
Abstract
The history of the North African Jewish community is ancient and complicated with a number of immigration waves and persecutions dramatically affecting its population size. A decade-long process in Israel of clinical-molecular screening of North African Jews with incurable autosomal recessive blindness led to the identification of a homozygous splicing mutation (c.95-2A > T; IVS2-2A > T) in RPE65, the gene encoding the isomerase that catalyzes a key step in the retinoid-visual cycle, in patients from 10 unrelated families. A total of 33 patients (four now deceased) had the severe childhood blindness known as Leber congenital amaurosis (LCA), making it the most common cause of retinal degeneration in this population. Haplotype analysis in seven of the patients revealed a shared homozygous region, indicating a population-specific founder mutation. The age of the RPE65 founder mutation was estimated to have emerged 100–230 (mean, 153) generations ago, suggesting it originated before the establishment of the Jewish community in North Africa. Individuals with this RPE65 mutation were characterized with retinal studies to determine if they were candidates for gene replacement, the recent and only therapy to date for this otherwise incurable blindness. The step from molecular anthropological studies to application of genetic medicine was then taken, and a representative of this patient subgroup was treated with subretinal rAAV2-RPE65 gene therapy. An increase in vision was present in the treated area as early as 15 days after the intervention. This process of genetically analyzing affected isolated populations as a screen for gene-based therapy suggests a new paradigm for disease diagnosis and treatment.
Introduction
Materials and Methods
Human subjects and clinical trial
The studies included 100 families of North African Jewish origin with the diagnosis of inherited retinal degeneration. Blood samples were obtained from the index patients and from other affected and unaffected family members for DNA analysis; patients underwent complete ophthalmic examinations. In 10 families with LCA (33 patients; see Fig. 1B), the c.95-2A > T (IVS2-2A > T) transversion in the RPE65 gene was identified in the homozygous state. Detailed measures of visual function and retinal structure were performed in two siblings from one of these families. Informed consent was obtained from all participants; procedures adhered to the guidelines in the Declaration of Helsinki and were approved by the institutional review boards from participating institutions. One of the two siblings with the RPE65 mutation was assessed with vision research studies before and after taking part in a Phase I clinical trial (registered in clinicaltrials.gov, NCT00481546) evaluating the safety of rAAV2-hRPE65.

Identifying and dating a founder mutation in North African Jewish families with autosomal recessive blindness.
Molecular studies
Genomic DNA was extracted from peripheral blood of all patients and family members using the FlexiGene DNA kit (QIAGEN, Valencia, CA) and published methods (Auslender et al., 2007).
Mutation analysis
Primers flanking studied exons were designed using Primer3 (
Estimation of mutation age
Analysis of mutation age was performed by the DMLE + program (version 2.2) (Reeve and Rannala, 2002). The population growth (r) was estimated by the following formula: T 1 = T 0e(gr), where T 1 represents the current population size, T 0 represents the ancestral population size, and g represents the number of generations. The estimated current population size (T 1) was determined as 670,000 (based on the Israeli Central Bureau of Statistics). The initial size of the population can be only roughly estimated by historical records, and therefore we used a range of 5,000–10,000 individuals. Using the above-mentioned formula, population growth (r) was calculated as X (for T 0 = 5,000) or Y (for T 0 = 10,000). For all calculations, we used a 25-year interval per generation.
Visual function and retinal imaging studies
Visual function
Absolute sensitivity of visual perception to flashed lights was determined using either localized or full-field stimuli. Techniques, methods of data analysis, and normal results have been described (Jacobson et al., 1986, 2009a; Roman et al., 2007a; Cideciyan et al., 2008). Dark-adapted sensitivity to the full-field stimulus was measured using blue and red, 200 ms-duration flashes with an LED-based, computer-driven stimulator (Colordome; Diagnosys LLC, Littleton, MA). The sensitivity difference (blue-red) to these two stimuli was used to assess whether rods, cones, or both photoreceptor systems mediated perception. Cone-mediated perception is expected to yield a much smaller sensitivity difference (blue-red, 3.1 dB) than rod-mediated perception (blue-red, 19.3 dB). Mixed mediation refers to values between those limits and indicates that rods are mediating blue detection and cones are mediating red detection (Roman et al., 2007a).
Photoreceptor layer topography
In vivo microscopy of the human retina was performed with a Fourier-domain (FD) optical coherence tomography (OCT) system (RTVue-100; Optovue Inc., Fremont, CA) as published (Jacobson et al., 2008, 2009b). Retinal cross sections were obtained along horizontal and vertical meridia centered on the fovea and extending 9 mm in either direction. Dense raster scans were also performed to sample an 18 × 12-mm region of the retina centered on the fovea. Postacquisition processing of data was performed with custom programs (MatLab 6.5; MathWorks, Natick, MA). For topographic analysis, the precise location and orientation of each raster scan relative to retinal features (blood vessels, intraretinal pigment, and optic nerve head) were determined using en face images of the fundus. Longitudinal reflectivity profiles (LRPs) making up the OCT scans were aligned by straightening the major RPE reflection (Huang et al., 1998; Jacobson et al., 2003). The outer photoreceptor nuclear layer (ONL) thickness was defined as the major intraretinal signal trough delimited by the signal slope maxima (thickness values <5 μm were considered not detectable). ONL thickness was related to colocalized psychophysical thresholds determined by dark-adapted static perimetry.
Modeling of the relationship between retinal structure and visual function
The relationship between photoreceptor structure and colocalized visual function was defined in patients using ONL thickness and rod-mediated sensitivity. Patient results were compared with an idealized model of the expected relationship for “simple” photoreceptor degenerations in which vision loss is thought to derive primarily from degenerative photoreceptor cell loss. The model assumes that absolute sensitivity of rod-mediated function near the visibility threshold is limited by quantum catch, and thus it is proportional to the product of the number of surviving photoreceptor cells and the length of their outer segments; both of these parameters are proportional to ONL thickness (Jacobson et al., 2005, 2007; Cideciyan et al., 2008). Thus, to a first approximation, loss of light sensitivity (in linear units) would be expected to be proportional to the square of ONL thinning.
Autofluorescence imaging
Spatial topography of the retinal pigment epithelium (RPE) health was estimated with recently developed near-infrared autofluorescence imaging (NIR-AF) methods (Cideciyan et al., 2007; Aleman et al., 2008; Gibbs et al., 2009). A confocal scanning laser ophthalmoscope was used for imaging (HRA2; Heidelberg Engineering GmbH, Heidelberg, Germany) with an NIR (790 nm) excitation light and a long-pass blocking filter for >804 nm. This NIR-AF signal is believed to be dominated by the melanolipofuscin in RPE and melanin in the RPE and choroid. Images were exported from the manufacturer's software and analyzed as described (Cideciyan et al., 2007).
Viral vector preparation and delivery
The adeno-associated virus (AAV) vector, AAV2-CB-hRPE65, as previously described (Jacobson et al., 2006), was packaged and purified according to clinical Good Manufacturing Practices (cGMP) by the two-plasmid cotransfection method at the University of Florida Human Applications Laboratory. In brief, HEK 293 cells were cotransfected with pDG packaging plasmid DNA, which encodes three adenoviral genes required for packaging (E2A, VA, and E4) and the AAV2 rep and cap genes, along with the AAV vector plasmid. Vector purification employed a three-column purification method with vector titer, expressed in vector genomes (vg), determined by dot-blot assay (Zolotukhin et al., 2002). The final vector reagent, as vialed and stored, passed testing for sterility, endotoxin, minimum vector titer, protein purity, correct capsid proteins, absence of visible foreign material, and pH. The vector also demonstrated in vivo potency (Roman et al., 2007b) shortly before clinical administration. The vector reagent was provided on a compassionate use basis.
The eye with worse visual function (in this case, the amblyopic left eye) was chosen for vector administration. Local retrobulbar anesthesia was administered and the eye then prepped and draped. A standard three-port 23-gauge vitrectomy was performed. The conjunctiva over the right-sided sclerotomy was dissected and the sclerotomy enlarged with a 20-gauge MVR blade so that the injection cannula (39 gauge; Synergetics Inc., O'Fallon, MO) could be easily inserted into the eye. A small retinotomy was performed with the tip of the cannula, and a volume of 300 μl of vector (containing 1.19 × 1011 viral particles) was introduced into the subretinal space at a slow rate and avoiding subfoveal delivery. At the end of the procedure, the 20-gauge sclerotomy site and the overlying conjunctiva were secured with 7.0 vicryl sutures. The other sites had no suturing. Subconjunctival antibiotics and steroids were administered. Topical antibiotics and steroids were used postoperatively, and tapered over the course of 28 days.
Monitoring of safety
Ocular safety was assessed by standard eye examinations at two baseline visits, daily for the first 5 days posttreatment, and on days 7, 10, 14, 21, 30, 60, and 90. Systemic safety was evaluated with a physical examination at both baseline evaluations, daily after administration for 5 days, and on days 7, 10, 30 and 90 postadministration. Routine hematology, serum chemistry, coagulation studies, and urinalysis were performed at baseline and at 1, 3, 10, 30, and 90 days. Serum samples were assayed for circulating antibodies to the AAV2 capsid proteins at baseline and on days 14 and 90, and the presence of AAV DNA in peripheral blood was tested by quantitative PCR at baseline an on days 1, 3, and 21 postoperatively as previously described (Hauswirth et al., 2008).
Results and Discussion
In our cohort of Israeli patients with hereditary retinal degenerations, there are 100 families (208 patients) of North African Jewish origin, mainly with autosomal recessive inheritance. The most common phenotypes are retinitis pigmentosa (RP), and an early-onset and severe retinal degeneration, termed Leber congenital amaurosis (LCA; OMIM #204100; Fig. 1A). With recent evidence of safety and efficacy of gene therapy in patients with LCA (Bainbridge et al., 2008; Cideciyan et al., 2008; Hauswirth et al., 2008; Maguire et al., 2008), we focused on this disease. In mutation analyses of LCA performed mainly in North American and European populations, at least 14 disease-causing genes and over 400 mutations have been identified; these genes and mutations accounted for about 70% of the cases (for reviews, see Stone, 2007; den Hollander et al., 2008). In contrast, in our LCA cohort of 43 patients, representing 18 families of North African origin (including Morocco, Algeria, Libya, and Tunis), three mutations in only three genes (RPE65, AIPL1, GUCY2D) were responsible for all cases (Fig. 1A).
The RPE65 (retinal pigment epithelium-specific-65-kDa) gene was the most prevalent cause of LCA at 56%, in contrast to <10% quoted in other studies (den Hollander et al., 2008), and in contrast to none among LCA patients of Ashkenazi Jewish descent in our cohort. Mutation analysis revealed a homozygous splice site mutation, c.95-2A > T (IVS2-2A > T), in the RPE65 gene in patients with LCA from 10 of the families (Fig. 1B–D; Table 1). This mutation has been reported previously in two other patients of unknown origin (Hanein et al., 2004; Simonelli et al., 2007). RPE65 encodes the isomerase of the retinoid cycle, the enzymatic pathway that underlies mammalian vision; mutations in RPE65 disrupt the retinoid cycle and cause LCA (Hamel et al., 1993; Redmond et al., 1998; Jin et al., 2005; Moiseyev et al., 2005; den Hollander et al., 2008; Cideciyan, 2010). The splice site mutation we identified was present neither in North African Jews with other retinal degenerations nor in LCA patients from other origins, suggesting a founder LCA mutation that is specific to the North African Jewish population. Haplotype analysis in seven of the patients, using SNP markers in the vicinity of RPE65, revealed a shared homozygous region (Fig. 1C), indicating a founder mutation. Using the DMLE program, the age of the founder mutation was estimated at between 100 and 230 generations ago, with a mean of 153 generations (Fig. 1E).
CF, counting fingers; FF, fix and follow light; HM, hand movements; LE, left eye; LP, light perception; NA, not available; RE, right eye.
When visual acuity data are available, age at exam is provided. Otherwise, current age is listed.
Spherical equivalent; average both eyes.
Similar in both eyes unless specified.
To estimate the carrier frequency of the c.95-2A > T mutation, we screened 179 origin-matched control individuals and identified two carriers (estimated carrier frequency of ∼1.1%). In the remaining eight families with LCA, we identified patients who are homozygous for the AIPL1 (arylhydrocarbon-interacting receptor protein-like 1) p.Val71Phe mutation in four families and patients who are homozygous for the GUCY2D (human photoreceptor guanylate cyclase 2D) c.389delC mutation in four families. One of the AIPL1 patients was also a carrier of the RPE65 mutation.
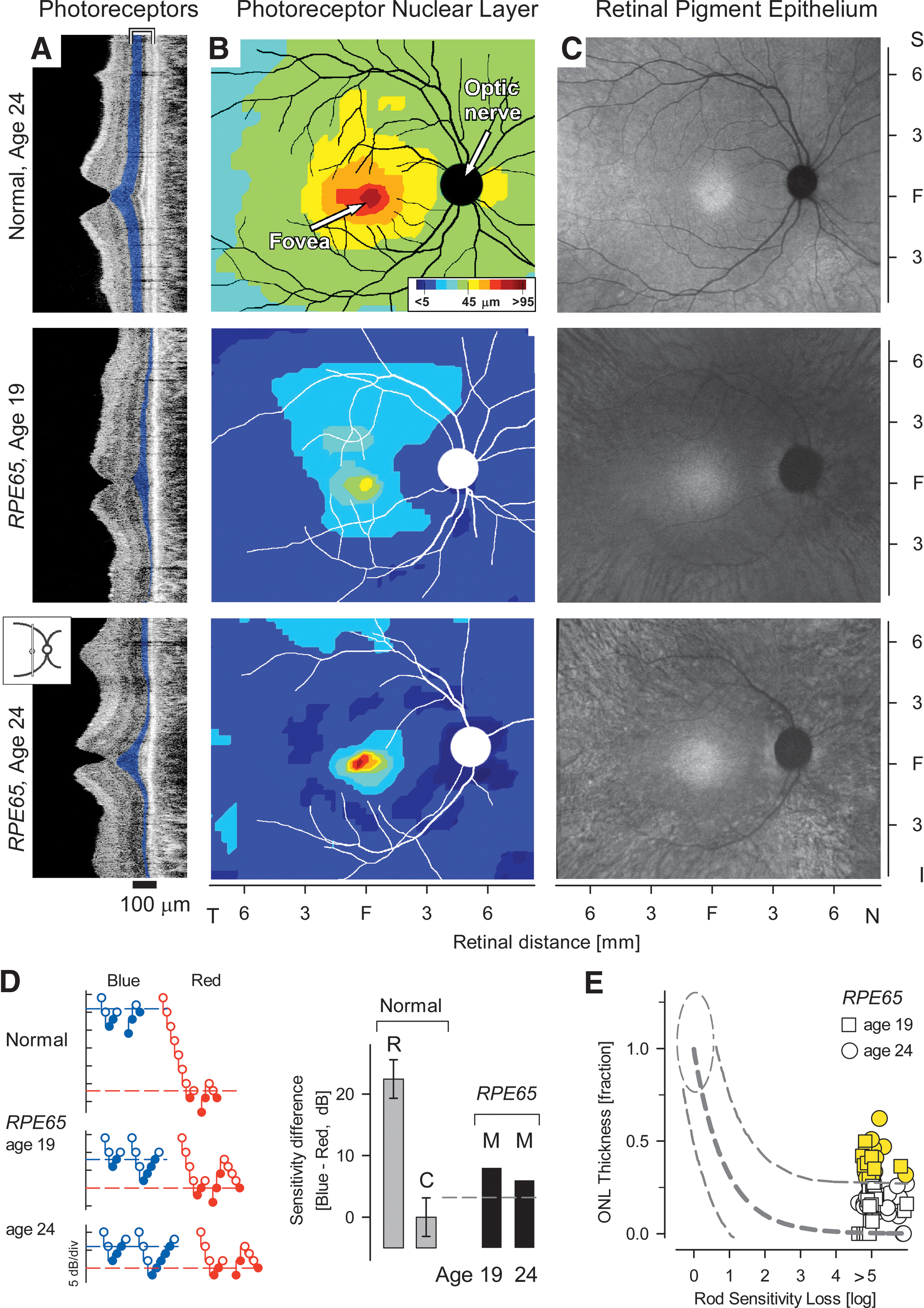
How does this founder mutation in RPE65 affect retinal structure and function? The mutation is likely to result in an aberrant mRNA transcript lacking exon 3, introducing a frameshift followed by a premature stop codon at amino acid 27. A mutant transcript such as this is likely to be recognized and degraded by the nonsense mediated mRNA decay mechanism, resulting in the lack of RPE65 protein expression. All affected individuals showed clinical manifestations of LCA (Table 1), including markedly impaired night vision, reduced visual acuity from an early age, and nystagmus; there was a spectrum of retinal abnormalities from mild pigmentary disturbances to atrophy. Electrophysiological responses, as measured by electroretinography, were nondetectable in all individuals tested, even at young ages. Most patients above the age of 30, as previously reported (Jacobson et al., 2007), had visual acuities at the light perception to counting fingers levels, with severely constricted visual fields. Aiming to study in detail the structure of the retina in patients who are homozygous for the c.95-2A > T mutation, we first quantified the photoreceptor layer (Fig. 2A and B). In a normal subject and in two affected siblings with RPE65-LCA (Family MOL0701, II:2 and II:3; Table 1 and Fig. 1B), retinal cross sections were obtained using in vivo microscopy (Jacobson et al., 2005). In normal subjects (Fig. 2A, upper) a foveal depression or pit and orderly retinal layers corresponding to nuclei, synapses, and axons are observed. The photoreceptor ONL (highlighted in blue) is thickest at the fovea centralis and thins with increasing distance from it. In both affected siblings (at ages 19 and 24), there is relative sparing of the ONL at the fovea, but diminishing ONL with increasing distance from the fovea (Fig. 2A, middle and lower panels). Photoreceptor layer topography across an expanse of central retina is also shown (Fig. 2B). In the 19-year-old with RPE65-LCA, there is a region with retained photoreceptors extending from the fovea into the superior retina; a less extensive region centered at the fovea is detectable in the 24-year-old subject.
RPE65-mutant retinas analyzed for photoreceptor and RPE disease and potential for treatment.
The topography of RPE integrity in the RPE65-LCA eyes was defined with NIR-AF imaging (Cideciyan et al., 2007; Aleman et al., 2008; Gibbs et al., 2009). In normal human eyes, NIR-AF images show a circular central region of homogeneous appearance and higher brightness extending from the fovea (Cideciyan et al., 2007). With increasing eccentricity, there is a decrease in brightness (Fig. 2C). NIR-AF topographies of the RPE65-LCA eyes showed a central region of higher intensity surrounded by a more peripheral region of lower intensity. In the 19-year-old individual, there was evidence of two zones of RPE demelanization surrounding the central ellipsoid of retained RPE. At the eccentricity of the optic nerve head, there was a wide homogeneous annulus of lower intensity likely corresponding to partially demelanized RPE cells. Further peripheral was a heterogeneous annulus probably representing loss of RPE cells and appearance of choroidal melanin structures. The two zones were also evident in the affected 24-year old individual, but loss of RPE cells and choroidal structures are more evident around the central preserved area.
Candidacy of individuals with this RPE65 mutation for treatment with gene replacement therapy depends on evidence of retinoid cycle blockade from RPE65 deficiency, i.e., that these RPE65-mutant retinas show greater photoreceptor layer thickness than predicted for the degree of visual loss (Jacobson et al., 2005; Cideciyan, 2010). Absolute sensitivity of visual perception to flashed lights was first determined using full-field stimuli and psychophysical methods (Fig. 2D and E; Roman et al., 2007a). Consistent with previous reports of the effects of human RPE65 mutations on vision (Jacobson et al., 2009), both individuals showed 4–5 log units of sensitivity loss to achromatic stimuli. Use of chromatic full-field stimuli revealed that rods and cones were contributing to the remnant visual function (Fig. 2D). The relationship between photoreceptor nuclear layer structure and colocalized visual function was then defined at selected extrafoveal locations along horizontal and vertical meridia (Fig. 2E; Jacobson et al., 1986; Cideciyan et al., 2008). Patient results were compared with an idealized model of the expected relationship for “simple” photoreceptor degenerations (i.e., not caused by RPE65 mutations; Jacobson et al., 2005; Cideciyan et al., 2008). In other words, in the idealized model, vision loss is thought to derive primarily from degenerative photoreceptor cell loss. At some retinal locations in the RPE65-LCA subjects (72/98 = 73%), the fraction of remaining ONL thickness was predictably related to loss of rod-based vision and was no different from the prediction of the model for non-RPE65 behavior. At other locations in the RPE65-mutant retinas (26/98 = 27%), however, there was a significantly greater amount of ONL preservation for the severity of visual loss (Fig. 2E, highlighted yellow). The latter result indicates that the Moroccan Jewish individuals with this founder RPE65 mutation are potential candidates for a recent and novel gene therapeutic intervention (Bainbridge et al., 2008; Cideciyan et al., 2008; Hauswirth et al., 2008; Maguire et al., 2008).
Patient II:2 from family MOL0701, described above, was enrolled in a Phase I clinical trial of ocular delivery of AAV2-based RPE65 gene replacement. Based on the photoreceptor topography of the left eye (similar to that in the right eye, Fig.2B), subretinal (between RPE and neural retina) injection of vector-gene was targeted to a superior retinal region with detectable ONL (Fig. 3A). After the injection, absolute visual sensitivity under dark-adapted conditions was sampled along the vertical meridian crossing fixation at the fovea and compared with the sensitivity obtained before the injection. Tested loci are shown overlaid on the infrared retinal image of the patient; location of the injection site and the estimated boundaries of the bleb formed by the injection are also shown for reference (Fig. 3B). By 15 days after the injection, sensitivities at three contiguous loci from 1.8 to 3 mm superior to the fovea (Fig. 3B, green circles, bracket) were significantly improved beyond the test–retest variability (3SD = 0.6 log10) obtained in the same patient on six sessions before the injection. At two of the loci, the extent of the improvement was also significant using previously published stricter criteria for change (0.8 log10) based on a population of RPE65-LCA patients (Cideciyan et al., 2008). At the most recent evaluation corresponding to 90 days after the injection, sensitivities at the three neighboring retinal loci remained significantly improved (ranging from 1 to 1.7 log10 units) compared with baseline. Sensitivities in the untreated eye were unchanged from baseline (data not shown). The increased light sensitivity in the treated eye (which was also self-reported by the subject) is thought to be driven by an increased synthesis of 11-cis-retinal chromophore due to wild-type RPE65 protein introduced into the RPE by expression from the AAV2 gene therapy vector (Cideciyan et al., 2008). Safety monitoring showed no vector-related serious adverse events and no systemic toxicities. There was no evidence of viral particles in the systemic circulation nor a significant rise in circulating antibodies to AAV serotype 2 capsid postoperatively.
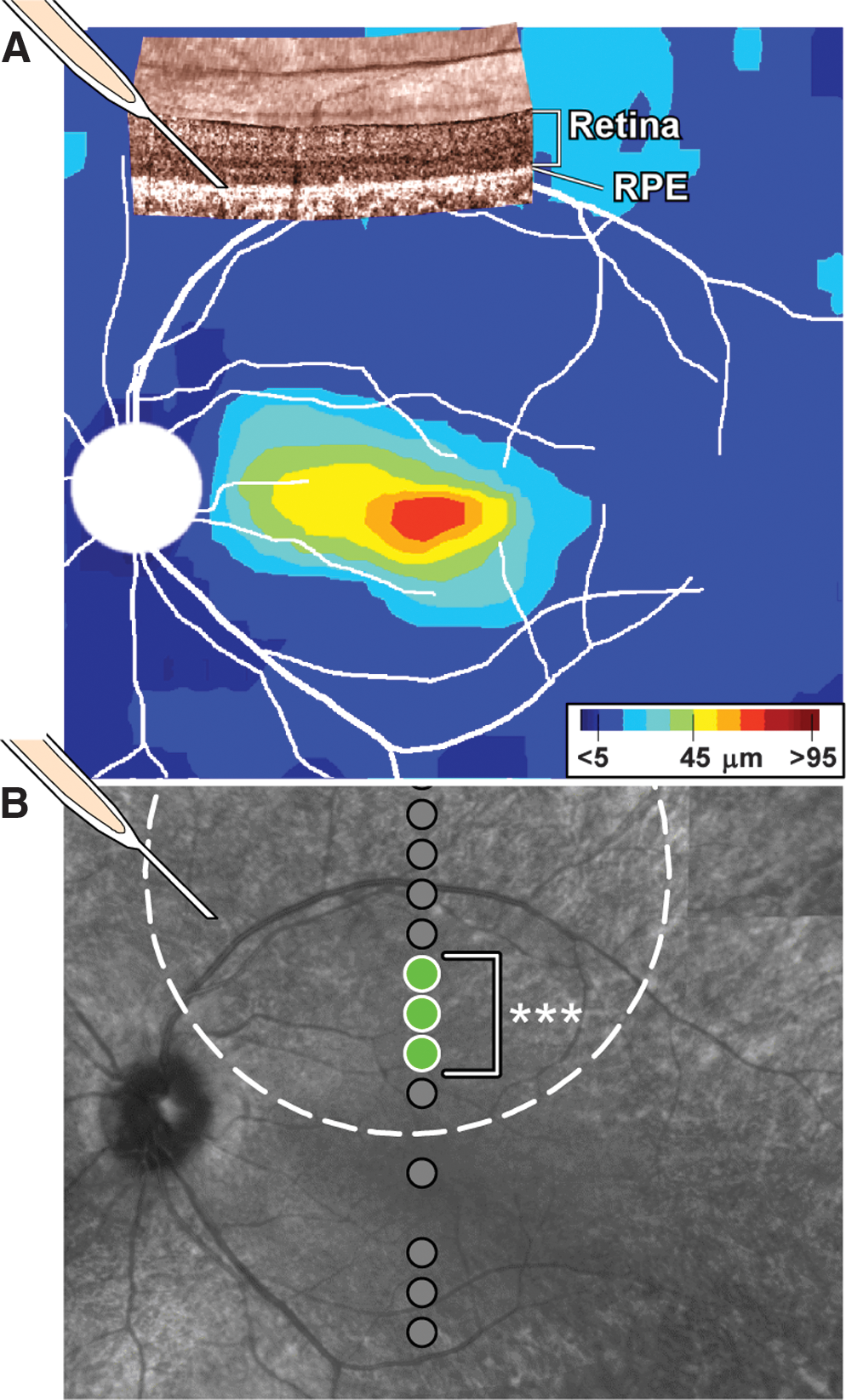
Retinal gene therapy in an individual with RPE65–LCA and the splice site mutation.
In summary, a unique combination of scientific, medical, genetic, and political analyses converged to provide treatment for a rare ocular disorder that was only recently considered incurable. The route to the medical treatment was nontraditional and emanated from a genetic analysis of dispersion of an ancient group of people that dated back centuries to their return to a present-day community. During that odyssey, they retained not only their ethnic and genetic identity but also their disease vulnerability. The advances made in identifying prevalent founder mutations in such defined populations using molecular genetic techniques, combined with the emergence of an effective gene therapy (at present only for the RPE65 gene), mandate modification of the way we approach patients from such communities, as the present report exemplifies. When a patient of North African Jewish descent is diagnosed with LCA, there is a probability of close to 100% that the genetic cause will be one of three specific mutations in three genes. There is ∼50% likelihood that the disease is caused by the IVS2-2A > T mutation in the RPE65 gene, which can be rapidly verified or excluded. Once identified, the RPE65-LCA patient undergoes in vivo retinal microscopy; areas with residual ONL that are amenable to treatment are mapped; and gene therapy can be administered to that area. An additional implication is that if there is a therapeutic response to gene therapy in one patient with a given RPE65 mutation, it becomes a “test case” for other patients with the same mutation, provided the patients have comparable photoreceptor topography and residual vision in the treated region of retina (Jacobson et al., 2008, 2009a) and, of course, considering factors such as age and success of surgical procedure. In the pre–gene therapy era for LCA, another RPE65 founder mutation, Tyr368His, was identified in an isolated Dutch community (Yzer et al., 2003), and the approach taken in the present study may now have applicability for this patient group. As other forms of gene therapy and other novel treatments for genetic diseases become increasingly available, application of this type of medical diagnostic and decision-making approach may become the standard of care in genetically defined subpopulations.
Footnotes
Acknowledgments
We thank the Macula Vision Research Foundation for supporting the clinical trial. The Yedidut Research Grant supported the molecular genetic parts of this study. We deeply appreciate the assistance of Dr. Ethan Galun, Dr. Linda Rasooly, and the staff of the Hadassah Medical Center GMP facility in the final pre-operative preparation of the viral vector, and the contributions of Dr. Anat Blumenfeld to the molecular genetic studies. Many thanks are due to Mr. Dan Blau, Mrs. Inbar Erdinset, Dr. Malgorzata Swider, and Mr. Israel Barzel for excellent engineering, technical, and fundus imaging assistance.
Author Disclosure Statement
W.W.H. and the University of Florida have a financial interest in the use of AAV therapies, and own equity in a company (AGTC Inc.) that might, in the future, commercialize some aspects of this work. The University of Pennsylvania, University of Florida, and Cornell University hold a patent on the described gene therapy technology (United States Patent 20070077228, “Method for Treating or Retarding the Development of Blindness”).