
Abstract
We investigated the therapeutic potential of the premature termination codon (PTC) readthrough-inducing drug PTC124 in treating the retinal phenotype of Usher syndrome, caused by a nonsense mutation in the USH1C gene. Applications in cell culture, organotypic retina cultures, and mice in vivo revealed significant readthrough and the recovery of protein function. In comparison with other readthrough drugs, namely the clinically approved readthrough-inducing aminoglycoside gentamicin, PTC124 exhibits significant better retinal biocompatibility. Its high readthrough efficiency in combination with excellent biocompatibility makes PTC124 a promising therapeutic agent for PTCs in USH1C, as well as other ocular and nonocular genetic diseases.
Introduction
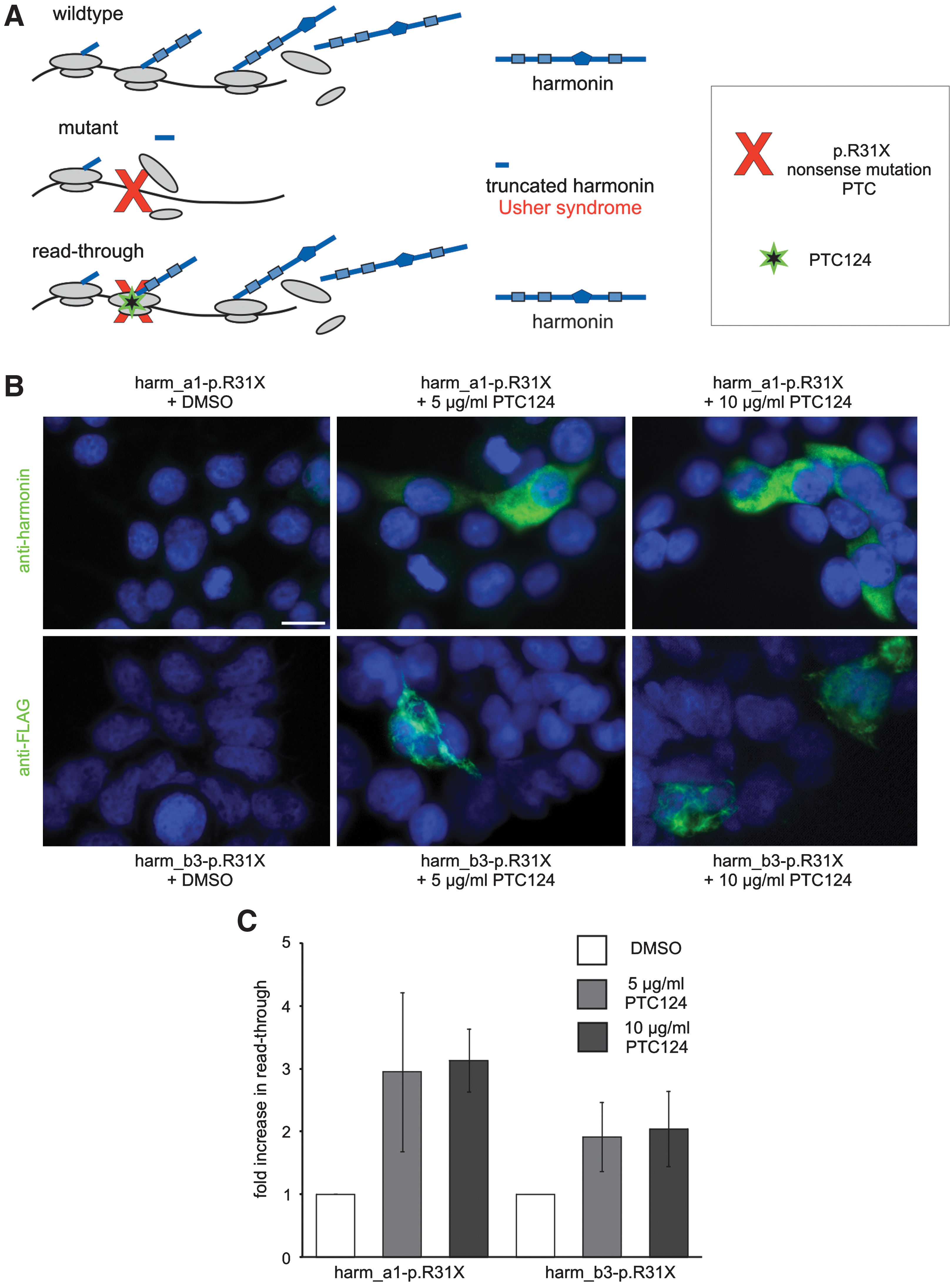
Indirect immunofluorescence analyses of PTC124-mediated readthrough of the USH1C nonsense mutation p.R31X in HEK293T cells. (
In earlier work aminoglycosides were shown to promote readthrough of PTCs, but also to induce severe side effects at therapeutically relevant concentrations (Linde and Kerem, 2008). In screens for readthrough agents with better biocompatibility, PTC124, structurally not related to aminoglycosides or any other drug, was identified (Welch et al., 2007). Chemical footprinting studies indicate binding of PTC124 to the 60S ribosomal subunit and thereby inducing readthrough of nonsense mutations (Finkel, 2010). The readthrough efficiency of PTC124 was successfully demonstrated in animal models of Duchenne muscular dystrophy (DMD) and cystic fibrosis (CF) (Welch et al., 2007; Du et al., 2008). Phase I and IIa trials demonstrated the effectiveness of PTC124 treatment combined with no drug-related serious side effects in children and adults (Hirawat et al., 2007; Kerem et al., 2008; Sermet-Gaudelus et al., 2010), paving the way for further clinical trials in, for example, patients with DMD, CF, and hemophilia.
Here we have investigated the readthrough efficacy of PTC124 on PTCs causing retinal diseases, in particular on the retinal degenerations related to Usher syndrome (USH), the most frequent cause of combined hereditary deaf–blindness (Reiners et al., 2006). Although the auditory deficit can be treated with cochlear implants (Damen et al., 2007), to date there is no effective treatment for the retinal phenotype of USH (Reiners et al., 2006). Approximately 12% of all USH cases are accounted for by PTCs (Baux et al., 2008). As proof of principle for USH-related PTCs, we focused in the present study on the p.R31X nonsense mutation in USH1C (p.R31X), which is one of the three known nonsense mutations in the USH1C gene (Zwaenepoel et al., 2001; Ahmed et al., 2002; Ebermann et al., 2007) (
The USH1C gene encodes the PDZ (postsynaptic density 95/disc large/zonula occludens-1)-containing scaffold protein harmonin, which is expressed as numerous alternative spliced variants (Verpy et al., 2000; Reiners et al., 2003). Harmonin isoforms a–c are classified by their modular composition of protein-interacting domains, namely PDZ, coiled-coil, and PST (proline, serine, and threonine-rich) domains. Their numerous interactions include the binding to all other USH1 and USH2 proteins, which specifies harmonin as a key organizer in the protein interactome related to USH (Reiners et al., 2006). Furthermore, the harmonin b isoforms directly bind to actin filaments inducing filament bundling (Boëda et al., 2002) and thereby link the USH protein network to the actin cytoskeleton (Reiners et al., 2006). In rodent and primate retinas, harmonin isoforms are expressed in both the outer and inner segments as well as the synapses of photoreceptor cells (Reiners et al., 2003; and M. Becker, T. Goldmann, K. Nagel-Wolfrum, N. Fuhrmann, M. Faust, C. Müller, J. Vetter, and U. Wolfrum, unpublished data). The p.R31X mutation of the USH1C gene introduces a PTC at the 5′ end (Zwaenepoel et al., 2001; Ahmed et al., 2002), which affects all known harmonin isoforms and results in a truncated harmonin polypeptide (∼3.4 kDa) lacking all scaffold function domains (Fig. 1A). The lack of harmonin scaffold function is thought to disrupt the USH protein network in photoreceptor cells, thereby leading to retinal degeneration (Reiners et al., 2005, 2006). Therefore, therapeutic attempts to treat USH1C in the retina should target the photoreceptor cells.
We demonstrated PTC124-mediated translational readthrough of the USH1C p.R31X mutation in cell culture, retinal explants, and in vivo. The recovered harmonin expression restored harmonin scaffolding function and actin filament-bundling activity. Furthermore, in comparison with the clinically approved readthrough-inducing aminoglycoside gentamicin, PTC124 revealed much better retinal biocompatibility. Our data conclusively demonstrate the capacity of PTC124 to treat the retinal phenotype of USH1C and other ocular and nonocular diseases.
Materials and Methods
Human material
Human donor eyes were obtained from the Department of Ophthalmology of the University Medical Center (Johannes Gutenberg University Mainz, Mainz, Germany). The guidelines of the Declaration of Helsinki were followed.
Animals
C57BL/6J mice were maintained on a 12 hr/12 hr light (200 lx)/dark cycle, with food and water ad libitum. Association for Research in Vision and Ophthalmology (ARVO, Rockville, MD) statements and institutional guidelines for animal care were followed.
Chemicals
PTC124, kindly provided by V. Maslenko (Exchemistry, Moscow, Russia), was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich, Deisenhofen, Germany). Gentamicin was purchased from Sigma-Aldrich. Monoclonal mouse antibodies to actin (clone C4) and to FLAG were obtained from Seven Hills Bioreagents (Cincinnati, OH) and Sigma-Aldrich, respectively. Polyclonal rabbit antibodies against harmonin (H3) were used as previously described (Reiners et al., 2003). Affinity-purified rabbit polyclonal antibodies against glial fibrillary acidic protein (GFAP) were obtained from Dako (Glostrup, Denmark). Rabbit antibodies against calbindin (Swant, Bellinzona, Switzerland) and protein kinase C (PKC)-α (Sigma-Aldrich) were previously characterized for retinal applications (Haverkamp and Wassle, 2000). Monoclonal rat antibodies against red fluorescent protein (RFP) were obtained from ChromoTek (Martinsried, Germany). Actin filaments were visualized with rhodamine–phalloidin (Sigma-Aldrich). Secondary antibodies conjugated to Alexa 488 were from Molecular Probes (Leiden, The Netherlands). 4′,6-Diamidino-2-phenylindole (DAPI; Sigma-Aldrich) was used for the visualization of nuclear DNA.
Plasmid clonings
The murine cDNA of harmonin a1 was amplified and inserted into the pCS2 + MT vector, encoding Myc tags (Rebibo-Sabbah et al., 2007); the pDest47 vector, encoding green fluorescent protein (GFP) (Invitrogen, Karlsruhe, Germany); or the pTER-mRFP vector, encoding monomeric red fluorescent protein (mRFP). cDNA of human harmonin b3 was subcloned into the pDest/C-SF-TAP vector, encoding S-FLAG (Gloeckner et al., 2007). All introduced tags were located at the C terminus of the harmonin isoforms. The p.R31X mutation was generated in all plasmids with a QuikChange Lightning site-directed mutagenesis kit (Stratagene, La Jolla, CA).
Cell culture and protein function analyses
HEK293T cells were grown in Dulbecco's modified Eagle's medium with GlutaMax supplemented with 10% fetal calf serum (Invitrogen) and 1% penicillin–streptomycin (Invitrogen) at 37°C and 5% CO2. Transfections were performed, with Lipofectamine LTX with PLUS reagent (Invitrogen), according to the manufacturer's protocol. Immunofluorescence analyses were done on methanol-fixed HEK293T cells as previously described (Overlack et al., 2008). For the analysis of harmonin b3-induced actin filament bundling, cells were incubated with rhodamine–phalloidin. Harvesting of cells and Western blot analyses were performed as previously described (Nagel-Wolfrum et al., 2004; Maerker et al., 2008). Western blots were analyzed with an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE). Glutathione S-transferase (GST) pull-downs were performed as previously described (Reiners et al., 2005; Maerker et al., 2008).
Retina culture and electroporation
Murine and human retinas were cultured as previously described (Reidel et al., 2006). Terminal deoxynucleotidyltransferase dUTP nick end labeling (TUNEL) was performed according to the protocol of the manufacturer (In Situ cell death detection kit; Roche, Mannheim, Germany). Plasmids were transferred into retinal explants by electroporation (ECM 830; BTX Harvard Apparatus, Holliston, MA) as previously described (Matsuda and Cepko, 2004). Retinas were cultured for 24 hr before adding the pharmacological compounds in fresh medium for an additional 72 hr.
In vivo electroporation and subretinal PTC124 injection
Retinas of postnatal (PN) day 0 mouse pups were electroporated as previously described (Matsuda and Cepko, 2004). Briefly, plasmids were injected subretinally followed by electroporation, using a pair of tweezer-type electrodes across the eyes. After 6 weeks, mice were anesthetized and 1 μl of PTC124 (2.5 μg/μl in DMSO) or DMSO as solvent control was injected subretinally. Three days after the injection, animals were killed. For fluorescence analyses, the eyes were removed, fixed with 4% paraformaldehyde, and embedded for fluorescence microscopy analysis. For quantification, confocal images were taken, applying the same set of imaging parameters. Fluorescence intensities of different regions of photoreceptor cells and fluorescence plot profiles were quantified with ImageJ software (National Institutes of Health, Bethesda, MD;
Microscopy
Light microscopy analyses of immunofluorescence and epifluorescence samples were performed with a Leica DM6000 B or a Leica SP5 confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany) and images were processed with Adobe Photoshop CS (Adobe Systems, San Jose, CA).
Statistical analysis
One-way t test analysis was applied to determine statistical significance.
Results and Discussion
PTC124-induced translational readthrough of p.R31X nonsense mutation in USH1C in cell culture
In the human USH1C mutation p.R31X, the CGA codon of an arginine (R) is altered to the premature stop codon UGA (X). Here, we analyzed the readthrough of this premature stop codon (PTC) exemplarily for the most abundantly expressed harmonin isoform a1 and the longer retinal harmonin isoform b3. For this, HEK293T cells were transfected with cDNAs encoding the mutated harmonin a1 (harm_a1-p.R31X) and mutated harmonin b3 (harm_b3-p.R31X), respectively. After adding PTC124 to the culture medium a dose-dependent increase in harmonin a1 and b3 expression was observed by immunofluorescence and Western blots (bands at 79 kDa for harmonin a1; 95 kDa for harmonin b3) applying anti-harmonin (H3) and anti-FLAG antibodies, respectively (Figs. 1B and 2A). In contrast, low harmonin expression was detected in DMSO-treated control cells (Figs. 1B and 2A), which probably results from spontaneous readthrough of the p.R31X nonsense mutation, previously reported for other correlated nonsense mutations (Rebibo-Sabbah et al., 2007).
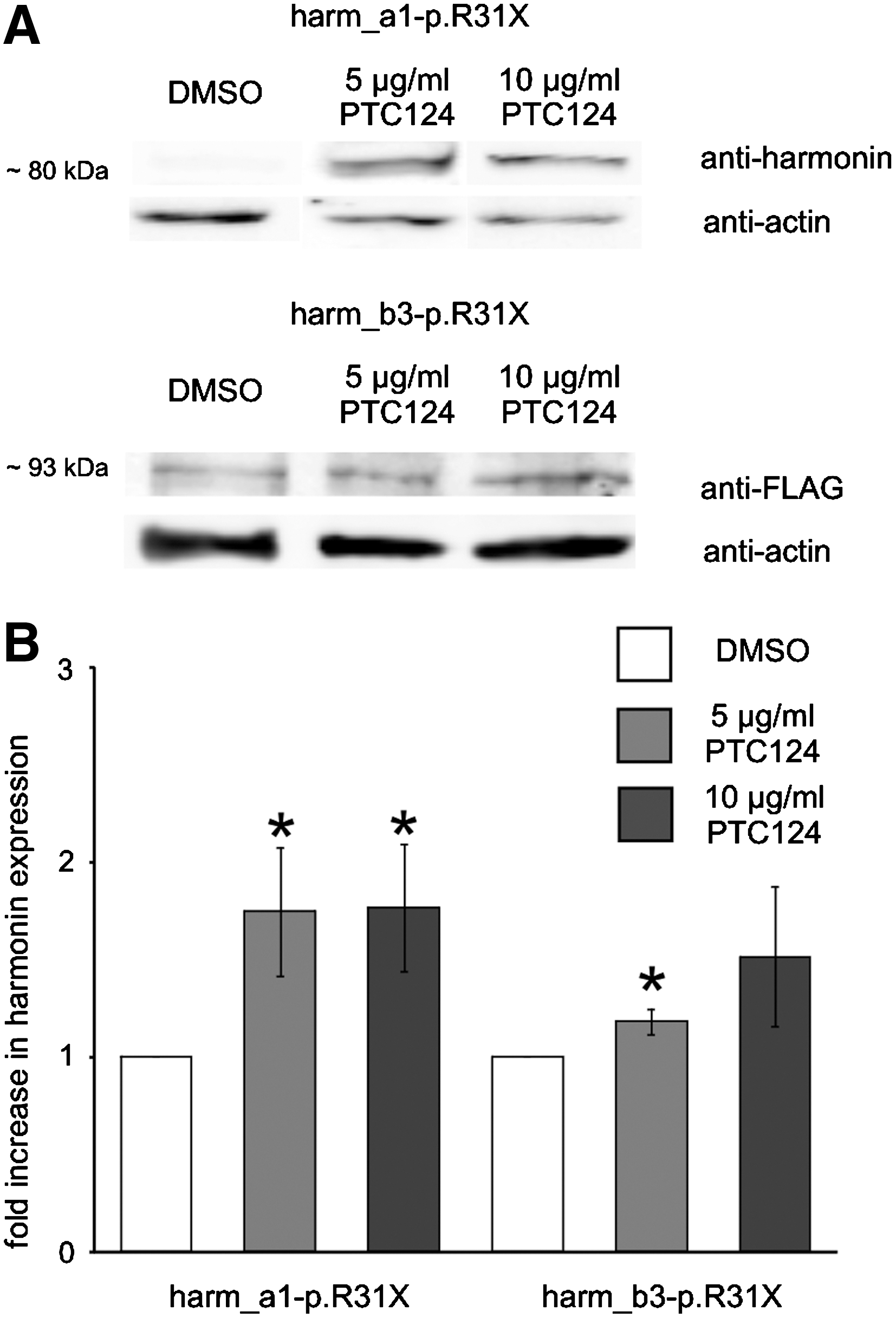
Western blot analyses of PTC124-mediated readthrough of the USH1C nonsense mutation p.R31X in HEK293T cells. (
Quantification of the number of immunofluorescently stained cells revealed a 3.1-fold increase in readthrough in harm_a1-p.R31X-transfected PTC124-treated cells and a 2.0-fold increase for harm_b3-p.R31X compared with control cells. The readthrough level obtained in Western blots revealed a 1.8-fold increase of readthrough in harmonin a1 p.R31X-transfected PTC124-treated cells and a 1.5-fold increase for harmonin b3 compared with control cells. The percentage of restored harmonin protein was calculated as the ratio of harmonin expression in p.R31X-transfected PTC124-treated cells to its expression in appropriate wild-type harmonin-transfected cells. This calculation indicated that PTC124 treatment induced 2.5% of the harmonin expression. Although the critical level of harmonin protein that is necessary to rescue retinal function in patients with USH1C is unknown, the PTC124-induced recovery of functional active harmonin might be sufficient to stop or at least slow the progression of the autosomal recessive USH1 disease, reestablishing a near-normal or at least clinically less severe retinal phenotype.
Translational readthrough might introduce an incorrect amino acid in the recovered harmonin protein at the p.R31X mutation (Linde and Kerem, 2008), which may interfere with protein function. To assess the restored properties of harmonin we analyzed the binding capacity of harmonin a1 to the PDZ-binding motif (PBM) in the cytoplasmic tail domain of USH2a isoform b (Reiners et al., 2005) and the actin filament-bundling activity of harmonin b (Fig. 3) (Boëda et al., 2002). In pull-down assays with GST-tagged USH2a cytoplasmic tail and cell extract of PTC124-treated cells, clear and single sustained harmonin a1 bands were visible, whereas from extracts of DMSO-treated control cells only faint bands were present (Fig. 3A).
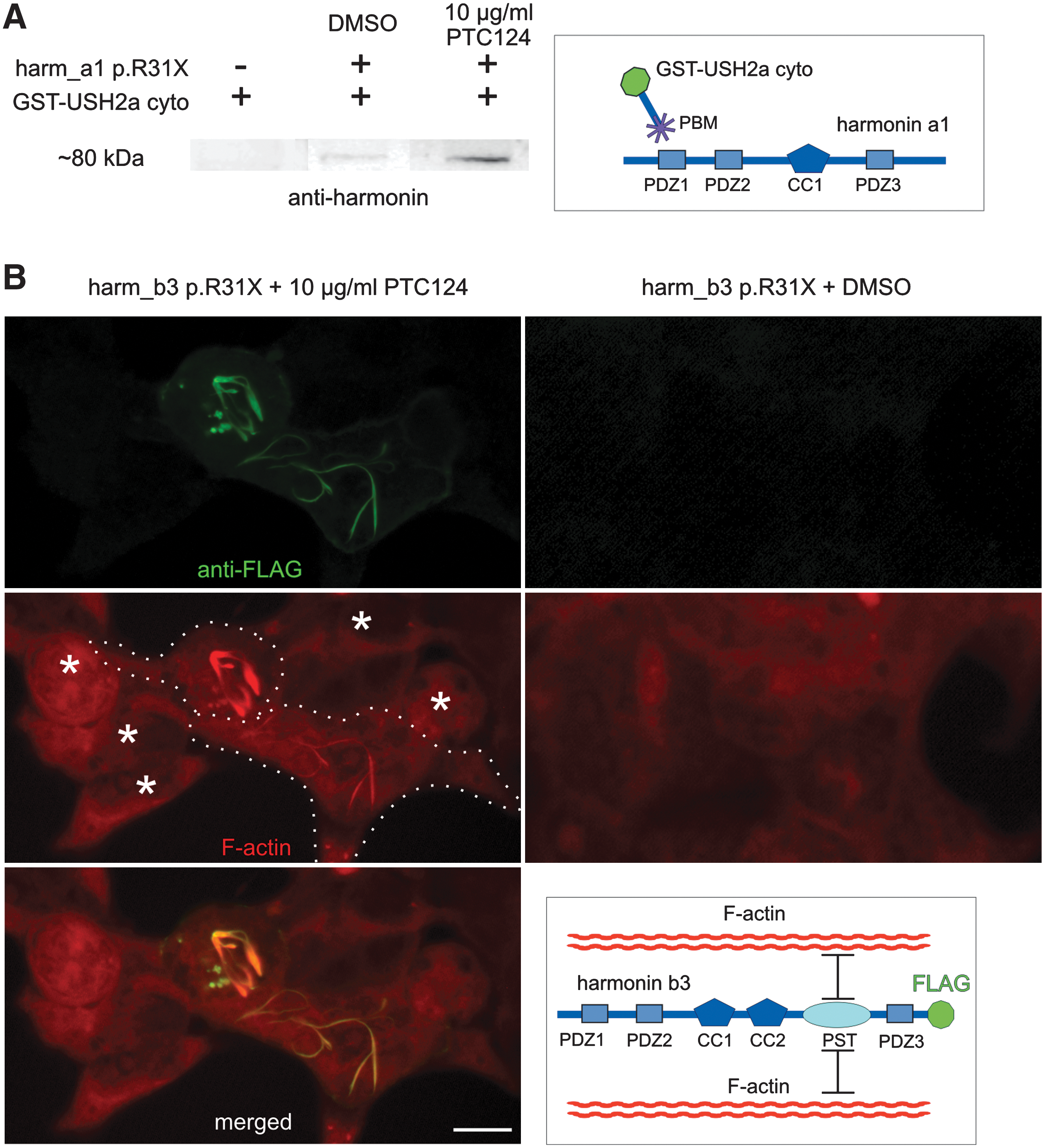
PTC124-mediated restoration of harmonin protein function. (
The actin filament-bundling capacity of recovered harmonin b3 was tested in cells transfected with FLAG-tagged harm_b3-p.R31X. Double labeling with anti-FLAG antibodies and rhodamine–phalloidin revealed actin filament bundling in PTC124-treated cells but not in DMSO controls (Fig. 3B). In summary, the translational readthrough of the p.R31X nonsense mutation of USH1C by PTC124 not only recovers the expression of full-length harmonin but also restores its cellular function as scaffold and actin filament-bundling protein. Thus functional recovery occurs although the randomly introduced amino acid at the stop codon is not necessarily the arginine of wild-type harmonin. These data are in line with previous genotype–phenotype correlations, which demonstrate that missense mutations in USH1C introducing a single incorrect amino acid in the protein sequence do not result in retinitis pigmentosa but cause nonsyndromic isolated deafness (Ahmed et al., 2002; Ouyang, et al., 2002; Reiners et al., 2006). In contrast, the p.R31X nonsense mutation in USH1C leading to the expression of truncated harmonin causes USH characterized by combined deaf–blindness. Therefore, PTC124-mediated readthrough of the p.R31X mutation in harmonin most probably will be sufficient for retinal function.
Readthrough of p.R31X mutation in organotypic retina cultures
We next investigated the readthrough potential of PTC124 in the retina, where harmonin isoforms are predominantly expressed in the photoreceptor cells (Reiners et al., 2003). For this, we electroporated murine retinal explants, introducing the mutated harmonin a1 C-terminally fused to a reporter encoding red fluorescent protein (harm_a1-p.R31X-mRFP). In DMSO-treated controls only a small number of mRFP-positive cells were detected in retinal explants transfected with harm_a1-p.R31X-mRFP (Fig. 4). These results were in accordance with our data on cultured cells, indicating spontaneous readthrough of the harm_a1-p.R31X nonsense mutation. In contrast, in retinal explants transfected with the mutated harm_a1-p.R31X-mRFP, treatment with PTC124 resulted in a significant increase in the number of mRFP-expressing cells. It was even higher than in explants treated with gentamicin (Fig. 4), a readthrough-inducing aminoglycoside previously adapted in animal experiments and clinical trials (Wilschanski et al., 2003; Guerin et al., 2008; Linde and Kerem, 2008; Moosajee et al., 2008).
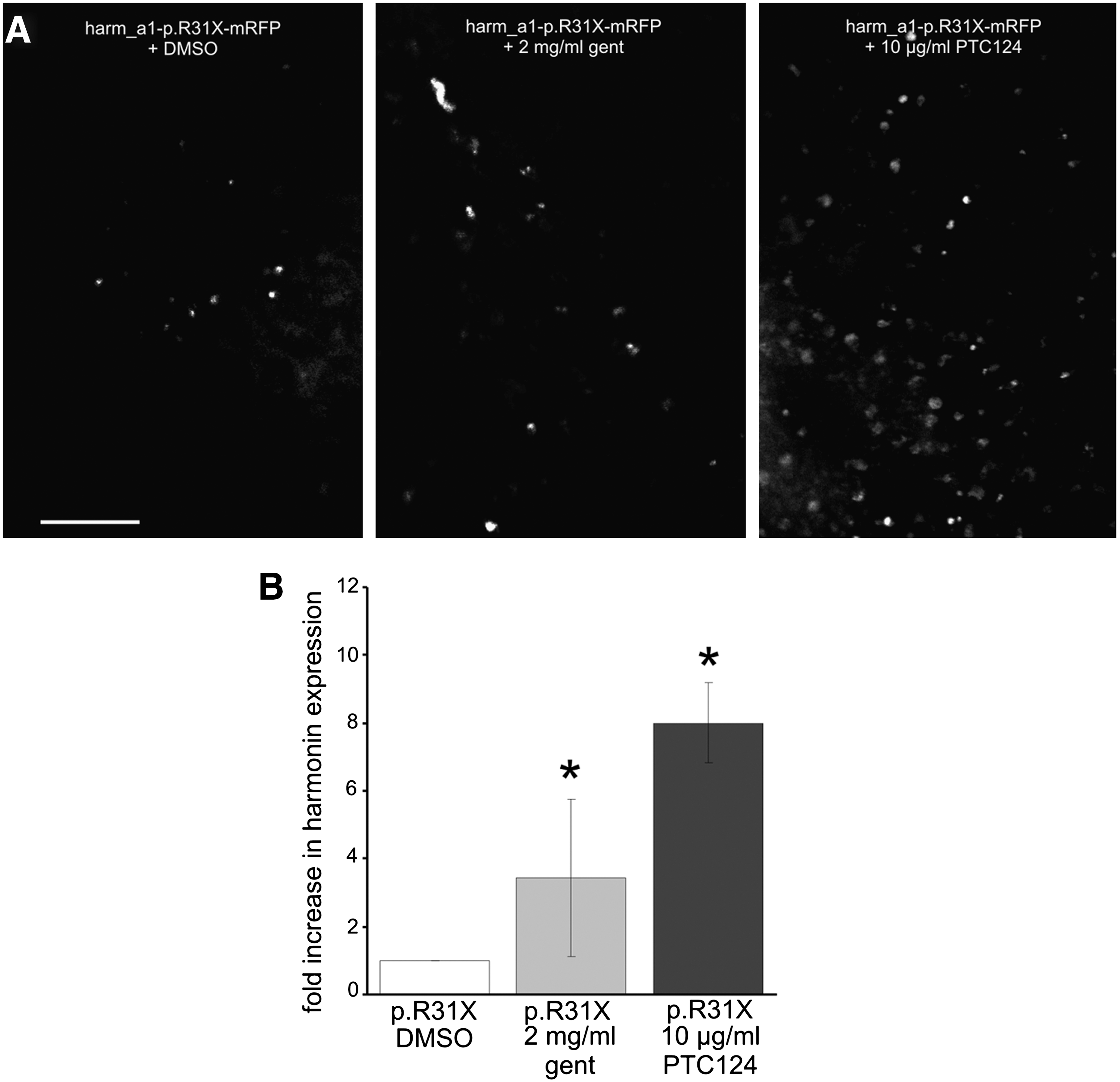
PTC124-mediated readthrough of the USH1C nonsense mutation p.R31X in organotypic retina cultures. (
To quantify drug-induced readthrough of the harmonin p.R31X nonsense mutation, mRFP-positive cells in seven randomly chosen regions of the whole mount were counted and calculated as fold increase over mRFP-expressing cells in DMSO-treated harm_a1-p.R31X-mRFP-transfected explants (Fig. 4B). This quantification revealed an 8.0-fold increase in readthrough of harm_a1-p.R31X-mRFP in PTC124-treated retinal explants, whereas the increase in gentamicin was lower (3.4-fold). These results indicate that PTC124 is twice as efficient in inducing readthrough of the harmonin p.R31X nonsense mutation in the retina than gentamicin. Previous studies demonstrated that systemic gentamicin treatment induced readthrough of a nonsense mutation in rhodopsin and thereby slightly improved retinal function (Guerin et al., 2008). Extrapolating the results from the latter study to the present data suggests that PTC124 treatment induces sufficient amounts of harmonin protein to combat retinal degeneration in patients with USH1.
Retinal biocompatibility of PTC124
An important concern for readthrough therapy is the compatibility of readthrough compounds in tissues and the organism (Linde and Kerem, 2008). Although systemic application of PTC124 has been shown to be nontoxic in animals and humans (Hirawat et al., 2007; Welch et al., 2007), retinal compatibility of PTC124 has not yet been evaluated. Here we analyzed the safety profile of PTC124 in the retina. We monitored potential histopathologies and retinal integrity by means of cell-specific molecular markers and possible cell death, applying TUNEL assays, a reliable tool with which to analyze any occurring cell death (Bramall et al., 2010). For this, organotypic retina cultures were incubated with various concentrations of PTC124 or the reference readthrough drug gentamicin.
Nuclei were stained with DAPI to monitor possible changes in the well-defined layered organization of the retina. Comparisons of PTC124-treated retinas with control treated retinas revealed no difference in the DAPI staining pattern, either in the outer nuclear layer containing the nuclei of photoreceptor cells or in the inner nuclear layer containing nuclei of the secondary retinal neurons (Fig. 5A). Furthermore, we did not find any decrease in the thickness of the outer nuclear layer, indicating that there is no effect of PTC124 treatments on the number of photoreceptor cells, often regarded as an indicator of retinal degeneration.
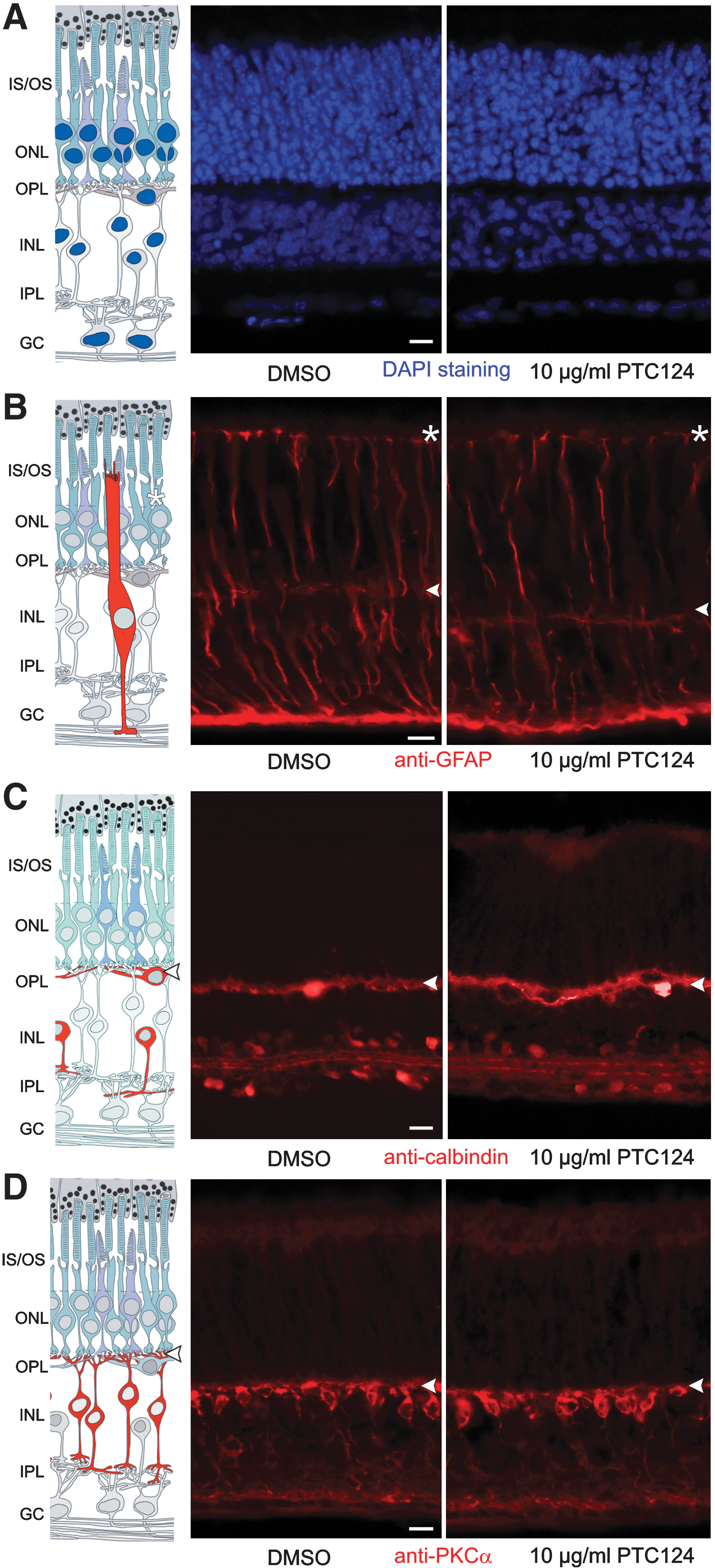
Histocompatibility and cytocompatibility of PTC124 in the retina. Retinal integrity and cellular composition of PTC124- and DMSO-treated murine retinal cultures were analyzed in cryosections. (
Effects of PTC124 treatment on specific retinal cells were monitored by indirect immunofluorescence, applying well-characterized antibodies as cell-specific molecular markers, namely anti-GFAP for Müller glia cells, anti-calbindin for horizontal cells and amacrine cells, and anti-PKC-α for bipolar cells. Our analyses revealed no apparent differences in the cell number and spatial organization of these retinal cell types of PTC124-treated compared with DMSO-treated retinas (Fig. 5B–D) or untreated retina cultures (Reidel et al., 2006; Orisme et al., 2010). In conclusion, these data indicate that PTC124 treatment does not alter the structural integrity of the neuronal retina and has no cytotoxic effect on retinal neurons and glia cells.
In addition, cell death was analyzed by TUNEL assays in treated retinas. Quantification of TUNEL-positive nuclei revealed only minor increases in cell death in murine retinal cultures treated with PTC124 compared with DMSO-incubated control cultures (Fig. 6A) or untreated retina cultures (data not shown). These data on murine retina cultures indicate that PTC124 is even more biocompatible than the novel designed aminoglycoside NB30, introduced as a potent investigational readthrough drug for retinal applications (Hainrichson et al., 2008; Goldmann et al., 2010). In contrast, quantification of TUNEL-positive nuclei revealed a more than 3.4-fold increase in dying cells after gentamicin treatment compared with control cultures (Fig. 6B). In human retinal explants cultured from donor eyes postmortem we observed 1.2% TUNEL-stained nuclei in DMSO-treated controls (Fig. 6), which is in line with cell death levels in untreated cultures (data not shown). Whereas the application of gentamicin slightly increased cell death (1.9-fold), nearly no difference from control cultures was found after PTC124 treatment of human retinal cultures (Fig. 6B).
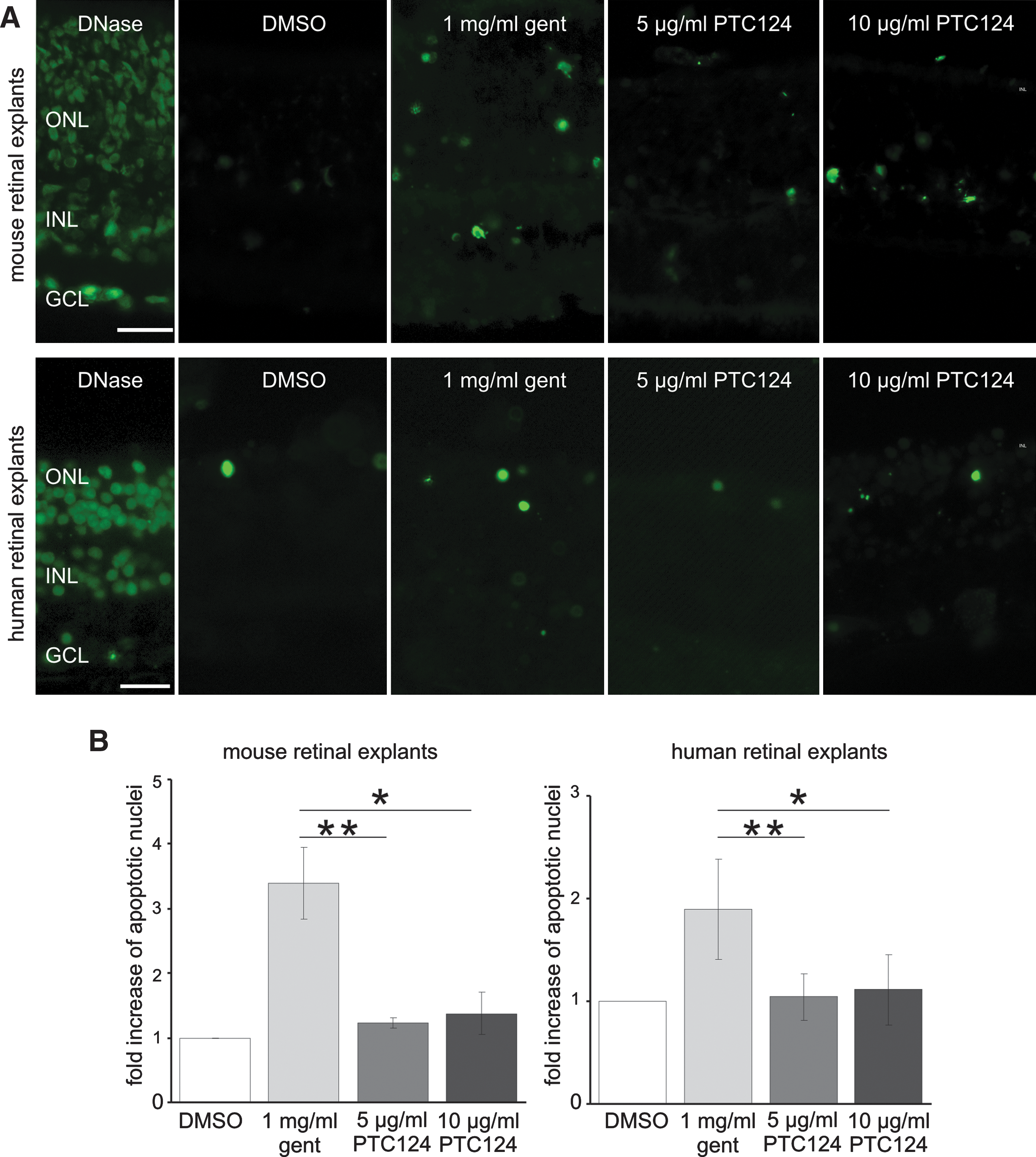
Biocompatibility of PTC124 in mouse and human organotypic retina cultures. (
These results clearly favor PTC124 as a readthrough-inducing drug for retinal applications compared with the clinically introduced reference gentamicin. Because of the dissatisfying safety profile of gentamicin in the retina we excluded the drug in further investigations. However, the excellent biocompatibility of PTC124 for retina further supports the previously stated high safety profile of PTC124 in humans (Hirawat et al., 2007).
Readthrough of p.R31X mutation in retina in vivo
Because application of PTC124 can restore harmonin expression in cell culture and ex vivo in organotypic retina cultures, we next tested the readthrough performance of PTC124 in the organism in vivo. For this we adopted an in vivo electroporation technique introduced by Matsuda and Cepko (2004) to investigate the functionality of PTC124 in the context of a mouse eye. We transferred harm_a1-p.R31X reporter constructs into the retina of newborn mice via electroporation. After 6 weeks PTC124 was injected subretinally into the electroporated mouse eye and readthrough was assessed by fluorescence microscopy analyses and Western blot analyses (Figs. 7 and 8). Fluorescence microscopy revealed hardly any GFP expression in the retinas of DMSO-injected control eyes. In contrast, in retinas treated with PTC124, apparent harmonin_a1-GFP expression was observed (Fig 7A). Quantification of the fluorescence intensity along photoreceptor cells showed an overall increase in GFP intensity after PTC124 treatment (Fig. 7B). In addition, separate measurements of photoreceptor compartments revealed a clear enhancement of fluorescence in the outer plexiform layer (photoreceptor synapses) and the inner and outer segments of photoreceptor cells (Fig. 7C), the subcellular compartments of the cell where harmonin is expressed in wild-type photoreceptor cells (Reiners et al., 2003). Our Western blot analyses demonstrated that PTC124 treatment recovered full-length harmonin a1 expression in harm_a1-p.R31X-mRFP-electroporated retinas (Fig. 8A). Quantification of the readthrough level obtained in the Western blot analyses revealed a 1.4-fold induction of readthrough in harmonin a1 (Fig. 8B). Present data prove in principle that PTC124-mediated readthrough of nonsense mutations is suitable to restore retinal protein expression in the organism. However, the specific mode of application of PTC124 into the eye will be reserved for further studies. These findings confirm the readthrough ability of PTC124 in vivo and highlight the potential for PTC124 as a treatment option for the p.R31X mutation and other retinal diseases. The relatively late onset of retinal degeneration in USH leaves a broad window for pharmacological interventions, compared with early-onset defects in the inner ear.
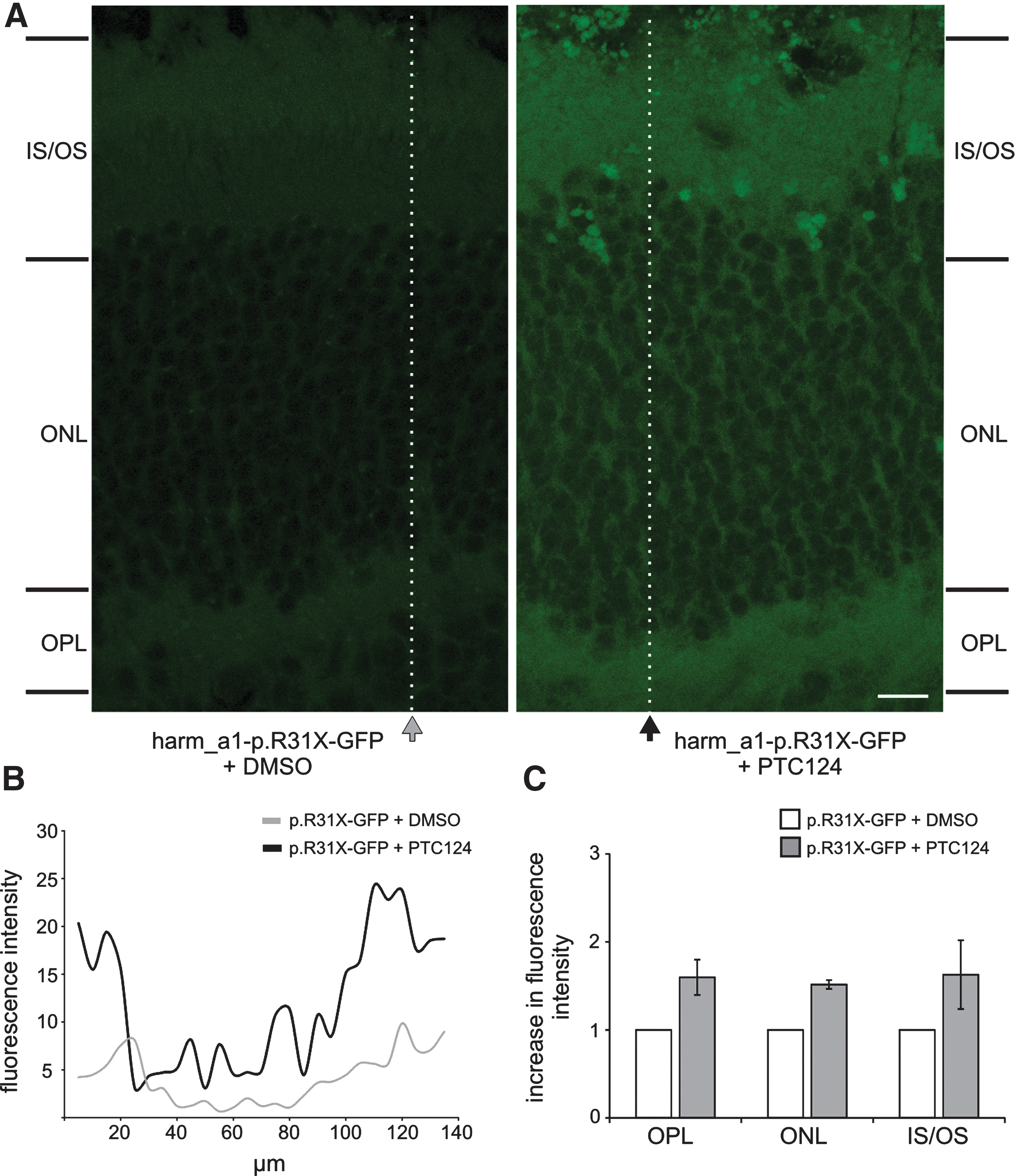
Confocal analyses of PTC124-mediated readthrough in retinas in vivo: retinas of harm_a1-p.R31X-GFP-electroporated mice. (
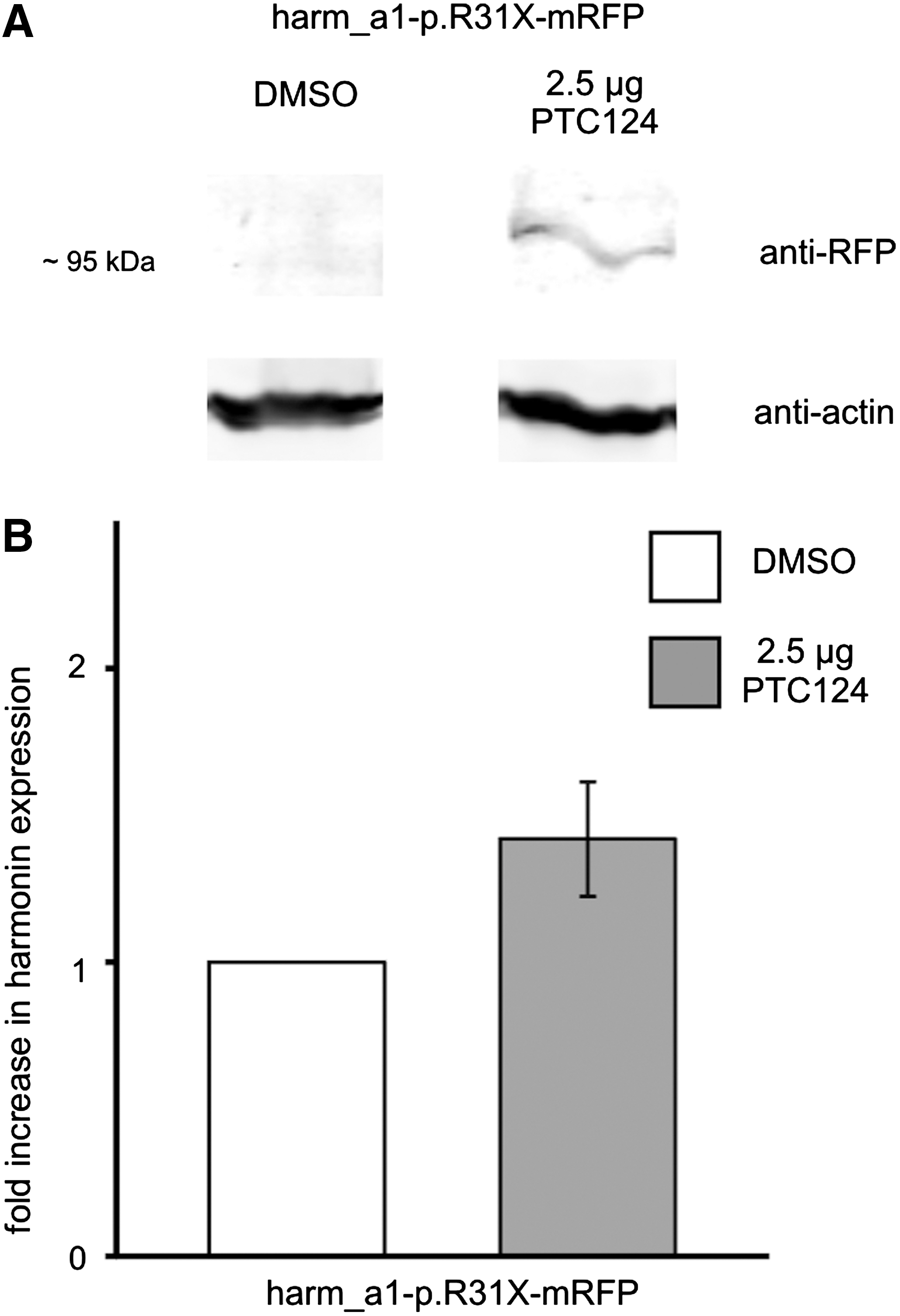
Western blot analyses of PTC124-mediated readthrough in retinas in vivo: retinas of harm_a1-p.R31X-RFP electroporated mice. (
Concluding remarks
The results obtained in this proof-of-principle study demonstrate the capacity of PTC124 for readthrough therapies of PTCs in retinal diseases as well as in other genetic disorders. PTC124 appeared to be highly biocompatible in the retina, in comparison with the aminoglycoside gentamicin. These gathered data emphasize the enormous potential of PTC124 as a readthrough therapy in the retina to combat nonsense mutation-based retinal disorders and other groups of genetic disorders with limited or no current therapeutic options, and raise hope for future clinical trials.
Footnotes
Acknowledgments
The authors thank Dr. Valdimir Maslenko for providing PTC124, Dr. Michiel van Wyk for critical reading of the manuscript, and Benjamin Spitzbarth and Fabian Möller for technical support. The authors also thank Dr. Susanne Haverkamp for kindly supplying the antibodies against calbindin and PKC-α. The research leading to these results has received funding from the European Community's Seventh Framework Programme FP7/2009 under grant agreement number 241955, Deutsche Forschungsgemeinschaft (DFG), and the FAUN-Stiftung, Nürnberg.
Conflict of Interest Statement
The authors declare no conflict of interest.