotective role through its expression of inhibitors that prevent activation of proinflammatory factors, as well as through its secretion of cytokines. The protective function of the cornea is regulated by the five cellular layers that comprise the cornea, including the epithelium, Bowman's membrane, stroma, Descemet's membrane, and endothelium. Direct accessibility of the corneal epithelium to the external environment facilitates more convenient vector delivery methods for gene therapy, such as noninvasive, topical administration; however, invasive vector delivery methods, including injection into the anterior chamber, have yielded greater transduction efficiencies (Klausner et al., 2007). Although gene therapy holds great potential in the treatment of inherited corneal endothelial diseases, as well as in the prevention of corneal allograft rejection (Tsai et al., 2002), the most extensive research concerning rAAV-mediated gene therapy in the cornea has been conducted in experimental models of acquired corneal endothelial disorders.
Acquired corneal diseases
Ocular neovascularization is a threatening condition in all of the tissues of the eye that it can affect, and particularly so in the avascular cornea. The transparent and immunoprotective nature of the cornea is compromised in corneal neovascularization, which can be induced by a wide variety of factors including inflammation, infection, and degeneration, and by both direct trauma to the cornea or indirect trauma to the limbus structure that borders it. Treatment approaches focus on the regulation of corneal angiogenesis to counter the development of new blood vessels from preexisting pathological vasculature. The homeostatic mechanism behind the regulation of corneal angiogenesis involves both pro- and antiangiogenic factors, many of which have been used in conjunction with rAAV-mediated gene therapy approaches. Specifically, the rAAV-mediated gene transfer of angiogenesis inhibitors, such as angiostatin and endostatin, has been shown to reduce and even inhibit corneal neovascularization (Cheng et al., 2007; Lai et al., 2007).
Cheng and colleagues observed a regression in corneal neovascularization due to experimental alkali burn-induced corneal angiogenesis in rats given a subconjunctival injection of rAAV-angiostatin. After establishing efficient rAAV-mediated transduction by expression of the GFP transgene, which encodes green fluorescent protein (GFP), rats were divided into two experimental groups and treated with either a blank rAAV control vector or an rAAV-angiostatin vector. In both cases, the viral vector was administered by subconjunctival injection 3 weeks before the induction of alkali burn-induced corneal angiogenesis. Corneal neovascularization was observed 1 week after injury induction and subsequently quantified by calculating the area of marked neovascular engorgement. The prolonged exposure of the limbal and conjunctival vasculature (the site at which corneal neovessels originate) to transgene expression was determined to account for the observed regression in corneal neovascularization of the rAAV-angiostatin-treated group (Cheng et al., 2007).
The corneal endothelium has presented challenges for gene therapy in terms of both efficient and prolonged gene transduction, which rAAV-mediated gene transfer has been able to overcome. Until more recently, however, in vivo regulation of transgene expression proved difficult even for rAAV-mediated gene therapy. In 2002, Tsai and colleagues demonstrated prominent transgene expression in the corneal endothelium in vivo when induced by inflammation. After pretreatment with rAAV vector containing the LacZ reporter gene, inflammation was induced by intravitreal injection of lipopolysaccharide (LPS) into the ocular anterior chambers of New Zealand White rabbits. A direct correlation was observed between transient ocular anterior segment inflammation induced by the lipopolysaccharide injection and increases in LacZ gene expression in the rabbit corneal endothelial cells. Results revealed a peak in inflammation 1 day after LPS injection; the inflammation concurrently activated transgene expression of LacZ in approximately 90% of corneal endothelial cells. Furthermore, a second LPS injection, given 60 days after the first, elicited a dramatic reactivation of transgene expression to levels once again nearing 90% of endothelial cells, even after transgene expression diminished as inflammation subsided after the first LPS injection. This finding of increased transgene expression after transient LPS-induced inflammation has expanded the implications of rAAV-mediated gene therapy for the treatment of other acquired corneal diseases including keratitis, anterior uveitis, and corneal graft rejection (Tsai et al., 2002).
In a more recent study by Tsai and colleagues, the effect of cell-specific and inducible expression systems on the level and timing of transgene expression in the treatment of experimental uveitis was investigated. Uveitis is a recurrent, intraocular inflammatory condition that can severely compromise vision. In previous gene therapy studies investigating the therapeutic potential of interleukin-1 receptor antagonist (IL-1Ra) as a treatment for uveitis, the efficiency of intraocular gene transfer by the chosen vector delivery systems was limited. Therefore, Tsai and colleagues employed the use of an rAAV vector encoding IL-1Ra cDNA to elicit transgene expression in the eyes of New Zealand White rabbits. The rAAV-IL-1Ra vector, as well as a control vector encoding the LacZ reporter gene, were administered intravitreally. The therapeutic potential of rAAV-IL-1Ra was assessed after induction of experimental uveitis by intravitreal injection of rAAV-IL-1α at both 10- and 100-day time points after rAAV-IL-1Ra delivery. Using methods of immunohistochemistry, ELISA, and RT-PCR, Tsai and colleagues witnessed recovery from experimental uveitis by transgene expression after a single administration of rAAV-IL-1Ra at both the 10- and 100-day time points (Tsai et al., 2009).
The demonstration of efficient rAAV-mediated transduction of corneal cells under both in vivo and ex vivo conditions has broadened the possible applications of rAAV-mediated gene therapy in corneal diseases to include inherited, iatrogenic, and metabolic diseases of the cornea (Liu et al., 2008). Work done by Liu and colleagues (2008) encompassed an investigation of the tropism of a variety of rAAV serotype vectors in organ-cultured human corneas. The efficiency of transduction was marked by expression of the GFP reporter gene, which was delivered via rAAV vectors. The observed transduction was extensive, reaching a variety of corneal cells including those of the epithelium and endothelium, and keratocytes. Their findings hold great promise for treating a wide spectrum of corneal disorders ranging from a group of corneal dystrophies in which the causative gene has been deduced to be a single mutation in the transforming growth factor-β-induced gene TGFBI/BIGH3, to other diseases in which the genetic components remain either unknown or are multifaceted. As demonstrated by the work of Liu and colleagues (2008), rAAV-mediated gene therapy can be used to target specific corneal cells, in which it can alter the expression levels of known mutant genes.
rAAV-Mediated Gene Therapy for Optic Neuropathies
The optic nerve is a unique ocular tissue because it originates in the eye yet functions in the nervous system by carrying the electrical impulses it receives from the retina to the brain. The axons of retinal ganglion cells converge into fiber bundles along the base of the inner retina, forming the optic nerve. Optic neuropathies encompass all conditions in which the optic nerve incurs any damage. Ocular rAAV-mediated gene therapy for the optic nerve involves both gene replacement and gene addition approaches, to which optic neuropathies such as Leber's hereditary optic neuropathy and glaucoma are amenable (Martin and Quigley, 2004; Colella et al., 2008).
Inherited optic neuropathies
Leber's hereditary optic neuropathy
Leber's hereditary optic neuropathy (LHON) is a common mitochondrial disease characterized by staggered, bilateral vision loss. As a maternally inherited disease, LHON primarily affects men during early adulthood. The disease is predominantly caused by three separate point mutations in genes that encode the subunits of nicotinamide adenine dinucleotide:ubiquinone oxidoreductase, or complex I, an enzyme involved in the oxidative phosphorylation pathway (Martin and Quigley, 2004). Guy and colleagues implemented rAAV-mediated gene therapy to replace the G11778A mutation, which encodes the ND4 subunit of complex I, and accounts for approximately 50% of all LHON cases. They used an allotopic expression system for rAAV-mediated gene transfer of this G11778A mutation to the mitochondrial genome, because as a viral vector, the rAAV vector cannot directly transfer exogenous genes to the mitochondrial genome. The allotopic expression system expresses a nuclear-encoded version of a mitochondrial gene that encodes a cytoplasmically expressed protein tagged with a mitochondrial-targeting peptide (Guy et al., 2002; Martin and Quigley, 2004). Although Guy and colleagues were successful in restoring the cellular respiration deficit caused by LHON in vitro, an animal model for LHON does not currently exist, which limits investigation of the rAAV-mediated allotopic ND4 gene therapy approach in vivo.
Optic neuritis
Optic neuritis is a condition that causes inflammation of the optic nerve. People suffering from multiple sclerosis, an autoimmune disease of the central nervous system, are often susceptible to progressive visual loss due to recurrent episodes of optic neuritis. Specifically, in optic neuritis, autoimmune-mediated oxidative injury targets oligodendrocytes, cells that function as neuroglia in the central nervous system and produce the myelin sheath that surrounds retinal ganglion cell axons. The axonal demyelination that results in optic neuritis contributes to axonal degeneration and subsequent neuronal degeneration. Qi and colleagues, in their extensive work, have used the experimental autoimmune encephalomyelitis (EAE) rat model of multiple sclerosis in conjunction with rAAV-mediated gene therapy to probe the mechanisms that underlie the autoimmune-mediated oxidative injury observed in optic neuritis. On the basis of prior findings elucidating the damage-inducing role of reactive oxygen species (ROS), including superoxide and hydrogen peroxide in the EAE disease model (Guy et al., 1998), Qi and colleagues used the ROS scavengers superoxide dismutase (SOD) and catalase to develop an antioxidant gene therapy approach (Qi et al., 2007b).
ROS are inducers of optic nerve demyelination and have also been found to interfere with the permeability of the blood–brain barrier in EAE. ROS scavengers counteract the oxidative damage done by ROS, with SOD functioning in the dismutation of superoxide to hydrogen peroxide. Catalase detoxifies hydrogen peroxide to water and oxygen (Qi et al., 2007a,b). The cDNAs for extracellular superoxide dismutase (ECSOD) and catalase were cloned into rAAV vectors, which were used to infect retinal ganglion cells of the rat EAE model by intravitreal injection. Relevant findings after transgene expression, using both the rAAV-ECSOD and rAAV-catalase vectors, included decreases in retinal ganglion cell loss by 29%, optic nerve demyelination by 36%, and axonal loss by 44% (Qi et al., 2007a). Earlier results obtained by Qi and colleagues also demonstrated an up to 78% reduction in optic nerve demyelination in the EAE model (Qi et al., 2007b). The promising results elicited by the rAAV-mediated gene transfer of extracellular superoxide dismutase and catalase serve to demonstrate the great therapeutic potential as well as highlight the advantageous features of rAAV-mediated gene transfer for optic neuritis. rAAV-mediated gene transfer allows for a direct treatment approach, as antioxidant genes were delivered directly to the oligodendrocytes, whereas previous treatment approaches, such as catalase protein delivery, have been limited by incomplete penetration through the blood–brain barrier. In addition, the long-term transgene expression enabled by rAAV-mediated delivery of the ROS scavenger genes promoted long-term suppression of optic nerve demyelination and subsequent axonal degeneration.
Acquired optic neuropathies
Optic nerve trauma
Optic nerve trauma can result from any condition in which the retinal ganglion cells undergo axotomy, including transection of the optic nerve, which can ultimately lead to both neuronal and retinal ganglion cell death. Retinal ganglion cells experiencing optic nerve trauma are deprived of brain-derived neurotrophic factor (BDNF), on which they depend for survival. To better understand how optic nerve trauma can be treated, experimental optic nerve transection models have been developed (Martin and Quigley, 2004). rAAV-mediated gene transfer has been used as part of the treatment approach for optic nerve transection in a murine model. The ability of the rAAV vector to sustain long-term gene transduction proved advantageous for the work of Cheng and colleagues, who investigated the effect of upregulation of TrkB expression on retinal ganglion cell survival. Cheng and colleagues used rAAV-mediated gene therapy to transfer the gene encoding TrkB, a BDNF receptor expressed by retinal ganglion cells, to the retinal ganglion cells in an optic nerve transection rat model. This group also supplemented TrkB transgene expression with direct administration of BDNF to the TrkB receptors. An increase in neuronal survival after optic nerve transection was observed (Cheng et al., 2002).
Glaucoma
Glaucoma is a progressive optic neuropathy that likely results from the interaction of multiple genetic and environmental factors. Although the predisposing genetic factors of glaucoma remain largely unknown, mutations in the gene encoding the myocilin protein have been found to cause autosomal dominant juvenile primary open-angle glaucoma, as well as nearly 3% of adult-onset open-angle glaucoma cases (Borrás et al., 2002). Many factors, including elevated intraocular pressure (IOP), can predispose an individual to developing the optic nerve head damage and eventual death of retinal ganglion cells characteristic of glaucoma. The implications for rAAV-mediated gene therapy in the treatment of glaucoma are wide-ranging, given that gene therapy targets for glaucoma can range from structures to cell types, including the trabecular meshwork, ciliary body, retinal ganglion cells, and Müller cells (Borrás et al., 2002).
Elevated intraocular pressure, which is widely known as the hallmark of glaucoma, is often accompanied by the accumulation of BDNF and its TrkB receptor at the optic nerve head. BDNF transport from the brain to the retinal ganglion cells of the inner retina is therefore interrupted, leading to BDNF deprivation of the retinal ganglion cells, and subsequent neuronal and retinal ganglion cell death. Previous work conducted by Ko and colleagues (2000) demonstrated the limited survival of retinal ganglion cells in an experimental rat model of glaucoma after intravitreal injection of BDNF in conjunction with an intraperitoneal injection of a nonspecific free radical scavenger. Recognizing the limitations of multiple intravitreal injections of BDNF, however, Martin and Quigley used rAAV-mediated gene transfer to transfect retinal ganglion cells, also in a rat model of glaucoma. Using intravitreal delivery of rAAV-BDNF, an rAAV vector in which the cDNA for BDNF was enclosed, a 38% rescue of retinal ganglion cells from BDNF deprivation was witnessed (Martin and Quigley, 2004). The ability of rAAV-BDNF to slow the rate of retinal ganglion cell death, and thereby the overall progression of glaucoma, in a rat model highlights the relevance of rAAV-mediated gene therapy as a potential treatment for both polygenic diseases and those with unknown etiologies.
Another rAAV-mediated gene therapy approach implemented for the treatment of experimental glaucoma involves caspase inhibitors. Activated caspase enzymes are intrinsic to the initiation and regulation of apoptosis in retinal ganglion cells, specifically caspase-8 and caspase-3. McKinnon and colleagues investigated the role of modulating the activation of these caspase enzymes in increased retinal ganglion cell survival and subsequent optic nerve survival. The gene encoding baculoviral IAP (inhibitor of apoptosis protein) repeat-containing protein-4 (BIRC4), a potent caspase inhibitor, was packaged into an rAAV vector and delivered to rat eyes by unilateral, intravitreal injection (McKinnon et al., 2002). Ocular hypertension was then induced in the treated rat eyes to simulate the elevated intraocular pressure characteristic of glaucoma. After a 12-week exposure to increased intraocular pressure, the rat optic nerve axons pretreated with rAAV-BIRC4 were counted and compared with balanced salt solution-treated control groups. On average, 50% of the optic nerve axons in rat eyes expressing the BIRC4 transgene had been protected, compared with the control glaucoma eyes. The survival of retinal ganglion cells by rAAV-mediated transgene expression of the BIRC4 caspase inhibitor demonstrates the therapeutic potential of interrupting apoptosis to treat glaucoma. As a chronic optic neuropathy, however, glaucoma likely requires transgene expression for longer than even the year-long expression that rAAV-mediated gene therapy can currently permit (McKinnon et al., 2002; Martin and Quigley, 2004).
rAAV-Mediated Gene Therapy for Retinal Degenerative Disorders
The retina is the ocular structure predominantly involved in generating vision, converting light into electrical impulses and transmitting these signals to the brain. Therefore, the most severe forms of visual impairment are generally attributed to disorders in which the retina is implicated. The hallmark of all retinal degenerative disorders is the progressive apoptotic loss of the rod and/or cone photoreceptor cells of the retina. Nearly all retinal degeneration is either inherited or gene-based, making gene replacement therapy for such ocular diseases a potentially viable treatment option. Ocular gene therapy approaches in the retina include gene replacement, gene silencing, and gene addition, all of which target defective genes encoding the expression of proteins vital to photoreceptor function (Colella et al., 2008; Smith et al., 2009). The timing of vector and/or gene delivery is significant in rAAV-mediated gene therapy for retinal degenerative disorders, because retinal degeneration can vary from early and severe to late and progressive. The retinal dystrophies that progress the fastest and have an earlier onset are the most difficult to treat, whereas slowly progressing degeneration has a wider therapeutic window (Surace and Auricchio, 2008; Smith et al., 2009). Certain retinal degenerative disorders have been found to be more amenable to treatment than others, such as Leber's congenital amaurosis, retinitis pigmentosa, and age-related macular degeneration. Retinal degenerative disorders affect various regions of the retina, which spans nearly the entirety of the interior ocular circumference; therefore the retina can be divided into the peripheral retina and the central retina.
Inherited degenerative disorders of the peripheral retina
Leber's congenital amaurosis
Leber's congenital amaurosis (LCA) is one of the most severe forms of an early-onset, inherited retinal degeneration and one of the most extensively studied disease models for retinal gene therapy. There are multiple forms of LCA that all share a common disease progression, featuring the onset of severe visual impairment from birth and a complete loss of vision by early adulthood (Smith et al., 2009). Mutations identified in at least 12 different loci, including those in the RDH12, RPGRIP, LRAT, and RPE65 genes, are currently thought to account for 50% of LCA cases. Mutations in RPE65, in particular, which cause a deficiency in production of the RPE65 enzyme, have provided the most successful example of gene therapy intervention in the treatment of an ocular disease. The RPE65 protein is localized in the retinal pigment epithelium and functions in visual cycle regulation by converting all-trans-retinoids to 11-cis-retinoids. RPE65 deficiency interrupts this process and causes rod photoreceptors to become dysfunctional, leading to photoreceptor degeneration. Animal models of LCA, such as the RPE65-deficient murine model, have greatly facilitated experimental intervention by rAAV-mediated gene replacement. More significant, however, were the results of rAAV-mediated gene therapy in the larger, spontaneous RPE65-null model of LCA in the Briard dog, which demonstrated a persistent improvement in vision over an 8-year period after only a single administration of rAAV vector. Rod photoreceptor function was restored after subretinal injection of either the rAAV2- or rAAV4-vectored canine RPE65 gene (Colella et al., 2008; Smith et al., 2009).
The promising results of preclinical research helped to launch LCA as the first ocular disease treated in gene therapy clinical trials. Since 2007, the safety and efficacy of rAAV-mediated gene therapy for treatment of the RPE65-deficient form of LCA have continued to be investigated in three separate phase I clinical trials. In each of the three clinical trials, a subretinal injection of rAAV2/2-vectored RPE65 was administered to three RPE65-deficient LCA patients, who were between the ages of 17 and 26 years and suffered from various degrees of visual impairment due to LCA. In addition, procedural differences between each of the three trials featured divergence in promoter type, either an RPE-specific RPE65 or chicken β-actin (CBA) promoter; in surgical protocols; and also in volume of rAAV vector used for injection. Changes in vision due to rAAV-mediated treatment in LCA patients compared with control patients were also evaluated by a variety of measures including pupillometry, microperimetry, and acuity testing (Bainbridge et al., 2008; Hauswirth et al., 2008; Maguire et al., 2008). Despite these differences, all three phase I trials revealed improvements in retinal sensitivity. Although preclinical experimentation suggested the greater amenability of younger subjects to rAAV-mediated gene therapy, the success of the three independent phase I trials has provided even stronger evidence of the need to investigate the specific therapeutic window for rAAV-mediated gene replacement in LCA patients (Bainbridge and Ali, 2008; Colella et al., 2008; Smith et al., 2009).
During the 1-year follow-up for the first LCA clinical trial to become a phase 1/2 safety and efficacy trial, none of the patients involved experienced adverse effects, and one patient demonstrated an unexpected gain in visual function. To better understand this patient's improved vision, investigators quantified the patient's foveal fixation in response to a series of dim targets that were contrasted with a range of luminances. One eye was used as a control, whereas the other eye was treated by rAAV-mediated gene therapy. Results revealed that foveal fixation was similar in both eyes; however, a shift in foveal fixation to the region of the retina occurred in the treated eye. The researchers surmise that gene therapy rescue of cone photoreceptors at the superotemporal region of the retina formed a pseudo-fovea, which accounted for the increased cone function and improved vision. This latest finding has introduced yet another application of rAAV-mediated gene therapy in the treatment of congenital blindness (Cideciyan et al., 2009).
Retinitis pigmentosa
Retinitis pigmentosa (RP) is one of the most prevalent inherited retinal diseases, affecting nearly 1 in 3000, and has a variety of inheritance patterns including dominant, recessive, and X-linked (Song et al., 2007). Initially, affected individuals present with night-blindness due to degeneration of their rod photoreceptor cells, which can lead to tunnel vision. Complete vision loss in an affected individual can arise once cone photoreceptor cells degenerate. Many of the genes encoding the numerous proteins involved in the phototransduction pathway are susceptible to mutation and lead to the defects in phototransduction in RP. For example, the X-linked form of RP, which accounts for 15–20% of all RP cases, is caused by a defect in the retinitis pigmentosa GTPase regulator (RPGR) gene. The RPGR protein product is thought to regulate the protein distribution in the connecting cilium by directing or restricting protein transport to the photoreceptor outer segment. Interestingly, this pathway is also implicated in the form of LCA caused by a defect in the RPGRIP gene by mislocalizing the RPGR protein, which has been found to anchor to the connecting cilium (Song et al., 2007; Smith et al., 2009).
The rapid degeneration of rod photoreceptors in the autosomal recessive form of RP is caused by null mutations in several genes encoding proteins involved in both phototransduction and photoreceptor outer segment regulation. In the murine model of autosomally recessive retinal degeneration, PDE6Brd1 is an allele of the gene that encodes the β subunit of the rod cGMP phosphodiesterase (βPDE), an enzyme vital to the phototransduction cascade. Meanwhile, another frequently used model for autosomal recessive RP is in mice containing a spontaneous mutation in the gene encoding the MER protein tyrosine kinase (MERTK), which is localized in the retinal pigment epithelium and is required for the phagocytosis of photoreceptor debris. Many other spontaneous retinal degenerative animal models exist for recessively inherited RP, which have all contributed to the understanding of autosomally inherited RP progression (Song et al., 2007; Smith et al., 2009).
While there are many challenges to treating early-onset, rapidly degenerating retinal dystrophies such as the X-linked and recessive forms of RP with gene replacement therapy, the mutational heterogeneity of autosomal dominantly inherited diseases provides the greatest challenge to implementing rAAV-mediated gene therapy, which has extensive potential as a treatment for monogenic diseases. Unlike the aforementioned primary gene defects for the X-linked and autosomal recessive forms of RP, no single causative gene has been identified for autosomal dominant RP. Rather, more than 200 mutations in the RHO gene, which encodes the photoreactive pigment absorbed by rod photoreceptor cells (rhodopsin, or RHO), have been found to account for the dominantly inherited form of RP. The complexity of treating dominantly inherited retinal disorders lies in the fact that several mutations in one gene can cause the same RP disease phenotype. Therefore, implementing a treatment that is independent of the mutation and that can still correct the resulting defect has been the focus of investigative efforts. Specifically, the successes in the implementation of gene replacement with gene suppression have renewed the possibilities for rAAV-mediated gene therapy in the treatment of RP (Chadderton et al., 2009). Although gene knockout strategies have long been used for gene suppression, the development of RNA interference (RNAi) technology has introduced the possibility for gene knockdown using short hairpin RNA (shRNA) and small interfering RNA (siRNA).
The limited assessment of retinal function afforded by earlier studies focusing on both mutation-specific and mutation-independent suppression of autosomal dominant RP in mouse models using either ribozymes or RNAi has driven the research efforts of Chadderton and colleagues. The researchers first established suppression of RHO in transgenic mice carrying a wild-type human RHO transgene after rAAV2/5 delivery of the RHO-targeting shRNA, shQ1, by subretinal injection. Chadderton and colleagues then studied the effects of rAAV-shQ1-mediated RHO suppression in conjunction with endogenous RHO gene replacement in transgenic mice carrying a Pro347Ser mutant human RHO transgene. rAAV-shQ1 and the control vector rAAV-shNT were injected into one of each pair of RHO mutant eyes, and retinal structure and function were assessed by histology and eletroretinography (ERG), respectively, at 10 weeks. Significant improvements in both histology and ERG were noted, with the retinal structure of Pro347Ser eyes treated with rAAV-shQ1 showing substantially greater outer nuclear layer thickness than rAAV-shNT-treated eyes at both 5 and 10 weeks postinjection. More importantly, average ERG values for rAAV-shQ1-injected Pro347Ser eyes were 2-fold greater than for rAAV-shNT-treated eyes at 10 weeks postinjection. The promising results of Chadderton and colleagues using the two-component strategy of gene suppression in conjunction with gene replacement outlines the great potential of this approach for many other animal models of dominantly inherited disorders such as autosomal dominant RP (Chadderton et al., 2009).
Mutations in genes that encode proteins in the connecting cilia of photoreceptor cells manifest as RP as well as systemically in syndromes such as Usher and Bardet-Biedl syndromes. These syndromes are also known as ciliopathies, because the resulting dysfunctional protein products of genetic mutations are localized in many, if not all, ciliated cells throughout the body. Consequently, in addition to RP, widespread syndromic defects result from ciliopathies. Individuals suffering from Bardet-Biedl syndrome can present with issues ranging from polydactyly and renal abnormalities to mental retardation and hypogenitalism (Song et al., 2007). Usher syndrome is the most common of the RP syndromes; affected individuals present with symptoms that include deaf/blindness and vestibular dysfunction. The syndrome consists of three subtypes, USH1, USH2, and USH3, which are categorized by their clinical phenotypes and causative genes. Cilial cells are generally implicated in the USH1 form of Usher syndrome; genes with USH1-associated mutations encode proteins important to the development and regulation of organs of the inner ear as well as to structural and functional integrity of the retina.
Defects in the MYO7A gene have been found to correlate with the USH1B form of Usher syndrome. Mutations in MYO7A are thought to account for the retinal degeneration associated with USH1B in humans, because MYO7A expression is localized in numerous cell types including retinal pigment epithelial and photoreceptor cells. In the naturally occurring Myo7a-deficient shaker-1 mouse model of USH1B, however, affected mice present solely with hearing loss. Although retinal degeneration is absent despite the Myo7a-deficieny of shaker mice, defective trafficking of melanosomes in the retinal pigment epithelial cells is thought to be attributable to the MYO7A mutation. Usher syndrome resulting from a defective MYO7A gene seems most amenable to rAAV-mediated gene therapy when the novel rAAV2/5 vector is used, as it facilitates the efficient transfer of larger gene constructs (Song et al., 2007; Smith et al., 2009).
Inherited retinal degenerative disorders of the central retina
Within the central retina, which is responsible for central vision, resides the macula. Central to the macula is the fovea, the area of the retina with the greatest density of cone photoreceptor cells. The macula functions mainly in visual acuity and color vision, and therefore degeneration of the portion of retinal pigment epithelium that nourishes macular photoreceptors or the macular photoreceptors themselves significantly reduces central vision.
Retinoschisis
Retinoschisis is a retinal dystrophy in which the structure, and subsequently the function, of the retina are greatly compromised. The X-linked recessive form of retinoschisis, known as juvenile retinoschisis, leads to the degeneration of the central retina. Specifically, the retina separates into several layers, primarily at the fovea, and can potentially lead to a retinal detachment. Juvenile retinoschisis is caused by mutations in the RS1 gene, which encodes retinoschisin, an extracellular protein integral to the retina for cellular adhesion and tissue stability (Dinculescu et al., 2005; Smith et al., 2009). Many studies investigating rAAV-mediated gene therapy in an RS1 knockout mouse model of human X-linked retinoschisis have demonstrated improvements in retinal function. Few studies, however, have shown restoration of the structural integrity of the retina. Min and colleagues found significant improvements in both retinal structure and function of 15-day old Rs1h-deficient mice after subretinal delivery of human RS1 cDNA via the rAAV5 vector. The structure and function of the retina in the Rs1h-deficient mice was restored to levels comparable to those in wild-type mice and was long-term, persisting for up to 1 year (Dinculescu et al., 2005; Min et al., 2005).
Achromatopsia
Achromatopsia is a rare autosomal recessive congenital disorder that can render patients completely colorblind. In addition, affected individuals often present with poor visual acuity, photophobia, and/or nystagmus (Song et al., 2007). In general, achromatopsia is characterized by slow progression of cone photoreceptor dysfunction that can lead to one of two known forms, complete or incomplete achromatopsia. The two forms bear phenotypic resemblance, with the only difference being that incomplete achromatopsia patients have slightly better visual acuity and cone function. The two forms, however, are often genetically heterogeneous: three different genes have been found to be implicated for achromatopsia-associated mutations. Of these three cone-specific genes (CNGB3, CNGA3, and GNAT2), only CNGA3 has been associated with both forms of achromatopsia (Song et al., 2007; Colella et al., 2008).
Although both murine and canine models of achromatopsia exist, results of rAAV2/5-mediated gene therapy interventions in the naturally occurring dog model of achromatopsia are still at the preliminary stage. However, the rAAV2-mediated gene replacement of GNAT2 in 2- to 3-week-old Gnat2-deficient mice has resulted in marked improvements in achromatopsic pathology. Cone electrophysiology and visual acuity, which are both directly dependent on cone photoreceptor function, were restored to wild-type levels for as long as 7 months after subretinal administration of rAAV2-GNAT2 in the Gnat2-deficient mouse model (Colella et al., 2008). Improving cone function is the goal of rAAV-mediated gene therapy for achromatopsia; it has been noted that success of gene therapy as a potential therapeutic may depend on the age at which a patient receives treatment (Smith et al., 2009).
Age-related macular degeneration
Age-related macular degeneration (AMD) is the leading cause of visual loss among the elderly population in many countries, including the United States. Although there is no known single causative factor of AMD, both genetics and the environment are thought to play predominant roles in development of the disease. Central vision loss is characteristic of AMD because the macula deteriorates as the disease progresses. Consequently, experimental investigation of AMD has been complicated by the fact that most of the mammalian models available lack the macular region of the retina. However, numerous murine models of AMD have greatly elucidated the physiology and genetics behind AMD (Rakoczy et al., 2006).
AMD is phenotypically heterogeneous, manifesting in one of two forms: the “dry,” nonexudative form or the “wet,” exudative form of AMD. Dry AMD is the more prevalent of the two forms and causes a milder phenotype, in which a yellowish-white substance called drusen is deposited just beneath the retina in the space between the retinal pigment epithelium and Bruch's membrane. The deposition of drusen causes death of the retinal pigment epithelial cells and subsequent degeneration of the photoreceptors. Eventually, geographic atrophy occurs as the retina thins and vision worsens. Wet AMD is easily differentiated from dry AMD; wet AMD progresses rapidly and results in much more severe vision loss due to neovascularization of the choroid. The associated visual impairment progresses rapidly once the blood leaks from the neovasculature into the subretinal region and thus damages the retina (Song et al., 2007). Although both the phenotypic and environmental heterogeneity inherent between the two forms of AMD contribute to notions suggesting that the two forms should be treated as two distinct diseases, genetic studies have linked polymorphisms in the complement factor H (CFH) gene to both the wet and dry forms of AMD (Chen et al., 2006).
Gene therapy approaches for the treatment of dry AMD are limited; however, the finding of a genetic variant in the gene encoding the Toll-like 3 (TLR3) receptor has expanded the possibilities for therapeutic intervention. Specifically, Yang and colleagues tested for an association between the functional TLR3 variant, which involves the substitution of phenylalanine for leucine at amino acid 412, with AMD. Their results demonstrated an association between the TLR3 variant and protection against the geographic atrophy indicative of dry AMD, which is thought to be mediated by suppression of retinal pigment epithelial cell death. However, no association was observed between the TLR3 variant and choroidal neovascularization (CNV), the phenotype of wet AMD (Yang et al., 2008). Several rAAV-mediated gene therapy approaches have been implemented in the treatment of the experimental model of choroidal neovascularization. rAAV-mediated gene therapy has been used in both the induction and inhibition of choroidal neovascularization in the rat disease model. First, induction of choroidal neovascularization was adapted in the development of the rat model of wet AMD through subretinal delivery of rAAV vector encoding vascular endothelial growth factor (VEGF), a proangiogenic factor. Subsequently, the injected rats presented with the common symptoms of exudative AMD, including subretinal neovascularization, photoreceptor degeneration, and blood leakage of the neovasculature (Wang et al., 2003).
rAAV-mediated gene therapy has also been used to inhibit experimental models of choroidal neovascularization in a variety of animal models including mice, rats, and monkeys (Mori et al., 2002; Lai et al., 2005). Mori and colleagues have observed a reduction in both the development of CNV, as well as the regression of already developed CNV in a murine model after subretinal and intravitreal delivery of rAAV vector encoding the pigment epithelial-derived factor (PEDF), an antiangiogenic, neurotrophic protein (Mori et al., 2002). The many preclinical studies using rAAV-mediated gene therapy, especially in the investigation of wet AMD, hold great promise for clinical trials using rAAV-mediated gene therapy. Clinical trials employing the use of alternative gene therapy methods to treat wet AMD are currently underway (Salehi-Had and Miller, 2008). One trial has focused on gene suppression of VEGF and its receptor, using siRNA targeting. Another trial employing adenoviral vector-mediated delivery of PEDF has been completed. As mechanisms of AMD pathology continue to be unveiled, such as the role of inflammatory pathways in the progression of AMD and the susceptibility of retinal pigment epithelial cells to damage, the potential for intervention by rAAV-mediated gene therapy for use in clinical trials has become clear.
Acquired retinal degenerative disorders
Retinal neovascularization
Retinal neovascularization is a common feature of many ocular diseases, including the neovascular form of age-related macular degeneration, as previously mentioned, and is caused by many of the same aforementioned factors. rAAV-mediated gene therapy for the treatment of retinal neovascularization in acquired diseases has far-reaching implications, because acquired diseases such as diabetic retinopathy and AMD account for the majority of cases of irreversible blindness in the world (Reich and Bennett, 2003). In a long-term study investigating the therapeutic potential of rAAV-mediated expression of the soluble VEGF receptor, sFlt-1, in inhibiting the angiogenic action of VEGF, Lai and colleagues observed a reduction in retinal neovascularization. Subsequent morphologic studies also suggested preservation of retinal structure, unlike the retinal damage that commonly accompanies retinal neovascularization by inducing photoreceptor loss (Lai et al., 2002). Similarly, Bainbridge and colleagues found significant and consistent inhibition of retinal neovascularization by rAAV-mediated gene transfer of sFlt-1 in their comparative study of sFlt-1 gene transfer using both adenoviral and rAAV vectors (Bainbridge et al., 2002).
Using these findings, Deng and colleagues investigated the therapeutic potential of various VEGF peptides expressed by rAAV vectors to inhibit VEGF-induced retinal neovascularization found in the mouse model of oxygen-induced retinopathy. Specifically, Deng and colleagues studied the expression of the short peptides encoded by exons 6 and 7 of the VEGF gene, which were known from previous work to confer antiangiogenic effects through direct interference with VEGF binding to its major receptors including its tyrosine kinase receptors, Flt-1 and KDR. The researchers employed methods of RT-PCR and Western blot analysis to confirm expression of the VEGF peptides in the eyes where rAAV vector was injected intravitreously. They found retinal neovascularization to be inhibited by 71–83% in the rAAV vector-injected eyes when compared with the contralateral control eyes, further demonstrating the potential of the rAAV-mediated transduction of antiangiogenic factors in the treatment of ocular neovascularization (Deng et al., 2005).
Gene therapy simultaneously targeted to retinal rod and cone cells
Although most of the rAAV-mediated gene therapy interventions for retinal diseases discussed here target either the rod or cone photoreceptors, it is useful to treat the cell types simultaneously as both are implicated in retinal disease pathology. Furthermore, mutations in specific rod photoreceptor genes can contribute to cone death as well as rod death, whereas ocular diseases caused by cone-specific mutations result only in cone death. To better understand the mechanism behind nonautonomous cone death, Punzo and colleagues studied the incidence of cone death in four different mouse models for retinitis pigmentosa. The researchers found a connection between cone survival and insulin release; nonautonomous cone death was triggered when cone photoreceptors were starved from a lack of endogenous insulin (Punzo et al., 2009).
Other research efforts seeking to treat both rod and cone photoreceptor cell death have unveiled rAAV-mediated ocular gene therapy approaches that target expression of both cell types. The work of Khani and colleagues introduced the first well-defined, compact promoter to drive transgene expression in both rods and cones. Khani and colleagues compared the promoter activity of the human rhodopsin kinase (hRK) gene, a gene that has previously been identified as both rod and cone specific, with the mouse opsin (mOps) promoter. Although both promoter types yielded significant rAAV-mediated transgene expression after subretinal injection, only the hRK promoter demonstrated expression in both rods and cones, whereas the mOps promoter was active only in rod photoreceptors. This primary finding holds great significance for rAAV-mediated treatment of numerous retinal diseases, which has previously been limited by the lack of well-defined, rod/cone-specific promoters (Khani et al., 2007). The same team of investigators demonstrated proof-of-principle of their 2007 findings in mouse models with defects in the AIPL1 gene. Mutations of this gene show allelic heterogeneity, with a null allele resulting in a presentation similar to that of human LCA in mice, and the hypomorphic allele manifesting similarly to human RP in mice. Sun and colleagues assessed the role of rAAV-mediated gene expression of AIPL1 using hRK to promote both rod and cone photoreceptor survival. Their results exhibited successful transgene expression in both rods and cones from the single hRK promoter (Sun et al., 2009).
Looking Ahead: Promising Prospects for Ocular Gene Therapies
Most recently, the therapeutic potential of rAAV-mediated gene therapy was yet again illustrated by Mancuso and colleagues, who studied the effects of rAAV-mediated intervention on colorblindness in adult primates. Colorblindness is one of the most prevalent X-linked recessive disorders in humans, and is a congenital condition in all male squirrel monkeys. Females of this species often have trichromatic color vision, meaning they possess all the necessary photopigments. The dichromatic male squirrel monkeys, however, lack either the L- or M-photopigment, preventing them from distinguishing particular wavelengths. Mancuso and colleagues tested two adult male squirrel monkeys for color vision deficits, using the Cambridge Colour Test. As expected, the two monkeys failed to discriminate between red-violet and blue-green. These same two dichromatic squirrel monkeys, lacking the L-opsin gene, were then given subretinal injections of an L-opsin-coding rAAV2/5 vector. The L-opsin transgene was coexpressed in a subset of endogenous M-cones in the primates, giving way to a shift in the spectral sensitivity of the M-cone photoreceptors. Mancuso and colleagues identified this very shift as the component underlying the transition from dichromatic color vision to trichromacy in the red-green colorblind adult primates, and that thereby corrected their colorblindness (Mancuso et al., 2009). Many advances in experimental research have also fueled the success of rAAV-mediated gene therapy as a potential treatment for numerous ocular diseases. One such advance is the possibility of regulating the activation and deactivation of rAAV-mediated transgene expression by pharmacologically inducible expression systems (Bainbridge et al., 2006; Klausner et al., 2007). Intrinsic to any gene-regulated gene delivery system is an inducible promoter and a trans-activator. An important consideration is the selection of the pharmacological agent used to induce any promoter system, several of which have already been identified as transgene expression regulators in eukaryotes. The development of pharmacologically regulated rAAV vectors has enormous implications for the treatment of a wide variety of ocular diseases. The near future holds even greater promise for rAAV-mediated gene therapy in the treatment of ocular diseases (Chtarto et al., 2003).
Footnotes
Author Disclosure Statement
No competing financial interests exist for any of the authors.