
Abstract
From the perspective of a pilot clinical gene therapy trial for Wiskott–Aldrich syndrome (WAS), we implemented a process to produce a lentiviral vector under good manufacturing practices (GMP). The process is based on the transient transfection of 293T cells in Cell Factory stacks, scaled up to harvest 50 liters of viral stock per batch, followed by purification of the vesicular stomatitis virus glycoprotein-pseudotyped particles through several membrane-based and chromatographic steps. The process leads to a 200-fold volume concentration and an approximately 3-log reduction in protein and DNA contaminants. An average yield of 13% of infectious particles was obtained in six full-scale preparations. The final product contained low levels of contaminants such as simian virus 40 large T antigen or E1A sequences originating from producer cells. Titers as high as 2 × 109 infectious particles per milliliter were obtained, generating up to 6 × 1011 infectious particles per batch. The purified WAS vector was biologically active, efficiently expressing the genetic insert in WAS protein-deficient B cell lines and transducing CD34+ cells. The vector introduced 0.3–1 vector copy per cell on average in CD34+ cells when used at the concentration of 108 infectious particles per milliliter, which is comparable to preclinical preparations. There was no evidence of cellular toxicity. These results show the implementation of large-scale GMP production, purification, and control of advanced HIV-1-derived lentiviral technology. Results obtained with the WAS vector provide the initial manufacturing and quality control benchmarking that should be helpful to further development and clinical applications.
Introduction
The Wiskott–Aldrich syndrome (WAS) is a candidate disease for gene therapy with HIV-derived lentiviral vectors (Galy et al., 2008). WAS is a rare primary immunodeficiency characterized by microthrombocytopenia, recurrent infections, and eczema and is associated with a high incidence of autoimmunity and of lymphoid malignancies (Ochs and Thrasher, 2006). Preclinical studies have shown the efficacy and safety of ex vivo gene transfer via autologous hematopoietic stem cells, using a third-generation SIN rHIV-1-derived lentiviral vector expressing the human WAS cDNA under the control of endogenous WAS promoter sequences and pseudotyped with the vesicular stomatitis virus glycoprotein (VSVg) (Dupre et al., 2006; Charrier et al., 2007; Mantovani et al., 2009; Marangoni et al., 2009; Zanta-Boussif et al., 2009). These results provided the rationale to develop a clinical gene therapy study to treat patients with severe WAS who do not have a donor for an allogeneic bone marrow transplant. In the perspective of clinical trials, we developed a process for the large-scale production of rHIV vectors according to good manufacturing practices (GMP).
There are few reports describing the production of rHIV vectors in GMP-applicable protocols and to our knowledge, only one that characterizes a batch of lentiviral vector released for clinical use in practical terms (Schonely et al., 2003). Current methods of rHIV clinical vector production generally rely on cotransfecting two to five plasmids encoding the accessory viral proteins or the cassette carrying the transgene of interest (Lu et al., 2004). To circumvent this complex process, several stable rHIV producer cell lines have been described, but none has yet found GMP application (Klages et al., 2000; Farson et al., 2001; Ikeda et al., 2003; Ni et al., 2005; Throm et al., 2009). Thus, transient transfection approaches continue to be employed and generally use 293T cells because of their superior productivity of rHIV particles, although no comparative study with other cells has been published to our knowledge. Several envelope glycoproteins can be used to pseudotype rHIV particles, but the vesicular stomatitis virus G glycoprotein (VSVg) is widely used because of its broad tropism (Burns et al., 1993). In addition, from a manufacturing standpoint, this pseudotype provides resistance to freeze–thawing and sufficient stability for the rHIV particles to withstand concentration or purification methods (Slepushkin et al., 2003; Transfiguracion et al., 2003; Kutner et al., 2009) and is therefore adequate for the manufacture of stable, well-characterized products under GMP conditions. High concentrations of rHIV particles are usually needed for efficient gene transfer in primary cells (Haas et al., 2000), requiring downstream processes for concentration of the viral stock. Relatively crude concentration methods such as ultracentrifugation are commonly employed but also concentrate cellular debris, membrane fragments, and proteins derived from the producer cells and from the culture medium. Such materials can be toxic to target cells, in particular to primary cells, and may cause inflammatory or immunogenic reactions when used in vivo (Selvaggi et al., 1997; Reiser, 2000; Tuschong et al., 2002; Baekelandt et al., 2003). Thus in addition to a concentration step, it is also imperative to purify the rHIV particles to obtain a nontoxic, more defined vector preparation (Scherr et al., 2002; Segura et al., 2007).
This paper herein reports the development of 50-liter scale production of rHIV with extensive purification steps using membrane-based techniques, ion-exchange chromatography, and gel-filtration chromatography. The process was applied to produce a vector developed for the treatment of patients with WAS. Characterization of the batches produced under GMP conditions, evaluation of their in vitro functionality, and some key safety parameters indispensable for their clinical use in comparison with research-grade preparations of lentiviral vectors are presented.
Materials and Methods
Cells
The HEK-293 cell line was derived from human embryonic kidney cells transfected with fragments of mechanically sheared human adenovirus type 5 and selected for characteristics of adenoviral transformation with early region 1 genes (E1A and E1B) (Graham et al., 1977). A clone of HEK-293 cells (so-called 293 cells) was kindly provided by Cell Genesys (South San Francisco, CA) for our study. The 293T cell line was derived from HEK-293 cells after transfection with a plasmid encoding the simian virus 40 (SV40) large T antigen (LTA) (the thermolabile LTA is under the transcriptional control of the Rous sarcoma virus long terminal repeat) and a subclone was selected for its high-yield performance in the production of lentiviral vectors by transient transfection (Naldini et al., 1996). GMP master and working cell banks of 293T cells were constituted for this study. In some experiments, 293FT cells (R700-07; Invitrogen, Cergy-Pontoise, France) were used. These cells were derived from HEK-293 cells (subclone 293F) after transfection with a plasmid encoding the SV40 LTA under the control of the cytomegalovirus (CMV) promoter. HCT 116 cells derived from a human colorectal carcinoma were obtained from the American Type Culture Collection (CCL-247; ATCC, Manassas, VA) and used for titration. The cell lines 293, 293T, and HCT 116 were cultured at 37°C, 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Invitrogen/GIBCO) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT). The B lymphoblastic cell line (B-LCL) 04W was derived from the blood cells of a patient with WAS after transformation with Epstein-Barr virus (EBV) and is described elsewhere (Charrier et al., 2007). The T lymphoid cell line C8166-45 (carrying but not expressing the human T lymphotropic virus [HTLV]-1 genome) originated from the NIH AIDS Research & Reference Reagent Program (Germantown, MD). The T lymphoid cell line C8166-45 and the B-LCL 04W cells were cultured in RMPI supplemented with 10% FBS, glutamine, and antibiotics.
Vectors and plasmids
The W1.6-huWASP-WPRE vector, described by Zanta-Boussif and colleagues (2009), was produced by transient transfection of 293T cells, using four plasmids consisting of either the pRRLW1.6-huWASP-WPREmut6-K or pCCLW1.6-huWASP-WPREmut6-K transfer plasmid combined with the pKLgagpol, pKRev, and pKG plasmids encoding, respectively, HIV-1 gag-pol genes, the HIV-1 rev gene, and unrelated vesicular stomatitis virus G glycoprotein. All plasmids carry the kanamycin resistance gene. During the development phase, the plasmids were produced with NucleoBond PC 10000 EF giga-gravity flow endotoxin-free columns according to the instructions of the manufacturer (Macherey-Nagel, Hoerdt, France). For the runs produced according to GMP, we used GMP-grade plasmids manufactured by COBRA (Keele, UK).
In some experiments, a control vector encoding enhanced green fluorescent protein (eGFP) was produced with a pRRL PGK-eGFP-WPRE transfer plasmid, using ultracentrifugation for purification, as previously described (Charrier et al., 2005).
Large-scale vector production in Nunc Cell Factory 10-tray stacks
On day 0, 24 Nunc Cell Factory 10-tray stacks (CF10; Thermo Fisher Scientific, Waltham, MA) were seeded with 293T cells. Culture and production medium was DMEM (GIBCO) plus 10% FCS (HyClone). Medium was changed 2 hr before transfection on day 3. Cells were transfected by a calcium phosphate method as reported previously (Graham et al., 1977) with the following amounts of plasmids per CF10: pPGK-GFP or pW1.6-huWASP (760 μg), pKLgagpol (500 μg), pKG (270 μg), and pKRev (191 μg). The medium was first changed on day 4. Supernatant was harvested 24 and 48 hr later. After pooling the harvested supernatants, the crude viral stock supernatant was filtered, clarified, and treated overnight at 4°C with nuclease (Benzonase, grade I, at least 1.1 × 106 U/mg; Merck Chemicals, Darmstadt, Germany), used at a final concentration of 5 U/ml. Viral particles were captured by DEAE ion-exchange chromatography (Tosoh Bioscience Division, Tokyo, Japan). After elution with 750 mM NaCl, the vector-containing eluate was concentrated approximately 20- to 30-fold by ultrafiltration to about 75 ml, using a hollow fiber cartridge (nominal cutoff, 750 kDa; GE Healthcare Life Sciences, Orsay, France) and diafiltered in phosphate-buffered saline (PBS). Concentrated and ultrafiltrated vector material was further purified by size-exclusion chromatography and formulated in X-VIVO 20 medium (Lonza, Basel, Switzerland). The vector was aliquoted and stored at −80°C.
Titration
Infectious titers were determined by quantitative polymerase chain reaction (qPCR) after infection of HCT 116 cells with serial dilutions of the vector and are expressed as infectious genomes (IG) per milliliter, as previously described by Charrier and colleagues (2007). The titration of lentiviral (LV)–eGFP was performed as described initially by Follenzi and Naldini (2002), using HCT 116 cells instead of HeLa cells, and the titer is expressed as transducing units per milliliter (TU/ml). Physical particle titers were determined by measuring capsid protein (p24) by ELISA (HIV-1 p24 core kit, NEK050B; PerkinElmer Life Sciences, Boston, MA).
Protein and DNA contaminant testing
Total protein content was measured by spectroscopy, using the Bradford protein colorimetric assay with a sensitivity of 10 μg/ml. Total DNA content was measured by spectrofluorimetry with PicoGreen (Invitrogen) with a sensitivity of 1 ng of DNA/ml. Levels of Benzonase were measured by ELISA (Merck) with a sensitivity of 200 pg/ml. Total host cell protein (HCP) levels were measured by ELISA with the HEK-293 HCP kit with a threshold of 300 pg/ml (Cygnus Technologies, Southport, NC). Bovine serum albumin was measured by ELISA with a sensitivity of 250 pg/ml (Cygnus Technologies).
Detection of replication-competent lentiviral particles by p24-decreasing test
The replication-competent lentivirus (RCL) assay was performed as described by Escarpe and colleagues (2003). The positive reference control was produced by cotransfection of a wild-type HIV-1 proviral plasmid lacking the accessory viral genes (ΔvifΔvprΔvpuΔnef) and another plasmid encoding the envelope glycoprotein, which must be the same as the one used for the pseudotyping of vectors to test. The sensitivity of the assay, in the context of the VSVg-pseudotyped vector production, was calculated to be approximately 1 median tissue culture infective dose (TCID50)/2 × 108 transduction units per T-175 flask as reported (Escarpe et al., 2003).
Detection of cellular DNA sequences in the vector
The presence of E1A and SV40 large T antigen sequences in the vector was tested by amplifying 64- and 112-bp sequences specific for E1A and SV40 large T antigen, respectively, using qPCR. In both cases, a noncorrelated internal control is spiked in the sample before the extraction step and its quantification allows the efficiencies of DNA extraction and PCR amplification to be normalized. The limit of quantification is 5000 copies/ml for both the E1A and SV40 large T antigen sequences.
Transfer of plasmid DNA (VSVg) and adenoviral (E1A and E1B) and SV40 (large T antigen) genomic DNA sequences to target cells
The transfer of DNA contaminants contained in vector batches to target cells was tested by qPCR after infection of C8166-45 cells with the vector and expansion of the cells in culture over a total of 29 days with six passages. For VSVg detection, the pMD.G plasmid was used to establish a standard curve. For the detection of E1A and SV40 large T antigen, the plasmids pTOPO_ALB_E1A and pTOPO_ALB_AgTSV40, containing one copy of a cellular gene (albumin) and one copy of either E1A or SV40 large T sequence, were used to prepare standard curves. The detection limit varied between 1000 and 20 copies of specific sequence per 100 ng of genomic DNA, depending on experiments.
Human CD34+ cell cultures
Umbilical cord blood (UCB) progenitor CD34+ cells were obtained by immunomagnetic selection (Miltenyi Biotec, Paris, France) and preactivated overnight by incubating 5 × 104 CD34+ cells in 0.2 ml of X-VIVO 20 medium (Lonza), supplemented with penicillin (50 U/ml), streptomycin (50 μg/ml), and 2 mM
Expression of the insert
A WAS protein (WASP)-negative B-LCL were infected with vector particles, using a concentration of 2 × 105 cells/0.2 ml per well in the presence of Polybrene (6 μg/ml) for 6 hr with various concentrations of vector ranging from 0.05 to 10 × 107 IG/ml, with the multiplicity of infection (MOI) ranging from 0.5 to 450. After transduction, cells were washed and incubated for 8–10 days, adding fresh medium every 3 days. The expression of WASP was measured by immunoblotting as previously described (Charrier et al., 2007).
Results
Initial specifications
The VSVg-pseudotyped WAS lentiviral vector, described elsewhere (Charrier et al., 2007; Galy et al., 2008), has been developed for the ex vivo transduction of patients' autologous CD34+ cells and leads to the genomic integration of a 5.9-kb expression cassette expressing the WAS protein (Fig. 1). From the perspective of manufacturing for clinical studies, the scale of production of such a vector was estimated from the following initial postulates. Approximately 5 × 109 particles of infectious vector would be needed per patient, based on an average 20-kg young patient receiving 5 × 106 CD34+ cells/kg transduced with a multiplicity of infection (MOI) arbitrarily fixed at 50. Aiming to produce a batch to treat several patients (at least five) and estimating that, because of quality assurance and regulations, the GMP quality controls would consume approximately one-half the volume of the batch, we set out to produce at least 5 × 1010 rHIV particles per batch. Because, typically, about 107 infectious particles per milliliter of VSVg-pseudotyped rHIV vector can be produced after transfection of 293T cells (Kutner et al., 2009), and considering that the yield of the purification and concentration steps could be about 10%, it was then estimated that 50 liters of viral supernatant stock should be manufactured per batch. Such a scale can be achieved by culturing the producer cells in Cell Factory 10-tray stacks (CF10), which enables the implementation of closed-circuit viral stock production schemes adaptable to GMP (Przybylowski et al., 2006). In addition, these vessels allow the direct upscaling of transfection conditions from tissue culture procedures, in a manner proportional to the surface area (Geraerts et al., 2005).
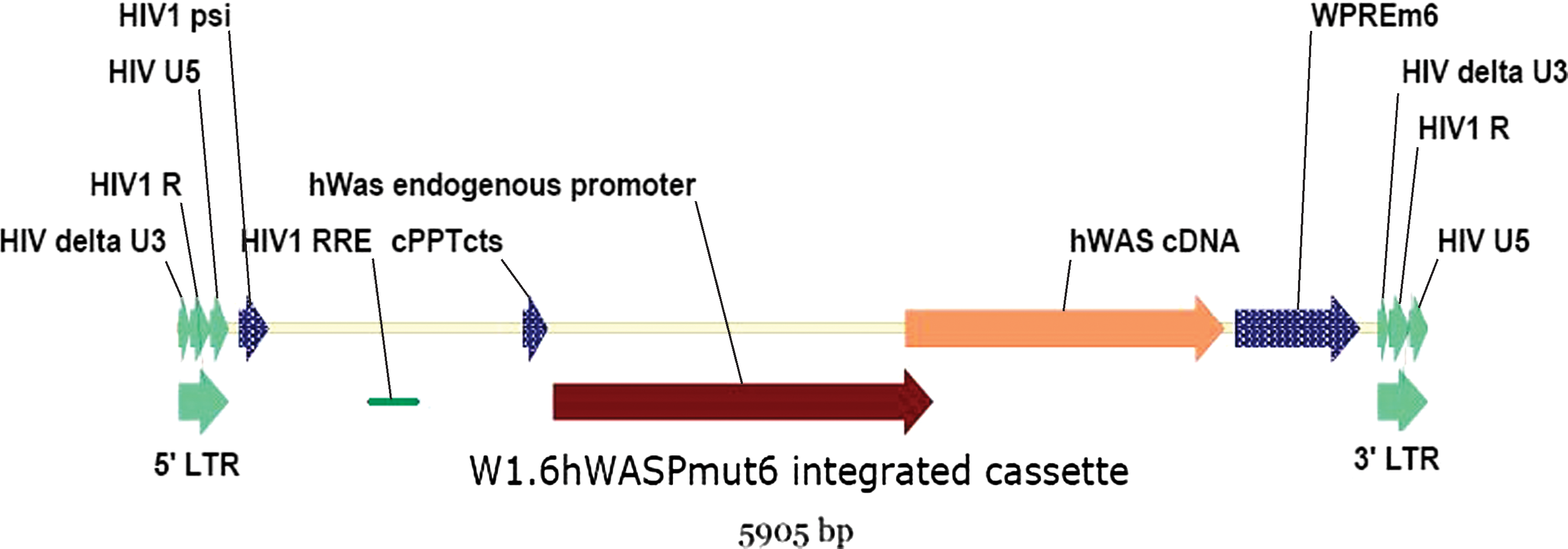
Schematic representation of the proviral sequence after integration of the WAS vector into target cells. HIV, human immunodeficiency virus; Psi, encapsidation sequence; PBS, primer-binding site sequence; LTR, long terminal repeat, 5′ or 3′; HIV R, HIV R region; HIV delta U3, U3 region with a deletion; RRE, Rev-responsive element; cPPT cts, central polypurine tract–central terminal sequence; hWAS, human WAS gene; WPRE m6, mutated woodchuck hepatitis virus posttranscriptional regulatory element (Zanta-Boussif et al., 2009). Color images available online at
Plasmid system for clinical vector production
Recombinant WAS–LV were produced by cotransfection of four plasmids encoding separately HIV-1 Gag-Pol viral proteins and enzymes, the HIV-1 Rev protein, the VSVg pseudotype, and the WAS gene transfer construct. Because the WAS vector is an advanced generation vector, the gene transfer cassette is deleted of all HIV accessory protein sequences and of tat. A hybrid 5′ LTR enables the Tat-independent production of the viral RNA (Dull et al., 1998). Such a heterologous 5′ LTR can be made by fusing the Rous sarcoma virus (RSV) or the CMV promoter to the HIV R-U5 elements, generating, respectively, a pRRL- or pCCL-type construct. To evaluate the impact of the 5′ LTR design on the production of the WAS vector, we compared the titers obtained in small-scale productions by quadritransfection with the pRRL-W-WAS or pCCL-W-WAS plasmid, differing only in the 5′ LTR region. Titers were established after concentration by ultracentrifugation. The pCCL system generated at least 2-fold higher titers than the pRRL system in small-scale productions performed in T-225 flasks [1.47 (±0.57) × 1010 vs. 0.84 (±2.2) × 1010 IG/ml] and particles of higher infectivity [1.8 (±0.6) × 105 vs. 1.1 ( ± 0.1) × 105 IG/ng p24]. Similar results were obtained in one CF10 flask experiment (5.5 × 1010 vs. 1.6 × 1010 IG/ml and 1.4 × 105 vs. 4.4 × 104 IG/ng p24). These results confirm previously reported observations (Dull et al., 1998) and prompted us to choose a pCCL design for the transfer vector.
Choice of producer cell line
Both HEK-293 cells (Lu et al., 2004) and their genetic derivative, 293T cells (Naldini et al., 1996), have been used for the production of lentiviral vectors by transient transfection. Although 293T cells are generally most widely used, the parental HEK-293 cell line may present an advantage in terms of safety as it lacks the SV40 large T antigen (SV40 LTA)-encoding gene, which is an oncogene. HEK-293 and 293T cells were therefore compared for the production of a lentiviral vector at small scale in T-75 flasks or in single CF10 flasks. A construct encoding eGFP was used for rapid evaluation. As expected, 293T cells had a significantly shorter doubling time than HEK-293 cells (19 vs. 25 hr). Visual inspection 24 hr after transfection showed higher percentages of eGFP+ cells in the 293T cell culture compared with HEK-293 cells, suggesting a higher transfection efficiency with 293T cells (data not shown). Under nonoptimized conditions, viral stock titers were higher with 293T cells than with HEK-293 cells (1.2 × 107 and 3.7 × 106 TU/ml in T-75 and CF10 systems, respectively, for 293T cells, and 2 × 106 and 9.9 × 105 TU/ml in T-75 and CF10 systems, respectively, for HEK-293 cells). Attempts to optimize vector production by HEK-293 cells in CF10 stacks did not sufficiently improve the production or titers to compare favorably with 293T cells. Under the best conditions, and using an eGFP construct, the total vector amount produced with 293T cells was about four times higher than that produced with HEK-293 cells (1.74 × 1010 vs. 4.59 × 109 TU). In addition, vector infectivity was superior by about 2-fold for 293T-based production than for HEK-293-based production. These results led to the decision to develop the lentiviral vector-manufacturing process on the basis of the use of 293T cells. The presence of SV40 large T antigen in these cells requires monitoring the levels of this specific contaminant in the vector preparation. For clinical vector production, a master cell bank of 293T cells was produced under GMP conditions and was certified.
Vector production
The process of rHIV lentiviral particle production results from several operations streamlined to reduce manipulations and to facilitate the adaptation of the process to a GMP environment. A published protocol based on calcium phosphate transfection of 293T cells and only two harvests (one per day) (Follenzi and Naldini, 2002) was scaled up to 24 CF10 stacks to produce 48–50 liters of viral stock. The concentrations and amounts of plasmids needed to produce the WAS vector were optimized in single flasks and then scaled up accordingly for 24 CF10 stacks. As in other published protocols, the concentration of the WAS transfer plasmid exceeds that of the other plasmids (Geraerts et al., 2005; Mitta et al., 2005). Matrix experiments varying the amounts of each of the four plasmids, from 0.5- to 3-fold the set amount, had no impact on the WAS LV titer except for a drop observed when the VSVg plasmid was used in high amounts (data not shown), thus confirming observations by others (Geraerts et al., 2005). In six full-scale WAS vector productions using the same process scheme, we obtained a raw viral stock titrating in the range of 1–5 × 107 IG/ml, with particles of good quality ranging in infectivity from 0.4 × 105 to 1.4 × 105 IG per nanogram of p24 (Table 1), but these harvested stocks also contained contaminants. In the four developmental runs the initial total protein concentration was 6.14 ± 1.47 mg/ml and the initial total DNA concentration was 1.97 ± 0.0 μg/ml, thus representing average values of 21.4 mg of protein and 6.9 μg of DNA per 108 IG in the starting material.
IG, infectious genomes; GMP, Good Manufacturing Practices.
Twelve liters processed.
The X-VIVO 20 formulation medium contains 1.12 mg of protein per milliliter.
Downstream process for vector purification
The downstream processing protocol had as its objectives the purification and concentration of the viral vector, the elimination of process- and cell-derived contaminants, and the formulation of the final vector preparation for clinical use. In preclinical studies the WAS phenotype could be effectively corrected when using the WAS lentiviral vector at concentrations of 0.5–1 × 108 IG/ml (Dupre et al., 2006; Charrier et al., 2007; Marangoni et al., 2009; Zanta-Boussif et al., 2009). We therefore set out to obtain a vector concentration of at least 2 × 108 IG/ml in the final processed product. From the perspective of GMP application, a multistep purification and concentration scheme was developed, combining several chromatographic and membrane-based process steps that had demonstrated their advantages in our hands and in prior published studies for retroviral or lentiviral vector purification (Scherr and Eder, 2002; Geraerts et al., 2005; Segura et al., 2006). This process was successfully applied to six large-scale production runs (Table 1). Whereas four of the large-scale runs were performed under standardized conditions during the development phase, the last two runs intended for clinical use were manufactured under GMP, using the same cells and plasmids as during development but now in the form of GMP-grade materials.
As a first step, the harvested viral stocks were clarified through low-retention membrane filters of decreasing pore size (0.8 to 0.45 μm). Subsequently, the filtered stock was treated with an endonuclease (Benzonase) overnight at 4°C to remove plasmid and other nucleic acid contaminants. About two-thirds of the infectious particles were recovered and the infectivity dropped by less than 2-fold (Fig. 2B) after this initial filtration and DNase treatment step, leading to about 85% reduction in DNA content of the product (Table 2). Anion-exchange chromatography, which is known to be an effective method to purify retroviral particles (Segura et al., 2006), was then used for capture. The bulk viral particles were pumped over a DEAE ion-exchange chromatography column, and the column was washed with PBS and step-eluted with NaCl in a volume of 2 liters. This process step led to the removal of greater than 99% of total proteins (Table 2). The eluate was desalted and concentrated approximately 20- to 30-fold by tangential flow filtration, which is a technique known to effectively concentrate VSVg-pseudotyped lentiviral particles with good yield (Geraerts et al., 2005). At this step, about 70 ml of product was generated by passage through a hollow fiber cartridge, leading to further reduction in protein and DNA contaminants (Table 2). Last, the concentrated viral suspension was equilibrated into the formulation medium (X-VIVO 20) by size-exclusion chromatography with a vector load volume-to-column volume ratio of about 0.15. The collected eluate of about 200 ml was sterile-filtered through a 0.22-μm capsule-filter, which was washed/chased with about 40–50 ml of vector formulation medium, generating about 250 ml of final product. At this stage the process had achieved a volume concentration of 200-fold and was purified as shown by an approximately 3-log reduction in total protein and DNA production contaminants with residual levels of 1.38 ± 0.3 mg of protein per milliliter and 1.38 ± 1.24 μg of DNA per milliliter (Table 2). The downstream processing reduced the global DNA load to levels of 0.6–3.2 μg/ml, providing 0.023–1.1 μg of DNA per 108 IG (Table 1). The most efficient steps to reduce the DNA load were the Benzonase treatment and the tangential flow filtration steps, eliminating 85.4 ± 1.93% and a further 14.5% (total reduction, 98.97 ± 0.83%) of the total DNA. For proteins, the reduction factor was >700-fold as evaluated globally. In this case, the most efficient step was the DEAE chromatography, eliminating 99.85 ± 0.09% of the total initial protein load (Table 2). Downstream steps showed no further significant improvement due to the use of a protein-containing formulation medium. At the gel-filtration step, the final vector preparation was formulated in X-VIVO 20 medium containing >1.12 mg of protein per milliliter, and therefore the residual protein level was elevated in the final product. For the development runs, the final product contained 1.38 ± 0.30 mg of total protein per milliliter (Table 2) (0.74 ± 0.29 mg of protein per 108 IG) and for the GMP runs, the levels ranged between 1.35 and 1.4 mg/ml (less than 0.1 mg of protein per 108 IG) (Table 1). When taking into account the addition of proteins due to formulation in X-VIVO 20 medium and deducting that value from the residual protein level, one can estimate that the process generated 0.12 ± 0.14 mg of protein per 108 IG in the case of the four development runs (Table 1). Considering an initial average value of 21 mg of protein per 108 IG in the harvested bulk stock, these values demonstrate that the process resulted in extensive protein removal and purification. Whether or not the proteins in the final formulation medium are taken into account, the final processed product contains, respectively, 0.74 ± 0.29 or 0.12 ± 0.14 mg of protein per 108 IG, which is far below the protein contamination level specification of <7 mg of protein per 108 IG proposed by Schonely and colleagues (2003).
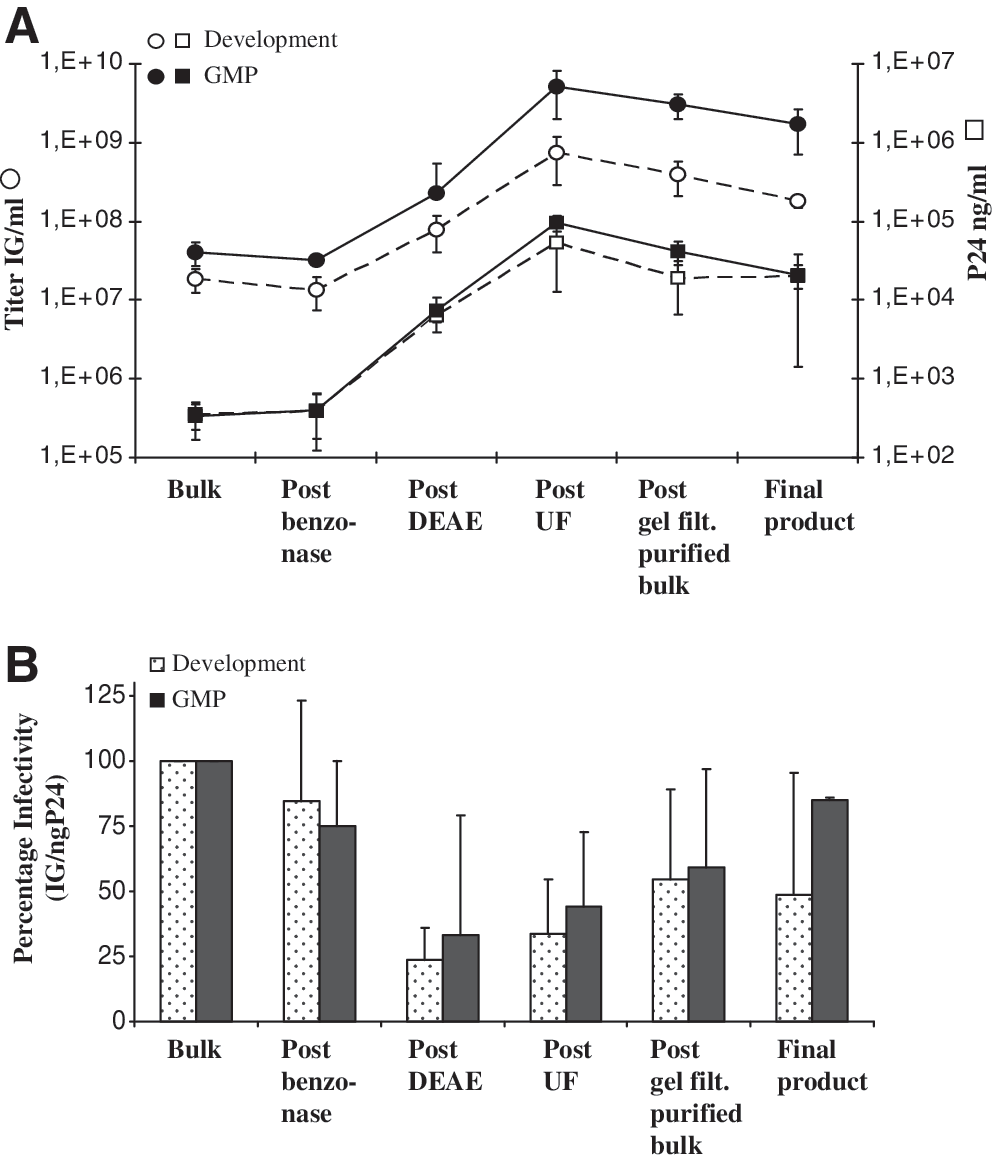
Analysis of the various steps of the downstream processing protocol under large-scale development and GMP conditions. (
TFF, tangential flow filtration.
The second and third columns show the expected volume and volumic concentration at each step of the process. The protein and DNA values measured during the four large-scale development runs were used to calculate reduction factors with actual volumes of product processed and obtained. The average initial total protein concentration was 6.14 ± 1.47 mg/ml, the initial total DNA concentration was 1.97 ± 0.0 μg/ml, the final total protein concentration was 1.38 ± 0.3 mg/ml (including the protein load added via the formulation medium), and the final total DNA concentration was 1.38 ± 1.24 μg/ml.
The X-VIVO 20 formulation medium contains 1.12 mg of protein per milliliter.
Altogether, with the six large-scale production runs, the process attained the initial specifications of 2 × 108 IG/ml, and the infectivity of the virions was generally higher than 1 × 104 IG/ng p24. The overall yield was low although generally over 10%. Greater than 5 × 1010 IG could be produced per batch, thus meeting our initial expectations. However, the two GMP runs exceeded those expectations, providing high vector titers (>109 IG/ml) with high infectivity (>4 × 104 IG/ngp24) and good yields (16–23%) and leading to a total production of 2–6 × 10 11 IG per batch (Table 1).
Concentration of vector during process: comparison between runs in development or GMP
During the process, the most important concentration steps were the ion-exchange chromatography and ultrafiltration steps leading to an approximate increase in infectious vector concentration by a factor of 67 ± 23-fold and 163 ± 113-fold, respectively, both for the runs performed in development and under GMP conditions (Fig. 2A). Physical particle titers increased accordingly at the same steps. A reduction in titer was observed in the final steps, due both to vector dilution and probably to a slight adsorption of the vector to the sterilizing membrane. Decreased infectivity (IG/ng p24 ratio) was noted after anion-exchange chromatography, which could be partially recovered in the ultrafiltration step. This can be explained by the elevated salt concentration necessary for vector elution, which seems to impact on the determination of the infectious titer by qPCR but not on the physical titer by ELISA. Indeed, the steps downstream of the DEAE chromatography step are characterized by a partial increase in the ratio of infectious genomes to nanograms of p24 (Fig. 2B). Differences between the development and GMP runs were noticed at two stages: first, at the start of the process, higher viral stock titers were obtained in the GMP runs; second, the tangential flow filtration step gave slightly higher yields in GMP than in the development runs (Fig. 2A). Overall, the quality of the vector (infectivity) during the development runs was reduced by about 40% when bulk and finished product values were compared, whereas under GMP conditions the downstream processing protocol kept the infectivity of the particles fairly constant (Fig. 2B).
Characterization of specific vector contaminants
The downstream processing protocol reduced global DNA and protein loads by greater than 1000- and 700-fold, respectively, as measured during in-process controls (Table 2). To facilitate the use of the finished product for the transduction of hematopoietic cells, it was formulated in X-VIVO 20 medium, which contains approximately 1.1 mg of protein per milliliter. Additional protein contaminants may also be present, originating from the production medium, producer cells, or the process, and may constitute a risk for using the product in patients. Levels of specific contaminants were therefore measured in the development runs to characterize the products and to provide the expected values and specifications for GMP runs. One of the process by-products is the endonuclease Benzonase, which is incubated at a concentration of about 5 ng/ml with the viral vector stock at the beginning of the process, thus representing the addition of about 2.5 × 105 units of Benzonase per batch. This exogenous enzyme, unnecessary for the biological activity of the vector, was removed during downstream processing. In the final purified and formulated product, residual Benzonase was undetectable in all batches produced in development or in GMP (Table 3). Bovine serum albumin is a constituent present at about 2.5 mg/ml in the viral production medium whereas less than 500 ng/ml of bovine serum albumin is measured in the final product indicating a removal factor of at least 5000-fold for this specific protein (Table 3). Total host cell proteins derived from the producer cells were measured with an HEK-293-specific ELISA and found in small amounts not exceeding 200 ng/ml.
GMP, Good Manufacturing Practices; LTA, large T antigen; SV40, simian virus 40; VSVg, vesicular stomatitis virus glycoprotein.
A specific product of the producer cells is the SV40 large T antigen (SV40 LTA), which is oncogenic for many cell types. Using Western blot analysis, it was possible to detect the presence of SV40 LTA in lentiviral vector preparations concentrated simply by ultracentrifugation of the viral stock (Fig. 3). In contrast, no residual SV40 LTA was detected with this method in vector purified by the multistep membrane and chromatographic process. Because all samples tested contained equivalent amounts of purified or ultracentrifuged particles, as shown by the HIV p24 and p17 immunoblot (Fig. 3), this confirmed that the purification process was able to remove this specific contaminant. To further assess the oncogenic potential of the preparation, we measured the presence of specific E1A or SV40 LTA DNA sequences that could have been potentially released from the producer cells during production. Using quantitative PCR, between 0.37 × 105 and 2.4 × 105 copies of E1A and between 0.69 × 105 and 4.2 × 105 copies of SV40 LTA were detected per milliliter of purified vector. With an average of 6.6 E1A copies per cell and 2.1 SV40 LTA copies per cell in 293T cells, and considering an arbitrary C value of 9.8 pg of DNA per 293T cell (considered to be triploid cells), the two purified GMP vector lots would contain between 0.05 and 2 μg of cellular genomic DNA per milliliter, which could represent as much as 70% of the total DNA content of the preparation as measured by fluorimetry (Table 1). The remainder of the DNA probably originates from plasmids used during cell transfection. To evaluate the significance of these residual E1A and SV40LTA sequences, we verified how much could be passively transferred to target cells on infection. We cultured sensitive indicator cells (C8166-45 cells) with the vectors and amplified the cell culture. At the first time point examined, 1 week after transduction, E1A and SV40 LTA sequence-specific signals were below detection in the culture by qPCR. Thus, the small amounts of residual E1A and SV40LTA eventually present in the purified vector would likely be insufficient to transform target cells. In the same experiments, we also verified the possible transfer of other DNA sequences, such as those from plasmids. The amplification of VSVg sequences (taken as reference for material of plasmidic origin), was not detected in cultures of indicator cells exposed to the purified vectors (Table 3), whereas ultracentrifuged preparations could transfer such sequences to cells in the first passages (data not shown).
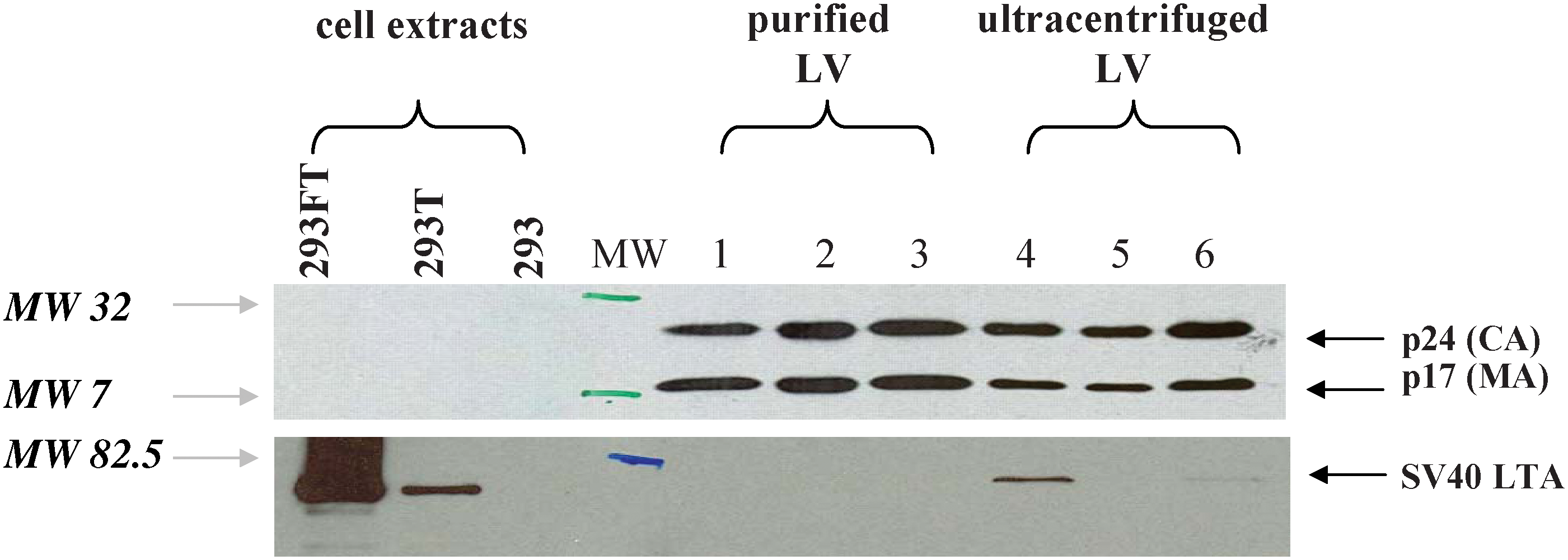
Absence of SV40 T antigen detection and identification of the viral proteins in the LV vector. Immunoblots of the purified viral vectors with antibodies specific for HIV p24 capsid and p17 matrix proteins (top blot; proteins of 24 and 17 kDa size; MW markers 32 and 7 kDa are shown) and specific for SV40 large T antigen (bottom blot, protein of 80–95 kDa; MW marker 82.5 is shown). Lanes 1–3 correspond to several preparations of rHIV WAS lentiviral vector purified and produced in development or in GMP conditions. Lanes 4–6 correspond to preparations of WAS or GFP rHIV vectors produced at small-scale and concentrated by ultracentrifugation. Cell extracts obtained from 293 and 293T cells were used as negative and positive controls. Cell extracts from 293FT cells were also added because these express SV40 T antigen at high levels. CA, capsid; LTA, large T antigen; LV, lentivirus; MA, matrix; MW, molecular weight. Color images available online at
Absence of replicative particles in WAS lentiviral vector
In spite of the many safety features of the lentiviral vector system, the absence of replicative recombinant HIV particles (replication-competent lentivirus, RCL) must be demonstrated under permissive conditions, using a sensitive assay. The absence of RCL was demonstrated in six batches of WAS lentiviral vector, including three small-scale batches, one large-scale development run, and two large-scale GMP batches. In all cases, 5% of the total volume of each batch was tested using the p24 decrease assay as described (Escarpe et al., 2003). A positive control, consisting of an attenuated strain of HIV-1 deleted of accessory genes and carrying both the HIV and VSVg envelopes, was used and gave a TCID50 corresponding to 10 to 20 fg of p24. Spiking controls confirmed that none of the vector batches tested inhibited the detection of RCL with this control. For all six batches of WAS LV tested, the p24 level was below the detection limit after amplification of C8166-45 target cells for 28 days. The largest amounts of infectious particles were tested in the two GMP batches and results indicated less than 1 RCL in 1.25–3 × 1010 IG, which corresponds to the administration of less than one RCL per patient, considering the doses that are planned to be used in the clinical trial.
Transduction and absence of toxicity of purified vector on CD34+ cells
The lack of toxicity of the WAS lentiviral vector was demonstrated by a lack of effect on cellular growth, viability, and differentiation after infection and culture of umbilical cord blood (UCB) CD34+ cells from normal individuals. Because WAS is a rare disease, such cells were used as surrogate representatives of the target cells in the clinical studies. Four large-scale development runs were tested on a total of 13 UCB CD34+ cell samples with vector concentrations ranging from 1 × 107 to 10 × 107 IG/ml. No effect of the vector was observed on the growth or viability of cells cultured in the presence of cytokines, and there was also no measurable impact on the clonogenicity of the hematopoietic progenitor cells compared with untransduced cells or cells transduced with a GFP-encoding ultracentrifuged vector (Fig. 4A). Similar results were obtained with the GMP runs (data not shown). The toxicity of the WAS vector was also assessed by measuring the differentiation of clonogenic progenitor cells after infection. Each of the two GMP runs were tested at two concentrations each and on three UCB CD34+ cell samples each and revealed no toxicity. Figure 4B shows that the WAS lentiviral vector batches produced under GMP did not affect the number of myeloid, erythroid, or primitive progenitor cells compared with untransduced cells, as was also observed with the vector produced during development (data not shown). The ability to transduce CD34+ cells was verified by measuring the average number of vector copies integrated into the genomic DNA of the cell population. A vector concentration dose-dependent effect on integrated copies was observed (Table 4A). At the highest concentration tested, 1 × 108 IG/ml, the average vector copy number was between 0.3 and about 1.2 per cell in the whole population, which is comparable to expected values and confirms the good infectivity of the vector produced under GMP.
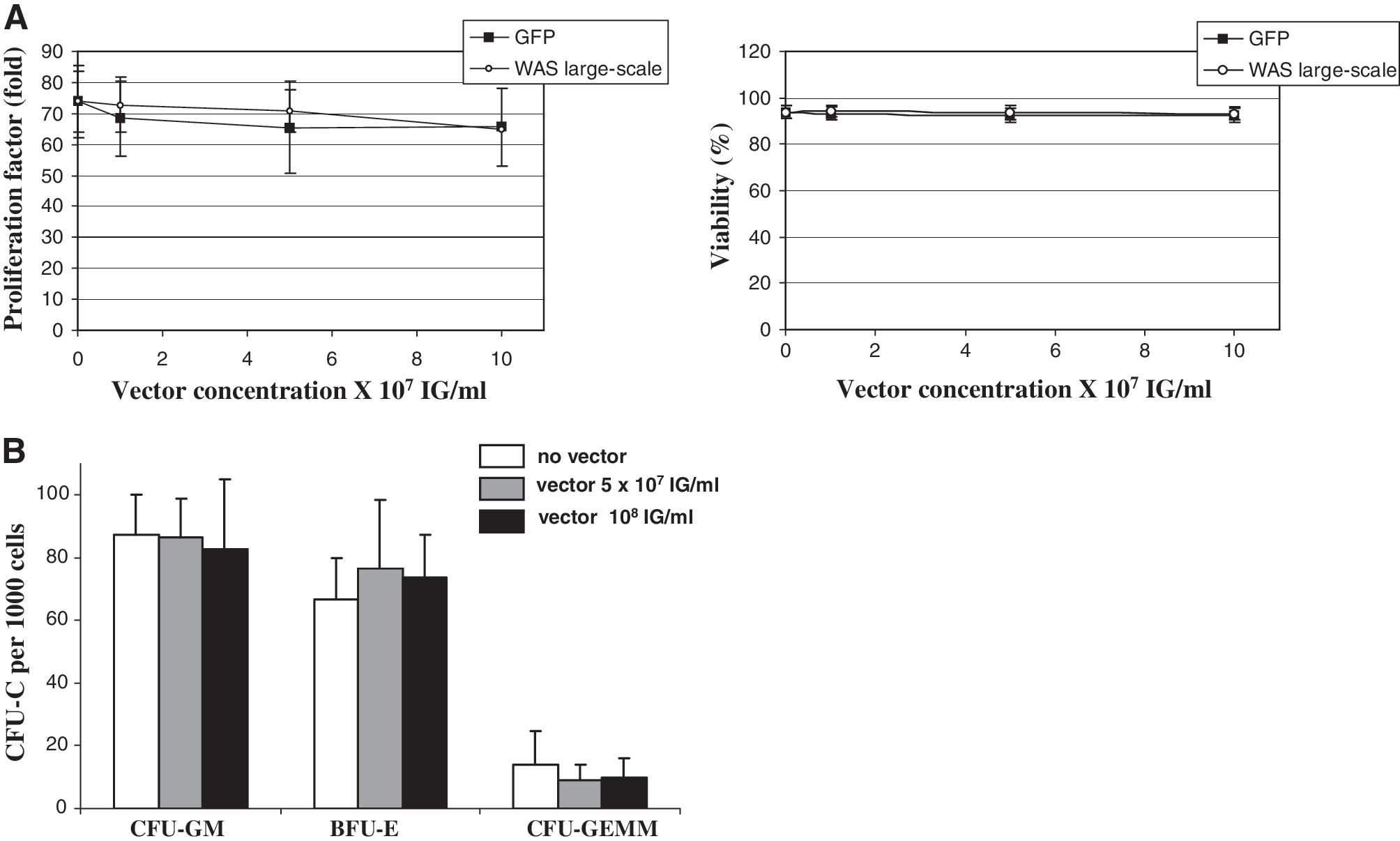
Absence of toxicity of the WAS vector on human umbilical cord blood CD34+ cells. (
GMP, Good Manufacturing Practices; IG, infectious genomes; UCB, umbilical cord blood; WAS B-LCL, Wiskott-Aldrich syndrome B lymphoblastic cell line.
Tested at 6.5 × 107 IG/ml.
Expression of insert
Expression of WAS protein from the WAS lentiviral vector was demonstrated after transduction of WASP-negative B-LCLs derived from patients with WAS as previously shown (Charrier et al., 2007). Concentrations of vectors ranging from 0.5 × 107 to 10 × 107 IG/ml resulted in the integration of vector copies in B-LCLs ranging from 0.1 to 2.9 copies per cell on average (Table 4B). A dose-dependent increase in transduction was seen particularly with the GMP batches. Western blots showed expression of WASP protein at the correct size and amounts increased in a vector dose-dependent manner, reaching full correction because levels similar to those found in B-LCLs from normal individuals were detected at 5–10 × 107 IG/ml (Fig. 5). WASP expression data were confirmed by fluorescence-activated cell-sorting (FACS) analysis (data not shown).

Expression of the insert after transduction with a GMP WAS lentiviral vector. The WASP-negative 04W B-LCL line was transduced with the indicated concentrations of the WASP vector (GMP run 1). After 8 days, cell extracts were immunoblotted with antibodies against WASP and actin. Positive controls consisted of B-LCLs obtained from normal individuals showing a ratio of 1.0 between WASP and actin levels. Results are representative of the two GMP batches tested. B-LCL, B lymphoblastic cell line; IG, infectious genomes; WAS, Wiskott–Aldrich syndrome; WASP, WAS protein; VCN, vector copy number.
Discussion
We herein describe the large-scale production and downstream process of an advanced generation SIN HIV-1-derived lentiviral vector that has been developed for a pilot phase I/II ex vivo gene therapy trial of Wiskott–Aldrich syndrome. The results show that concentrated highly purified, nonreplicative and biologically active recombinant particles can be manufactured, and this has important implications for the efficacy and safety of gene therapy in patients. The development of the process and the initial results obtained under GMP conditions provide an important point of reference for lentiviral technology but also show that the process is still under development and that further efforts will be needed for its improvement and validation in pharmaceutical terms.
In spite of variable yields, the manufacturing process was adapted to GMP and was reproducibly and successfully implemented at a full 50-liter scale as shown here with a total of four batches produced during development. Two consecutive batches of WAS vector were produced with GMP-grade materials and under controlled GMP conditions. A total amount of 2.6 × 1011 and 6.2 × 1011 IG, representing 4.16 and 6.76 mg of p24, respectively, was produced per GMP batch (Table 1). Titers greater or equal to 109 IG/ml were achieved, thus exceeding the specifications of at least 2 × 108 IG/ml in the final product. This should permit the treatment of several patients per batch, thus fulfilling our initial expectations in titer and amount of particles per batch.
An extensive panel of analytical tests has been performed on both GMP vector batches, in accordance with the guidelines of the European Medicines Agency (EMA) on the manufacture of lentiviral vectors (CPMP/BWP/2458/03) and in compliance with European Pharmacopoeia Monograph 6.6-5156 on Retroviridae-derived vectors for human use (
This property was confirmed by efficient transduction of primary CD34+ cells, because an average of 0.3 to 1 copy of vector per cell was integrated (considering the whole cell population), using the highest vector concentrations tested (Table 4A). These values may appear to be relatively low but as such, these low vector copy numbers should limit the risks associated with genotoxicity of integrative vectors while providing therapeutic benefit as suggested from murine preclinical studies in models of WAS (Marangoni et al., 2009). Similar values of transduction are reported in other ex vivo hematopoietic stem cell gene therapy models such as for sickle cell disease (Levasseur et al., 2003) or metachromatic leukodystrophy (Biffi et al., 2004). An average vector copy number per cell value inferior to 1 after transduction of CD34+ cells was also achieved in a clinical trial for the treatment of adrenoleukodystrophy patients (Cartier et al., 2009). The purified WAS vector induced expression of normal levels of the genetic insert. The integrity of the vector was further verified by sequencing the provirus integrated in the indicator cells (data not shown). The product was not toxic, as it did not affect the viability, growth, or differentiation potential of umbilical cord blood CD34+ cells, which can be considered as surrogates for the patients' cells. Although these tests are useful indicators, the actual toxicity and transduction efficiency of the vector remain to be tested in clinical conditions with patients' cells.
This paper therefore describes the first batches of lentiviral vector produced for Wiskott–Aldrich syndrome but is also among the first reports of extensively purified and characterized batches of HIV-derived lentiviral vectors for clinical application. Such results provide a significant point of reference for the manufacture of lentiviral vectors for clinical application by describing a protocol for a production and downstream process leading to acceptable yields of a purified, concentrated, and biologically active product. Several points appear to be critical in this operation. The producer cells are an important factor because they determine in part the yield and quality of the initial viral stock, which is a critical element, as we have seen with the GMP runs. The choice of 293T cells was based entirely on the production of superior yield and infectivity of particles compared with HEK-293 cells, but other derivatives such as 293FT cells were tested and not found to be superior in this application (data not shown). This is consistent with the fact that 293T cells are the most commonly used producer cells for rHIV lentiviral vector production (Kutner et al., 2009) and facilitates the translation of preclinical efforts toward GMP by providing continuity in the experimental conditions and data. Conditions for the transfection of the producer cells, such as plasmid ratios, cell density, and times of harvest of the viral stocks, were based largely on the initial protocol described by Naldini and colleagues (1996), and have been adapted to large-scale work. Similar results were found by us and others (Segura et al., 2007) concerning the need to use relatively low levels of the VSVg-encoding plasmid but excess amounts of transfer plasmid and high expression system in the 5′ LTR of the transfer plasmid. Another critical point is the extensive downstream processing of viral stocks, which not only increased the concentration of the product but also improved its safety. Studies with gammaretroviral vectors have shown that by-products such as bovine proteins can prime immune responses in patients treated with unpurified vectors even in the context of the use of the product ex vivo and for the treatment of immunodeficient patients (Tuschong et al., 2002). Furthermore, it has been shown that it is critical to purify VSVg-pseudotyped lentiviral vectors to remove aggregates, cellular debris, and/or components of the tissue culture medium that generate inflammatory responses when administered in vivo in animals (Baekelandt et al., 2003). The downstream processing protocol that we have developed is similar to a large-scale protocol reported elsewhere (Slepushkin et al., 2003; Lu et al., 2004), although the order of certain steps was inverted. The downstream processing was feasible and removed, overall, almost 3 logs of protein and DNA contaminants, which reduced potentially oncogenic sequences, enzymes, and plasmidic DNA. We could not detect the presence of SV40 LTA protein by immunoblotting. In addition, preliminary experiments aiming to determine SV40 LTA by ELISA seemed to confirm that the levels were below detection by this method as well (data not shown; M. Radrizzani, personal communication). We have previously performed a study of the protein composition of purified lentiviral particles, using two-dimensional gels and matrix-assisted laser desorption and ionization time-of-flight
The overall recovery of the process described by others (Slepushkin et al., 2003) is 30% of the starting material, which seems to be higher than in the process that we report here. The difference can be explained by the supplementary size-exclusion chromatography step as well as by differences in the choice of separation materials that we have used, such as chromatographic matrix or ultrafiltration cartridges. With this process, the expected minimal amount of vector needed per batch was obtained in most of the standardized runs operated during the process development and technology transfer phase. The vector concentration of the final developmental batches (>2 × 108 IG/ml) was comparable to that published for other large-scale protocols using chromatographic, membrane-based or ultracentrifugation methods (Scherr et al., 2002; Slepushkin et al., 2003) (L. Couture, communication ASGT, Boston/USA, 2008 and F. Dupont, communication CONSERT LabCourse, Evry/France, 2008). However, it remains difficult to precisely compare these different approaches based on the infectious titers in the absence of standardized methods. Operating under controlled GMP conditions had a positive effect. Higher and less variable yields were obtained in two consecutive batches operated under GMP conditions, using GMP-grade materials. The vector concentration of GMP batches, >109 IG/ml, was much better than in any published results and seems to be due essentially to the 2- to 2.5-fold increase in IG concentration in the bulk product and also to improved conditions at the ultrafiltration step, with probably a slight improvement in the final sterilizing filtration step as shown in Fig. 2. At this point, we demonstrate the feasibility of implementing this process at large scale and in GMP. Although such a process is performing adequately, further improvements could be made to the process efficiency, yield, and robustness. Our results therefore provide an important biotechnological point of reference to set the first specifications for the quality control of future runs and as a stepping stone for future improvements in lentiviral manufacturing.
There is little indication in the scientific literature for setting release specifications (except for Schonely et al., 2003) and therefore our results provide data in that regard. We have used the large-scale production runs, performed during the technology transfer to the GMP production unit, to establish specifications for the manufacture of clinical products. We set more stringent specifications than those reported previously (Schonely et al., 2003). Significant differences concern the specifications for the residual endotoxin (<100 vs. <10 EU/ml; data not shown) and the residual Benzonase levels (<100 vs. <0.2 ng/ml). The residual protein levels, whether or not taking into account the final formulation medium (respectively, 0.74 ± 0.29 or 0.12 ± 0.14 mg of protein per 108 IG), were significantly below the protein contamination level specification of <7 mg of protein per 108 IG proposed by Schonely and colleagues (2003). With respect to infectious titer (IG/ml), our specification, >2 × 108 IG/ml, is 20 times higher than that fixed by Schonely and colleagues (2003). In the future, additional implementation of the process for the manufacture of additional batches of the WAS vector or of other types of lentiviral vectors will be required to assess the reproducibility of the operations in GMP and to adapt the specifications accordingly.
The results presented in this paper provide a point of reference for future developments aiming to adapt or to improve the process. We have identified production of the initial viral stock as critical to the success of the operation, and therefore it may be possible to improve this step further compared with the current method. We have used calcium phosphate as a transfection reagent because of the simplicity of the product and its adaptability to GMP. Several groups have proposed the use of polyethylenimines as robust transfection reagents (Devitt et al., 2007; Kuroda et al., 2009). These reagents can also be used with suspension-adapted HEK-293 cells, which can be grown in bioreactors and produce titers of 108 TU/ml in the starting viral stock (Ansorge et al., 2009). Although the current process effectively removes DNA, this contaminant is still present in the microgram per milliliter range in the final product, and therefore future efforts should attempt to achieve a more complete removal of contaminating DNA by optimizing the Benzonase digestion step, which is the major DNA-removing step in the process. Additional analytical tests are also needed to better characterize the contaminants. Such improvements will probably be needed from the perspective of drug licensing and for applying the rHIV gene transfer technology in vivo. Further optimization, in particular of the ion-exchange chromatographic and ultrafiltration/diafiltration steps, will lead to reduced vector loss and improved yields. Other groups have developed the use of heparin affinity chromatography to successfully purify VSVg-pseudotyped rHIV vectors (Segura et al., 2007). However, the full characterization and biological testing of these purified vector particles remain to be reported.
In summary, the characterization of large-scale batches of WAS lentiviral vectors (non-GMP and GMP) has proven the comparability with research-scale vector batches with respect to functionality and higher quality features and showed that the manufacturing process was able to provide the quality and yield of vector preparation that is needed for clinical use. A phase I/II clinical trial for the treatment of patients with WAS is currently being planned.
Footnotes
Acknowledgments
The authors are grateful to their colleagues at Genethon: Loïc Millot, Sandrine Boissinot, Frédéric Barnay-Toutain, Stéphanie Rundwasser, Séverine Leroy, Graziella Griffith, and Stéphanie Bucher-Laurent for practical contribution to this study. The authors also acknowledge the sustained support from Soraya Bachir-Chérif, Ariane Kobrine, Nicolas Delaunay, and Didier Caizergues concerning regulatory affairs and quality assurance issues during this project. Many thanks to Mehdi Gasmi and Philippe Moullier for careful review of the manuscript.
Author Disclosure Statement
Dr. Marina Radrizzani and Mrs. G. Vallanti are employed by MolMed; Dr. Muriel Audit is employed by Genosafe.