
Abstract
Gene therapy for dominantly inherited diseases with small interfering RNA (siRNA) requires mutant allele-specific suppression when genes in which mutation causes disease normally have an important role. We previously proposed a strategy for selective suppression of mutant alleles; both mutant and wild-type alleles are inhibited by most effective siRNA, and wild-type protein is restored using mRNA mutated to be resistant to the siRNA. Here, to prove the principle of this strategy in vivo, we applied it to our previously reported anti–copper/zinc superoxide dismutase (SOD1) short hairpin RNA (shRNA) transgenic (Tg) mice, in which the expression of the endogenous wild-type SOD1 gene was inhibited by more than 80%. These shRNA Tg mice showed hepatic lipid accumulation with mild liver dysfunction due to downregulation of endogenous wild-type SOD1. To rescue this side effect, we generated siRNA-resistant SOD1 Tg mice and crossed them with anti-SOD1 shRNA Tg mice, resulting in the disappearance of lipid accumulation in the liver. Furthermore, we also succeeded in mutant SOD1-specific gene suppression in the liver of SOD1G93A Tg mice, a model for amyotrophic lateral sclerosis, using intravenously administered viral vectors. Our method may prove useful for siRNA-based gene therapy for dominantly inherited diseases.
Introduction
A major problem encountered in our strategy to silence the mutant allele with RNAi in SOD1G93A Tg mice was that expressed siRNA failed to specifically recognize the point mutation, resulting in suppression of the wild-type allele in addition to the mutant allele. In order to treat dominantly inherited diseases using this RNAi strategy, mutant allele-specific suppression is necessary, especially when the genes in which mutation causes diseases have normally an important role. Anti-SOD1 shRNA Tg mice that demonstrate marked suppression of endogenous wild-type SOD1 also exhibit a fatty liver, similar to that observed in SOD1 knockout mice (Sasaguri et al., 2009). siRNA can be designed to discriminate between single nucleotide alterations by targeting the mutation itself or disease-linked polymorphisms (Gonzalez-Alegre et al., 2003; Miller et al., 2003, 2004; Li et al., 2004; Dykxhoorn et al., 2006b; van Bilsen et al., 2008). In a systematic analysis investigating the design of single nucleotide-specific siRNA, mismatches located in the central and 3′ regions of the guide strand (especially at positions 10 and 16 from the 5′ end) provided a high efficacy of single nucleotide discrimination between mutant and wild-type alleles (Schwarz et al., 2006). Introducing a mismatch into the seed region of siRNA was also shown to enhance discrimination (Ohnishi et al., 2008). We have also reported on the design of siRNA that demonstrates relative discrimination of a mutant allele possibly resulting from a change in the RNA secondary structure (Li et al., 2004).
Despite these design strategies, discrimination of mutant and wild-type alleles is not always complete. In addition, the cleavage efficiency of the mutant allele is not necessarily maximal, as the selection sites used in the design of siRNA are limited to the siRNA-related region. More than 125 different mutations in the SOD1 gene in familial ALS have been identified to date (Pasinelli and Brown, 2006). We have designed mutant allele-specific siRNA for G93A and A4V SOD1 (Yokota et al., 2004), but not for G37R SOD1. To overcome these problems, we proposed a novel method for allele-specific suppression by siRNA where both mutant and wild-type alleles are inhibited by most effective siRNA and where wild-type protein is restored using wild-type mRNA modified to be resistant to the siRNA. The amino acid sequence encoded by modified mRNA is the same as that of native mRNA, whereas the nucleotide sequence of mRNA targeted by siRNA is altered (Kubodera et al., 2005). A similar method of mutant allele-specific siRNA design was reported by another group in the same year (Xia et al., 2005). Here, we examined this strategy in vivo by applying the method to the rescue of the anti-SOD1 shRNA Tg mice phenotype and to the selective suppression of the mutant allele in SOD1G93A Tg mice.
Materials and Methods
Construction of expression vectors
Construction of anti-SOD1 shRNA expression vector has been reported previously (Saito et al., 2005). The shRNA sequence (sense sequence position 536–555, NM_000454) of this vector was common to the siRNA target region in both human and mouse SOD1 mRNA. siRNA-resistant human SOD1 expression vector, in which the human wild-type SOD1 genome is mutated so that it is not cleaved by the anti-SOD1 siRNA but is translated to the same amino acid sequence of native human SOD1, was made by site-directed mutagenesis (Stratagene, La Jolla, CA). The human SOD1 promoter and wild-type SOD1 genomic DNA were kindly provided by Dr. Masashi Aoki.
For recombinant adeno-associated virus (rAAV)-mediated delivery of shRNA targeting SOD1 and siRNA-resistant mouse SOD1 cDNA, the anti-SOD1 shRNA expression cassette containing human U6 promoter (shRNA) or that followed by the siRNA-resistant mouse SOD1 expression cassette containing cytomegalovirus (CMV) promoter (siRNA-resistant) was cloned into the plasmid containing adeno-associated virus (AAV) serotype 2 inverted terminal repeats (pAAV-MCS) (Stratagene). For genome copy titration of rAAV vectors, the human growth hormone poly(A) signal was inserted downstream of the shRNA cassette.
Viral vector production
The rAAV pseudotyped-8 (rAAV-2/8; AAV-2 inverted terminal repeat, AAV-8 viral capsid) vectors were produced using the adenovirus-free triple transfection method (Stratagene). The AAV vector plasmid (pAAV), the packaging plasmid (P5E18-VD2/8; a gift from Dr. James M. Wilson, University of Pennsylvania, Philadelphia, PA), and a helper plasmid (pHelper; Stratagene) were co-transfected into human embryonic kidney 293 cells under calcium phosphate precipitation. At 6 hr after transfection, the culture medium was replaced with fresh medium, and the cells were incubated for 48 hr. The cells were then harvested from the culture dishes and pelleted by centrifugation, resuspended in phosphate-buffered saline (PBS), and subjected to three rounds of freeze–thawing. Cell debris was then pelleted by centrifugation at 1,200 g for 15 min. AAV vectors were purified using ammonium sulfate precipitation and iodixanol (Axis-Shield, Norton, MA) continuous gradient centrifugation. Genome titers of the AAV vectors were determined by quantitative polymerase chain reaction (PCR) using the TaqMan system (Taymans et al., 2007). The following primers and probes targeting the poly(A) signal were used: 5′-CAGGCTGGTCTCCAACTCCTC-3′ and 5′-GCAGTGGTTCACGCCTGTAA-3′ served as the primer set, and 5′-TACCCACCTTGGCCTC-3′ served as the probe.
Animals
Animal experiments were performed in accordance with the Guidelines for Animal Experimentation of Tokyo Medical and Dental University and were pre-approved by the local ethics committee (protocol 0090104). The generation of anti-SOD1 siRNA Tg mice has been described previously (Saito et al., 2005). To produce siRNA-resistant human SOD1 Tg mice, the siRNA-resistant human SOD1 expression vector was injected into fertilized mouse eggs. Double Tg mice were generated by crossing anti-SOD1 shRNA Tg mice with siRNA-resistant human SOD1 Tg mice. Genotypes of the mice were determined by PCR analysis using genomic DNA from the tail tip. PCR was carried out using the following primer sets: 5′-CATCAGCCCTAATCCATCTGA-3′ and 5′-CGCGACTAACAATCAAAGTGA-3′ for siRNA-resistant human SOD1 Tg mice and 5′-CTTGGGTAGTTTGCAG-3′ and 5′-CAGGAAACAGCTATGAC-3′ for anti-SOD1 shRNA Tg mice.
AAV injection
SOD1G93A Tg mice were intravenously administered a single dose of 1 × 1012 vector genomes of rAAV2/8-shRNA or -shRNA/resistant SOD1 vectors via the tail vein. All mice were sacrificed 3 weeks post-injection. Mice were deeply anesthetized with pentobarbital sodium and perfused with cold PBS. Tissue samples were then collected and snap-frozen in liquid nitrogen for analysis.
Western blot analysis
Protein samples were extracted from tail, liver, brain, and spinal cord and homogenized in buffer containing 0.1% sodium dodecyl sulfate (SDS), 1% Triton X-100, 1% deoxycholate, and 1 mM phenylmethylsulfonyl fluoride. Equal amounts of extracted protein were then mixed with Laemmli sample buffer, denatured, and separated on 15% SDS-polyacrylamide gel electrophoresis. After transfer to a polyvinylidine difluoride membrane (Bio-Rad, Hercules, CA), blots were probed with anti-SOD1 polyclonal antibody S-100 (Assay Designs, Ann Arbor, MI) or anti-β-actin monoclonal antibody (Sigma, St. Louis, MO) and then visualized using enhanced chemiluminescence. Densitometric analysis was performed using Image J application software with the amounts of SOD1 being normalized for β-actin.
Quantitative reverse transcription-PCR
Total RNA was extracted from liver samples using Isogen (Nippon Gene, Tokyo, Japan), and 1 μg of total RNA from each sample was reverse-transcribed to cDNA using the High Capacity RNA-to-cDNA kit (Applied Biosystems, Carlsbad, CA). cDNA was used for quantitative reverse transcription-PCR (qRT-PCR) using the TaqMan system and the ABI Prism 7700 Sequence Detection system (Applied Biosystems) according to the manufacturers' instructions. The following primers and probe were used to quantify mouse and human SOD1: 5′-GGTGCAGGGAACCATCCA-3′ and 5′-CCCATGCTGGCCTTCAGT-3′ for the mouse primer set, with 5′-AGGCAAGCGGTGAACCAGTTGTGTTG-3′ for the mouse probe; and 5′-CCACACCTTCACTGGTCCATTA-3′ and 5′-CGACGGCCCAGTGCA-3′ for the human primer set, with 5′-TTCCTTCTGCTCGAAATTGATGATGCCC-3′ for the human probe.
Measurement of SOD1 activity
Each liver sample was homogenized in 5 volumes (wt/vol) of homogenization buffer containing 0.25 M sucrose, 20 mM Tris-HCl, and 1 mM EDTA and centrifuged at 78,000 g for 60 min. The supernatant was carefully removed and analyzed. To inactivate Mn-SOD, the sample was treated with 2% SDS at 37°C for 30 min. After cooling to 4°C, 0.1 volume of 3 M KCl was added, and the mixture was centrifuged at 20,000 g for 10 min to remove excess SDS. The supernatant was then assayed for SOD activity using the SOD Assay Kit-WST (Dojindo, Kumamoto, Japan) according to the manufacturer's instructions.
Histological study
For histological observation, formalin-fixed, paraffin-embedded liver sections (4 μm thick) were stained with hematoxylin and eosin, and frozen liver sections (8 μm thick) were stained with Sudan III using standard protocols. To quantify hepatic lipid accumulation, the density of lipid droplets (minimal diameter, >2 μm) was measured on the visual fields of a light microscope.
Serum alanine aminotransferase
Blood was collected from the animals via a retro-orbital plexus bleed, and the alanine aminotransferase (ALT) levels in the serum were measured using the ultraviiolet method. Measurements were conducted at Nagahama Life Science Laboratory (Shiga, Japan).
Statistical analysis
Statistical significance was assessed between groups using Student's t test or one-way analysis of variance. Significance was defined as p < 0.05.
Results
Generation of siRNA-resistant SOD1 Tg mice
We applied a selective suppression RNAi strategy to rescue the side effects resulting from downregulation of endogenous SOD1 in anti-SOD1 shRNA Tg mice. First, we attempted to generate Tg mice that express wild-type human SOD1 modified to be resistant to the siRNA. The nucleotide sequence encoded by siRNA-resistant SOD1 was altered to encode the same amino acid sequence as that of native SOD1 (Fig. 1a). Two strains of high- and low-copy siRNA-resistant SOD1 Tg mice were obtained in which the expressed human SOD1 protein levels differed on western blot analysis (Fig. 1b). We also quantified the expression of human and mouse SOD1 mRNA by qRT-PCR using TaqMan probes specific for their respective SOD1 mRNA. The expression level of human SOD1 mRNA in low-copy siRNA-resistant SOD1 Tg mice was approximately one-fifth that of endogenous mouse SOD1 mRNA, whereas that in the high-copy siRNA-resistant SOD1 Tg mice was four times higher than that of mouse SOD1 mRNA (Fig. 1c).
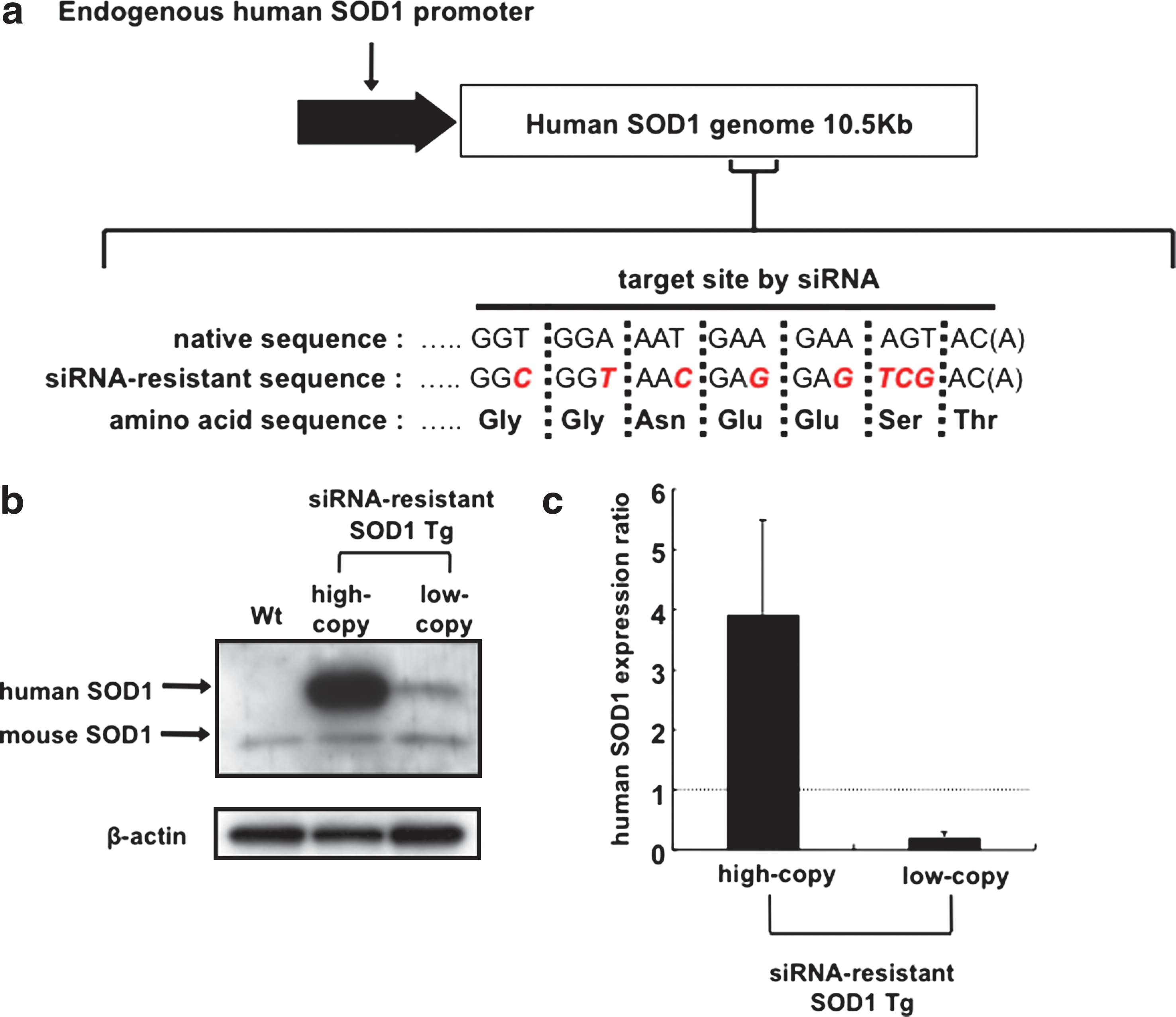
Generation of siRNA-resistant SOD1 Tg mice. (
Generation of double Tg mice that express both anti-SOD1 shRNA and siRNA-resistant SOD1
We next attempted to generate double Tg mice by crossing high-copy siRNA-resistant SOD1 Tg mice with anti-SOD1 shRNA Tg mice. In the liver, brain, and spinal cord of double Tg mice, human SOD1 protein was robustly expressed, whereas the endogenous mouse SOD1 protein remained suppressed (Fig. 2a). The corrected band density of SOD1 in wild-type littermates and double Tg mice is shown in Fig. 2b. In double Tg mice, the expression level of human SOD1 protein was almost equal to that of siRNA-resistant high-copy SOD1 Tg mice, indicating that human SOD1 was not inhibited by siRNA (Fig. 2c). The expression level of human SOD1 mRNA in double Tg mice was five times higher than that of endogenous mouse SOD1 mRNA observed in wild-type littermates (Fig. 2d), whereas that of mouse SOD1 mRNA was markedly decreased in anti-SOD1 shRNA Tg mice and double Tg mice by 98.0% and 96.1%, respectively (Fig. 2e). In contrast, the enzymatic activity of SOD1 in the liver of double Tg mice was almost equal to that of the wild-type littermates (Fig. 2f).
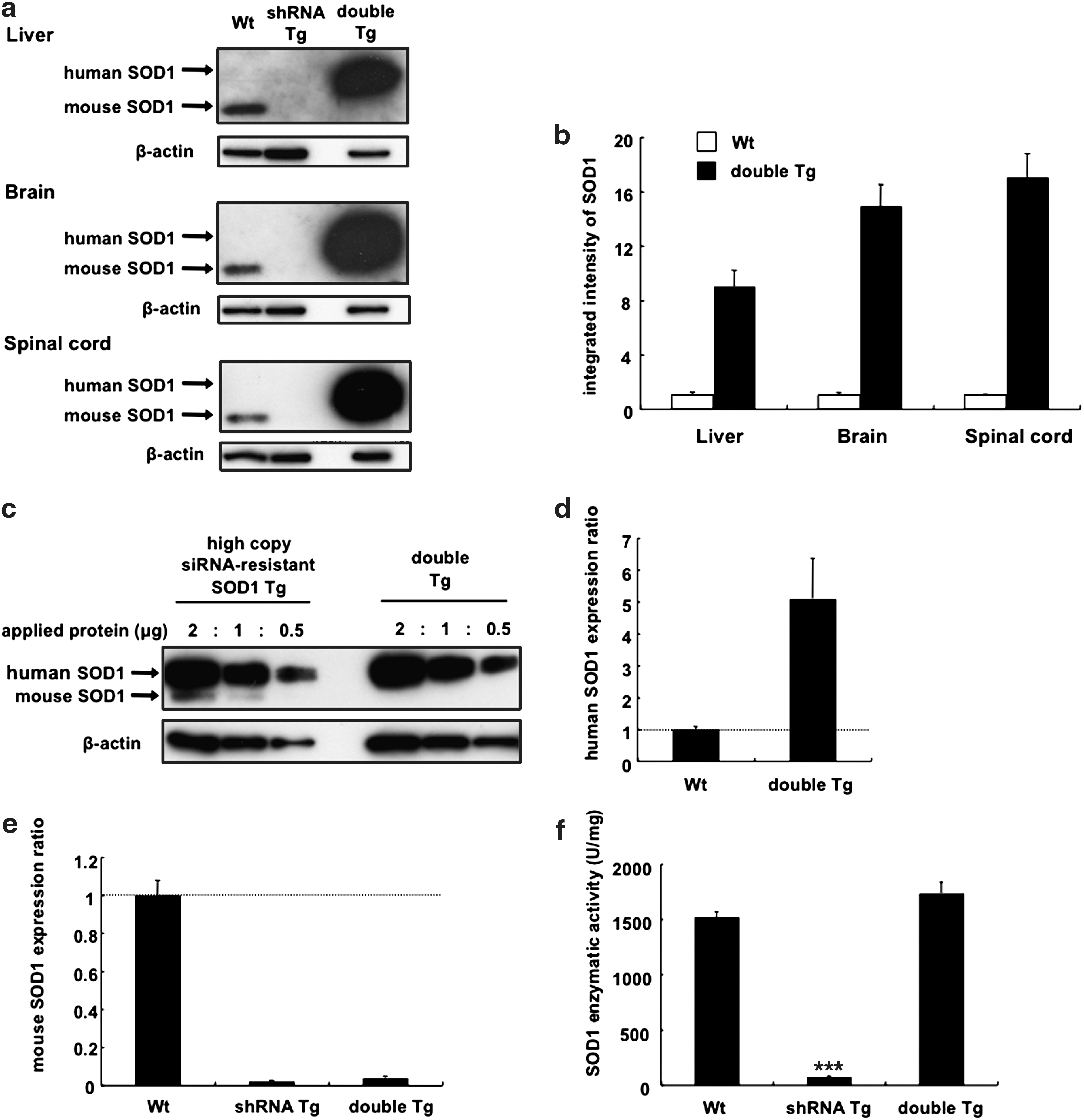
Restoration of wild-type SOD1 expression in double Tg mice. (
Rescue of liver dysfunction in double Tg mice
SOD1 is a major antioxidant, and SOD1 knockout mice exhibit abnormalities such as reduced fertility and enhanced susceptibility to axonal injury and cerebral ischemia (Reaume et al., 1996; Matzuk et al., 1998; Kawase et al., 1999). Furthermore, hepatic lipid accumulation has also been found in SOD1 knockout mice (Uchiyama et al., 2006). Thus, it was considered that oxidative stress enhanced hepatic lipid accumulation by impairing lipoprotein secretion due to the degradation of apolipoprotein B in hepatocytes. Similarly, we observed a significant increase in ALT (Fig. 3a) and the presence of numerous small lipid droplets in the liver of anti-SOD1 shRNA Tg mice (Fig. 3b and c).

Disappearance of liver dysfunction in double Tg mice. (
In double Tg mice, serum ALT levels were recovered to within the normal range (Fig. 3a). Moreover, the number of lipid droplets in the liver of double Tg mice was decreased to levels similar to that of the wild-type mice as observed on Sudan III staining (Fig. 3b and c). The liver abnormalities identified in anti-SOD1 shRNA Tg mice disappeared in double Tg mice, indicating that loss of wild-type SOD1 function was recovered by the expression of siRNA-resistant SOD1.
Vector-mediated delivery of both anti-SOD1 shRNA and siRNA-resistant SOD1
To achieve conditional in vivo knockdown of the target gene with this strategy, we used vector-mediated delivery with rAAV. In order to introduce both shRNA and siRNA-resistant mRNA to each cell in vivo, we generated a construct that dually expressed anti-SOD1 shRNA and siRNA-resistant SOD1 cDNA (pAAV-shRNA/resistant SOD1), as well as shRNA against SOD1 alone (pAAV-shRNA) (Fig. 4a).
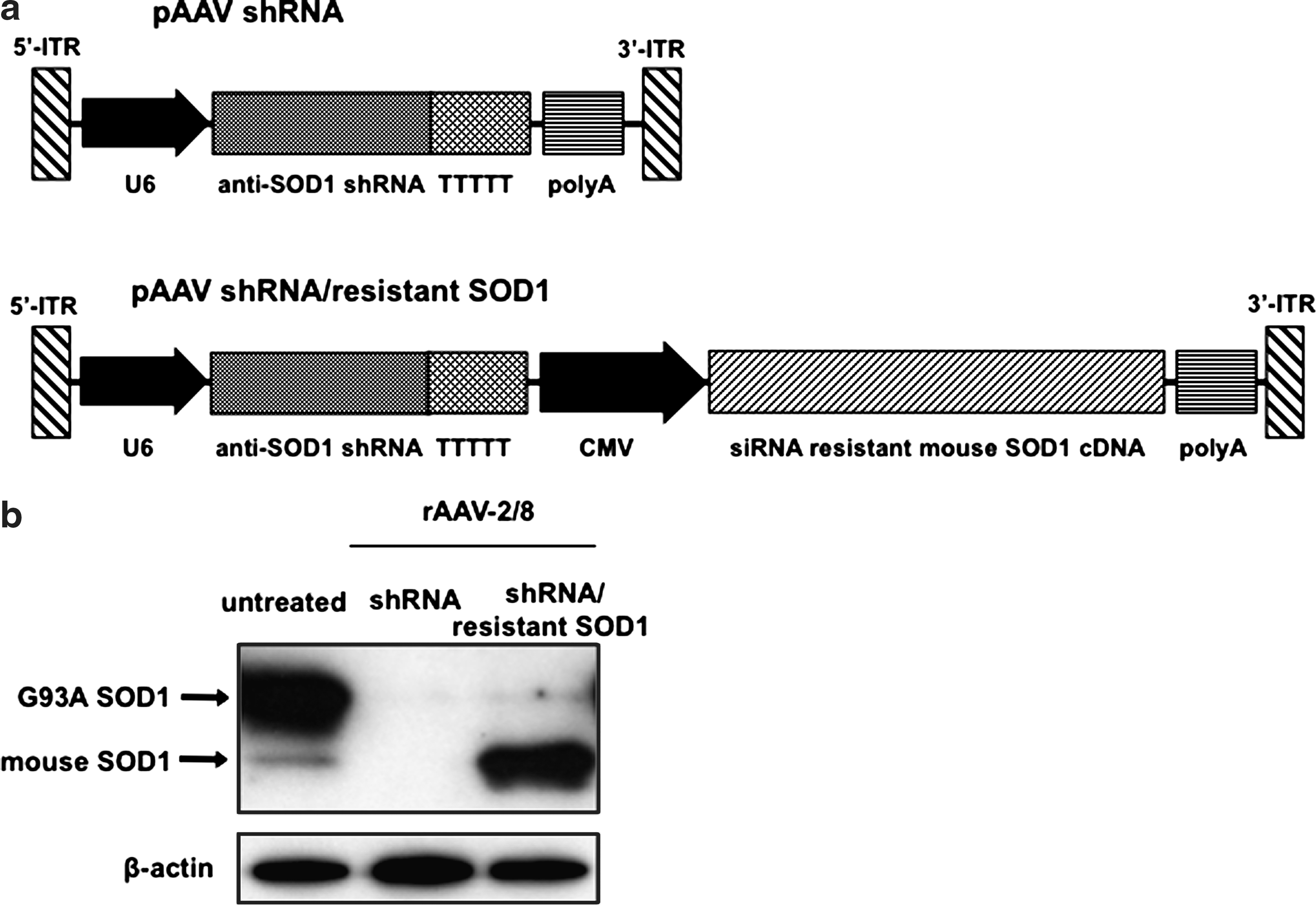
Vector-mediated delivery of anti-SOD1 shRNA and siRNA-resistant SOD1 cDNA. (
SOD1G93A Tg mice were intravenously injected with 1 × 1012 vector genomes per mouse of rAAV-2/8-shRNA or -shRNA/resistant SOD1. Three weeks later, we found that the SOD1G93A Tg mice injected with rAAV-2/8-shRNA demonstrated significant inhibition of both mutant G93A SOD1 and endogenous mouse SOD1 proteins in the liver as assessed by western blot analysis. On the other hand, in SOD1G93A Tg mice injected with rAAV-2/8-shRNA/resistant SOD1, levels of wild-type mouse SOD1 protein were much increased under inhibition of mutant G93A SOD1 protein (Fig. 4b).
Discussion
Our initial reports on our RNAi strategy raised several problems, including (1) the requirement of both siRNA and restored wild-type protein to be delivered to every cell, (2) differences in function that may exist between the endogenous and exogenously expressed proteins, and (3) the control of expression levels of the restored wild-type protein (Kubodera et al., 2005).
In order to express both siRNA and restored wild-type protein in each cell, we constructed a cassette that dually expressed anti-SOD1 shRNA containing U6 promoter and siRNA-resistant wild-type SOD1 cDNA containing CMV promoter in rAAV vectors. Following systemic intravenous injection of this rAAV vector into SOD1G93A Tg mice, mutant G93A SOD1 protein in the liver almost disappeared as seen in mice injected with rAAV vectors expressing anti-SOD1 shRNA alone. In addition, the wild-type mouse SOD1 protein was also restored. As both promoters work ubiquitously, two transcripts should be expressed in each cell. Recently, another vector construct that simultaneously expressed transgene and shRNA was reported (Samakoglu et al., 2006). In this construct, a promoter-less lariat-embedded shRNA sequence was inserted within the intron of the PolII-driven protein-coding transgene, generating efficient shRNA from the processed primary transcript.
In the double transgenic mice, the enzymatic activity of restored siRNA-resistant SOD1 was similar to its endogenous levels, and the side effects observed in the anti-SOD1 shRNA Tg mice disappeared without any other additional side effects. Therefore, the expression level of restored siRNA-resistant SOD1 appeared appropriate for our purpose. However, the level of the restored SOD1 enzymatic activity was much less than expected compared with the overexpressed siRNA-resistant SOD1 mRNA in the double Tg mice. As SOD1 functions in its dimeric form, chimeric dimerization of human and mouse SOD1 may not function as a mouse homodimer. Alternatively, there may be differences in post-translational modifications between recombinant human and endogenous mouse SOD1 in the mouse liver that affects enzymatic activity.
Using rAAV-mediated gene delivery, the protein level of overexpressed wild-type mouse SOD1 in the liver was much greater than the endogenous level. It has been reported that human wild-type SOD1 transgenic mice, unlike mutant SOD1 transgenic mice, do not develop motor abnormalities and paralysis (Gurney et al., 1994; Ripps et al., 1995), while aged mice overexpressing wild-type SOD1 show minor motor abnormalities (Jaarsma et al., 2000). In addition, increased wild-type SOD1 accelerates the phenotype of an ALS mouse model with mutant SOD1 (Deng et al., 2006; Wang et al., 2009). α-Synuclein, amyloid precursor protein, and peripheral myelin protein-22 are known to cause autosomal dominant disease in the presence of duplication or triplication of a gene locus (Harding, 1995; Singleton et al., 2003; Rovelet-Lecrux et al., 2006), indicating that wild-type protein expression levels should be strictly controlled. The inducible expression system represents one of the possible techniques that can be used to regulate gene expression. A few gene expression systems that can be regulated with a steroid hormone-dependent and tetracycline-dependent transcriptional switch have been reported (Goverdhana et al., 2005; Manfredsson et al., 2009). However, a precise method for tuning the levels of proteins expressed from transgene has not been established.
The major targets of our RNAi strategy are dominantly inherited diseases in which the causative gene normally plays an important role. Recent studies have indicated that mutations in several dominantly inherited diseases, including polyglutamine diseases, prion disease, and Alzheimer's disease, contributed to pathology through both a loss- and gain-of-function (Van Raamsdonk et al., 2005; Harris and True, 2006; Thomas et al., 2006; Shen and Kelleher, 2007; Lim et al., 2008). For example, in the case of spinocerebellar ataxia type 1, which is one of the polyglutamine diseases and caused by the expansion of a glutamine-encoding CAG repeat in the ataxin-1 gene, ataxin-1 protein forms at least two distinct native complexes. Polyglutamine expansion alters the proportion of the mutant protein participating in the formation of these complexes. One complex then causes disease via a gain-of-function mechanism, whereas the other complex concomitantly causes a loss-of-function (Lim et al., 2008). Our RNAi strategy for allele-specific suppression is suitable for these cases, as concomitant loss of wild-type protein function can be restored in addition to inhibiting the toxicity of the mutant protein. The optimal restored level of the wild-type protein, however, may differ depending on the mechanism of concomitant loss-of-function in each disease.
In conclusion, we present an efficiency of our RNAi strategy for allele-specific suppression in vivo, by preventing the side effects due to downregulation of endogenous wild-type protein using Tg mice and furthermore by the mutant allele-specific gene suppression using intravenously administered viral vectors. Although the restored protein level should be specifically determined for each disease, our in vivo results indicate that our RNAi strategy is promising for gene therapy of dominantly inherited diseases, especially those exhibiting concomitant loss of wild-type protein function.
Footnotes
Acknowledgments
We thank Dr. Masashi Aoki for providing human SOD1 promoter and cDNA and Dr. James M. Wilson for providing the AAV packaging plasmid. This work was supported in part by grants from the Ministry of Education, Science, Culture, Sports, and Technology, Japan (18659256 and 20790610), the Ministry of Health Labor and Welfare, Japan (2212065), and the 21st Century COE Program on Brain Integration and its Disorders from Tokyo Medical and Dental University.
Author Disclosure Statement
No competing financial interests exist.