
Abstract
It has been reported that certain polypeptides derived from aggregation-prone cellular proteins can turn soluble green fluorescent protein (GFP) into aggregates. Here we report our finding that a short peptide derived from a viral gene, ICP10 of herpes simplex virus (HSV)-2, also possesses such a property. A sequence as short as 13 amino acids from the middle region of the gene can convert GFP into an aggregation-prone toxic protein once it is fused to the C terminus. Moreover, this short peptide can direct a surrogate tumor antigen into the autophagosome/lysosome degradation pathway, drastically increasing both MHC class I and class II antigen presentation. The simultaneous induction of both arms of the T cell immune response to the tumor antigen effectively protects the immunized animals from tumor challenge. Designated VIPA (i.e., viral inducer of protein aggregation), this unique viral sequence may represent an attractive candidate as a molecular adjuvant for cancer immunotherapy and for other immunologically preventable diseases.
Introduction

A polypeptide within the ICP10 protein of HSV-2 induces protein aggregation. (
Here we report our accidental discovery of yet another non-RR function associated with vR1, that is, the ability to convert soluble green fluorescent protein (GFP) into aggregates. This finding was made during our construction of the oncolytic HSV-2 mutant, FusOn-H2, in which the GFP gene was inserted into to the N-terminal region of the ICP10 gene (Fu et al., 2006). We found that a sequence as short as 13 amino acids from the middle region of the ICP10 gene, designated VIPA for viral inducer of protein aggregation, could convert GFP into an aggregation-prone form once it is fused to the C terminus. Moreover, the N-terminal region of ICP10 contains a signal sequence that dictates the location of aggregate formation.
Traditionally, major histocompatibility complex (MHC) class I presents endogenously synthesized antigens through proteasomal processing whereas MHC class II presents antigens taken externally by lysosomal degradation. However, under certain conditions, intracellular antigens can gain access to the lysosomal degradation pathway. One such example is by autophagy, which serves as one of the major intracellular protein degradation systems and is frequently induced by protein aggregation (Mizushima and Klionsky, 2007). To examine whether the aggregation induced by VIPA can guide antigens to lysosomal degradation for enhanced presentation, we linked a surrogate tumor antigen (chicken ovalbumin, OVA) together with GFP and VIPA. Addition of VIPA to the fusion protein indeed led to aggresome formation as revealed by GFP visualization. The immune response to OVA was greatly elevated, which led to efficient rejection of tumor cells that had been transduced with the OVA gene. These data suggest that VIPA can function as an adjuvant by guiding an antigen to the lysosomal degradation pathway by inducing protein aggregation.
Materials and Methods
Cells
Vero cells (African green monkey kidney fibroblasts), 293-T cells, human tumor cell lines Skov-3 (ovarian cancer cells) and U20S (osteosarcoma cells), and murine melanoma B16 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). B16-OVA was a kind gift from S.-Y. Chen (Center for Cell and Gene Therapy, Baylor College of Medicine, Houston, TX). The cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS).
Plasmid construction
The construction of pcDNA-PKF-2 has been described (Fu et al., 2006). It contains a GFP gene that is fused in-frame to the amino acid 403–774 region of ICP10 and the fusion gene is driven by the cytomegalovirus (CMV) promoter (see Fig. 1A). The construction strategy of the following five plasmids is shown in Fig. 2A (see below). Briefly, pcDNA-PKF-M3 (containing the amino acid 403–548 region of ICP10) was constructed by deleting the 3′ region of ICP10 sequence from pcDNA-PKF-2 by digestions with PshAI and XbaI, followed by religation. pcDNA-PKF-M4 (containing the amino acid 403–415 region of ICP10) was constructed by further deletion of the 3′ region of the ICP10 sequence from pcDNA-PKF-M3 by digestion with HpaI, followed by religation. pcDNA-PKF-M5 was constructed by removing the entire ICP10 sequence from pCDNA-PKF-2. pcDNA-PKF-M1 was constructed by introducing a stop codon between the GFP and ICP10 sequences in pCDNA-PKF-2. pcDNA-M4-PKF was constructed by inserting 13 amino acids (TPYHLREYATRLV) of the ICP10 sequence at front of GFP. The construction of pCDNA-CDD-M4 and pCDNA-PK-M4 is depicted in Fig. 4A (see below). Briefly, pcDNA-CDD-M4 was constructed by replacing the amino acid 403–774 ICP10 sequence in pcDNA-PKF-2 with the N-terminal amino acid 303–415 sequence, and pcDNA-PK-M4 was constructed by replacing the region with the N-terminal amino acid 1–415 sequence. pGFP-M4-OVA was constructed by cloning a truncated OVA fragment (amino acids 138–386) from the pOVA plasmid (kindly provided by Dr. Z. You, University of Pittsburgh School of Medicine) into pcDNA-PKF-M4 to fuse it in-frame with the other two components. All plasmid DNA was obtained by alkaline lysis of bacterial culture and purified by passage through a QIAGEN-tip 500 column (Qiagen, Valencia, CA).
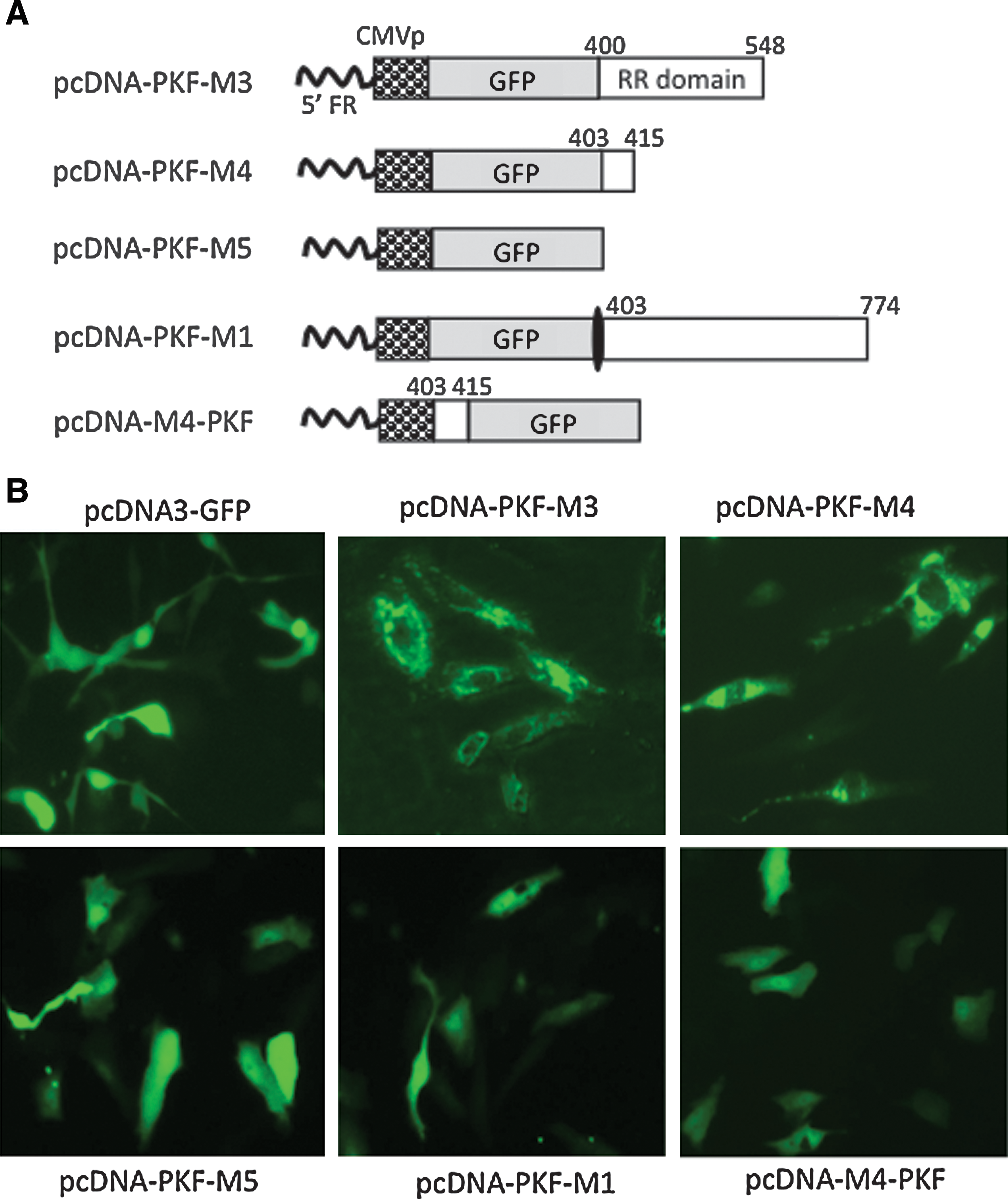
Defining the region of vR1 that induces protein aggregation and its location effect. (
Cell transfection and visualization of aggregate formation
Cell seeded in 6-well plates were transfected with plasmid DNA, using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Micrographs were taken 24–72 hr after DNA transfection, using a Zeiss microscope (NA 1.4; Carl Zeiss, Thornwood, NY). For Hoechst staining, Hoechst 33342 (Sigma-Aldrich, St. Louis, MO) was added directly at a final concentration of 5 μg/ml to the transfected cells at the indicated time. For immunohistochemical staining, transfected cells were washed three times in phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde in PBS for 10 min at room temperature. Cells were then permeabilized for 10 min with ice-cold methanol at 4°C. After washing, the cells were treated with 5% goat serum in 0.3% Triton X-100/PBS (staining buffer) for 30 min at room temperature. The cells were then incubated with rabbit anti-human primary antibodies to LC3B and vimentin (Cell Signaling Technology, Danvers, MA) or with rabbit anti-ovalbumin antibody (Abcam, Cambridge, MA) in staining buffer overnight at 4°C. Last, the cells were washed three times for 5 min each, and stained with a Texas red-conjugated goat anti-rabbit secondary antibody (Abcam) for 2 hr at room temperature.
Animal vaccination and host immune responses
All animal experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Houston (Houston, TX). Immune-competent female C57BL/6 mice (4–6 weeks old) were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were vaccinated by subcutaneous injection of either 1 × 105 B16-OVA cells or B16 cells transfected with pcDNA-M4-OVA or pOVA. All cells were irradiated before they were injected into the animals. A second vaccination was given 7 days later. On day 21 after the first vaccination, mouse spleen cells were harvested and purified with Ficoll (Sigma-Aldrich).
To separate CD4+ cells, Ficoll-purified spleen cells were passed through murine CD4+ T cell enrichment columns (BD Biosciences, San Jose, CA).
For high-throughput separation, CD4+ cells were collected and plated into 96-well filter plates (Millipore, Bedford, MA) that had been precoated with anti-interferon (IFN)-γ monoclonal antibody (BD Biosciences). The cells were seeded at a density of 4 × 105 cells per well for the IFN-γ enzyme-linked immunospot (ELISPOT) assay as described previously (Li et al., 2007a,b). Briefly, after overnight incubation at 4°C, cells (seeded in triplicate) were stimulated with OVA MHC-II peptide (positions 323–339, ISQAVHAAHAEINEAGR) at 50 μg/ml for 36 hr. After washing, biotinylated anti-IFN-γ antibody (BD Biosciences) was added to each well and the plates were incubated for another 1 hr at 37°C. A streptavidin–alkaline phosphatase conjugate was added to the wells, and after 1 hr of further incubation, the chromogenic alkaline phosphatase substrate was added. The colorimetric reaction was terminated within 5 to 20 min by washing with tap water. After drying, the spots were counted. An MHC-I-restricted ELISPOT assay was similarly done. Briefly, purified splenocytes were seeded at a density 2 × 105 cells into triplicate wells, and stimulated with OVA MHC-I peptide (positions 257–264, SIINFEKL) at 50 μg/ml for 24 hr. After washing, biotinylated anti-IFN-γ antibody (BD Biosciences) was added to each well and the plates were incubated for another 1 hr at 37°C. A streptavidin–alkaline phosphatase conjugate was added to the wells, and after 1 hr of further incubation, the chromogenic alkaline phosphatase substrate was added. The colorimetric reaction was terminated within 5 to 20 min by washing with tap water. After drying, the spots were counted.
Therapeutic evaluation of vaccinations on tumor growth
Freshly harvested B16-OVA cells (2 × 105) were injected into the left flanks of C57BL/6 mice, which were then randomly distributed to three groups (n = 8 mice per group). Three days after tumor cell implantation, the mice received, via a single subcutaneous vaccination into the right flank, either 1 × 105 B16 cells or B16 cells transfected with either pGFP-M4-OVA or pOVA. All cells were irradiated before they were injected. Mice were observed for 3 weeks, during which time tumor growth was monitored periodically by measuring two perpendicular tumor diameters with a caliper. Tumor volume was calculated according to the following formula: tumor volume (mm3) = (length [mm]) × (width [mm])2/2. For the survival study, mice were monitored for 40 days.
Statistical analysis
All data were normally distributed, and the Student t test (two tailed) was used to determine the statistical significance (p < 0.05) of various comparisons. Results are reported as means ± SD.
Results
Middle region of vR1 encoded by HSV-2 ICP10 can convert soluble GFP into aggregates that localize predominantly in the cytoplasm
In our previous studies, we constructed a mutant HSV-2 deleted for the N-terminal 400-amino acid region of vR1, which was subsequently used as an oncolytic agent (Fu et al., 2006). The mutagenesis was designed in such a way that a copy of the GFP gene would replace the N-terminal 400-amino acid region, to fuse in-frame with the remaining RR domain. For this purpose, a plasmid (pCDNA-PKF-2) was constructed in which the GFP gene was fused in-frame with a portion of the ICP10 gene (Fig. 1A), which was then used for homologous recombination to generate the mutant virus (Fu et al., 2006). During the experimental process, we frequently noticed that cells transduced with pCDNA-PKF-2 showed a drastic change in pattern of GFP distribution, from usually even distribution across the entire cell body to aggregate formation localized predominantly in the cytoplasm (Fig. 1B). The aggregates localized mainly around the nucleus, forming typical pericentriolar aggresome-like structures.
To more precisely define the region within vR1 that bears this activity, we made several additional constructs that contain different truncations of this region (Fig. 2A). In pcDNA-PKF-M3, the region was truncated in the C terminus from amino acid 774 to 548. In pcDNA-PKF-M4, this region was further truncated so that only a 13-amino acid residue was retained. The vR1 sequence was completely removed in pcDNA-PKF-M5. In pcDNA-PKF-M1, a stop codon was introduced into the immediate start of this region so that the downstream amino acid 403–774 ICP10 sequence would not be translated from this construct. As shown in Fig. 2B, further truncations in either pcDNA-PKF-M3 or pcDNA-PKF-M4 did not abolish aggresome formation, although the patterns of aggresomes were slightly different from that of pcDNA-PKF-2. On the other hand, removal of the entire vR1 sequence or insertion of a stop codon to prevent translation of the vR1 sequence completely abolished aggregation formation, with a GFP distribution indistinguishable from that of pcDNA3-GFP. Among all the constructs that have been tested, pCDNA-PKF-M4 appears to be the most potent in inducing aggregation formation. Thus, the 13-amino acid residue, designated VIPAM4, was chosen for making other constructs for further studies as detailed in the following sections. One such study was to examine the positional effect of VIPA, in which VIPAM4 was positioned at the N terminus of GFP as shown in Fig. 2A. Despite its strong ability in inducing aggregation formation when it was placed in the C terminus of GFP, VIPAM4 completely lost the ability to generate protein aggregates once it was fused to the N terminus of GFP (Fig. 2B).
The data presented previously were obtained when the expression plasmids were transfected into Vero cells. To determine whether aggregation formation also occurs in other cell types, we transduced several of these constructs into cells of various tissue origins, most of them being tumor cells. A similar pattern of GFP aggregation formation occurred in all the cells tested (data not shown). Thus, we conclude that the induction of protein aggregation is a generalized phenomenon associated with this region of ICP10. In addition, we noticed that the induced aggregate formation seems to be toxic to the transfected cells, as many of the cells showing aggregation formation became visibly cytopathic within 48–72 hr of transfection (Fig. 3A, indicated by white arrows). The cytopathic effect of the aggregation formation on the transfected cells is more clearly visible in the micrograph on the right-side side of Fig. 3A, which was taken from a sparsely seeded cell monolayer at a higher magnification. The GFP-positive cells (indicated by white arrows) clearly show a cytopathic effect (cells are rounded up and shrunk), whereas the untransfected cells (indicated by light blue arrows) show typical normal cell morphology. Cell death did not seem to be apoptotic, as Hoechst dye staining did not reveal any nuclear condensation (Fig. 3B). In actuality, the nuclei of cells with aggregation formation were even less condensed than those of untransfected cells (Fig. 3B, indicated by white arrows).
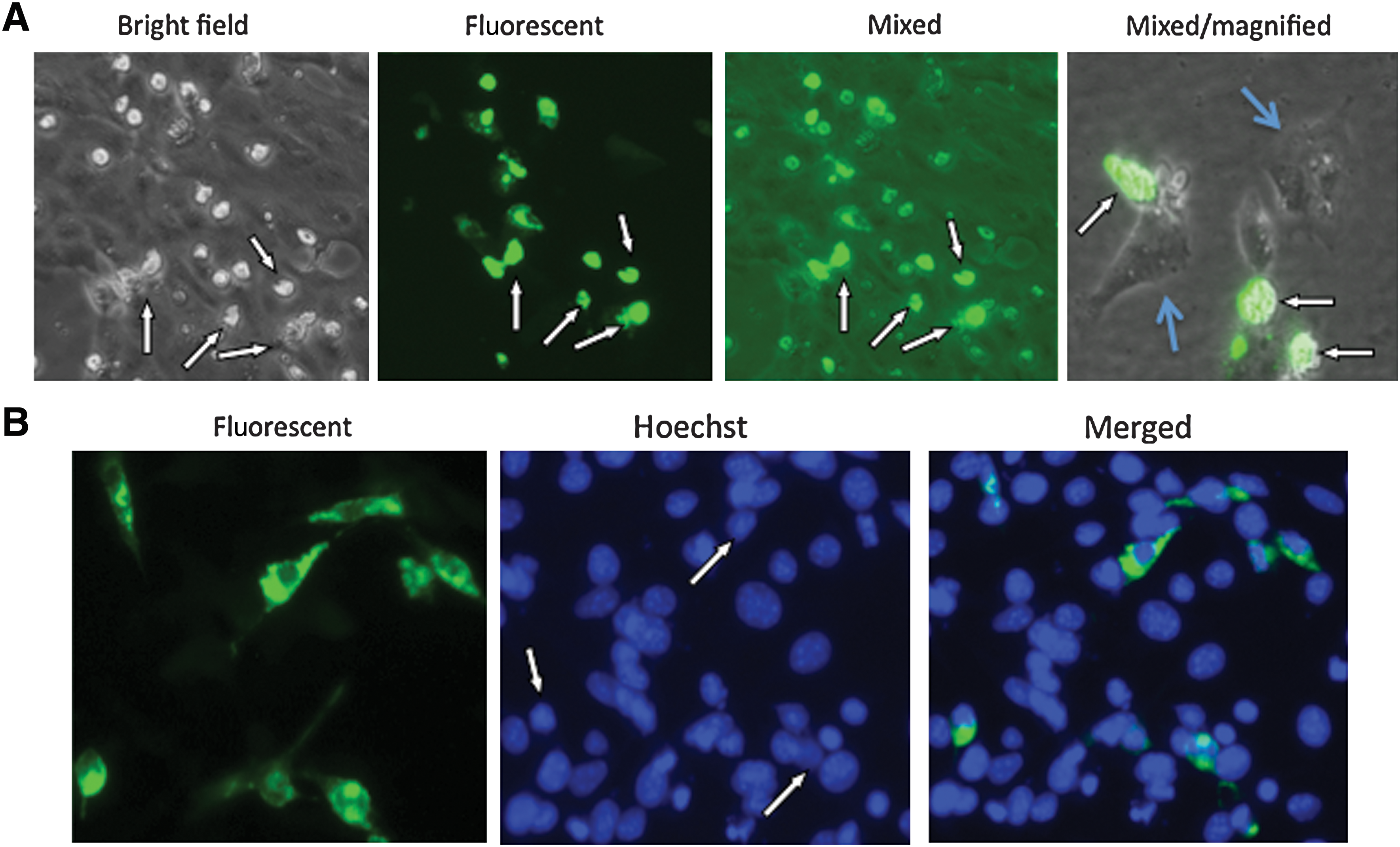
Cytopathic effect of VIPA (viral inducer of protein aggregation)-induced aggregate formation. Vero cells were transfected with pcDNA-PKF-2. (
The N-terminal region of vR1 contains a signal sequence that dictates the localization of GFP aggregation formation
To further define the region that can induce aggregation formation, we made several additional constructs. As illustrated in Fig. 4A, they contain, in addition to the 13-amino acid VIPAM4, extensions into the N-terminal 400-amino acid region. An additional 100 amino acids into the NH2 domain of ICP10 did not alter the ability of VIPAM4 to induce aggregation formation, nor did it affect the pattern of the aggregates (Fig. 4B). However, inclusion of the entire NH2 domain completely changed the localization of the aggregates, from predominately cytoplasmic, to almost entirely nuclear (Fig. 4B). These data suggest that the NH2 domain within the amino acid 1–300 region contains a signal sequence that dictates the location of aggregate formation.

A region in the NH2 domain of vR1 dictates localization of aggregate formation. (
VIPA-induced protein aggregates are associated with autophagosomes
Studies have suggested that autophagy is a major degradation pathway for protein aggregates. Abnormal polypeptides that escape proteasome-dependent degradation and aggregate in cytosol can be transported via microtubules to an aggresome, an organelle where aggregated proteins are stored or degraded by autophagy. Clearance by autophagy occurs by sequestration of the target organelle/protein into double-membrane structures called autophagosomes. Autophagosomes subsequently fuse with lysosomes, discharging their contents for proteolytic degradation by lysosomal hydrolytic enzymes. Among the autophagy-related genes, Atg8 (LC3 is the mammalian homolog) was found to localize to the developing autophagosome (Tanida et al., 2004). We conducted an experiment to determine whether VIPA-induced protein aggregation leads to autophagosome formation by staining the cells for LC3. The results show that LC3 was indeed colocalized with aggresomes (Fig. 5). We also stained the cells for vimentin, as this intermediate filament protein has also been shown to be intimately associated with aggresome formation (Johnston et al., 1998). The results showed that vimentin staining also colocalized with VIPA-induced aggresomes (Fig. 5).
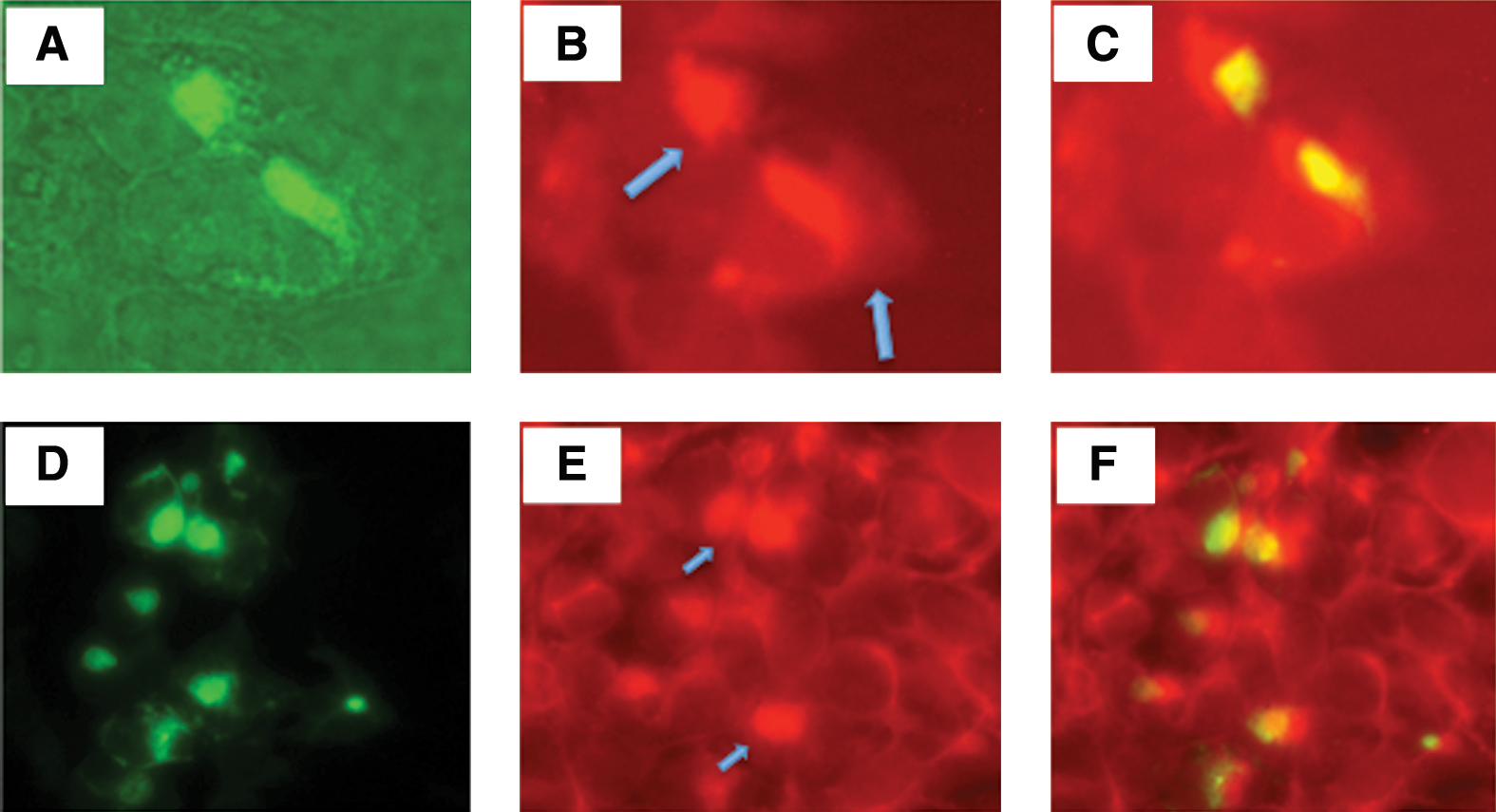
VIPA-induced protein aggregation leads to autophagosome formation. Vero cells were transfected with pcDNA-PKF-M4 and stained by immunohistochemistry for LC3 or for vimentin. Micrographs (
VIPA incorporated into a fusion gene can guide a surrogate tumor antigen into aggregate formation
To determine whether an antigenic polypeptide could be guided to autophagosomes by VIPA, we constructed another expression plasmid, pGFP-M4-OVA. We chose chicken ovalbumin (OVA) as the testing antigen, as it has been widely used as a surrogate tumor antigen for both in vitro and in vivo studies (Hornung et al., 1995). In addition, both class I and class II antigenic epitopes have been well defined for OVA (Carbone and Bevan, 1989). As depicted in Fig. 6A, in this expression plasmid, GFP, VIPAM4, and OVA are fused in-frame and the fusion gene is driven by the CMV promoter. Murine B16 melanoma cells transfected with pGFP-M4-OVA showed protein aggregation formation that is identical to that of cells transfected with pcDNA-PKF-M4 (Fig. 6), indicating that the presence of the OVA sequence did not affect the ability of VIPA to induce aggregate formation. We also stained the transfected cells for OVA expression. OVA antigen is clearly expressed and colocalizes with the aggregates, suggesting that it has been guided to the aggresomes by VIPAM4.

VIPA can guide a surrogate tumor antigen into autophagic pathway. (
Enhanced antigen presentation and antitumor effect after VIPA-directed antigen aggregate formation
Studies suggest that autophagic degradation may facilitate MHC class II presentation of endogenous antigens, as the by-products of autophagy-mediated lysosomal antigen degradation are primarily loaded onto MHC class II molecules (Gannage and Munz, 2009). To examine whether VIPA-induced aggresome formation can guide OVA into lysosomal degradation for enhanced antigen presentation, we vaccinated mice with the expression constructs described previously. For reliable and more abundant expression of the introduced antigens, we explored a cell delivery approach. A syngeneic cell line (B16) was transfected with OVA-expressing plasmids, pOVA or pGFP-M4-OVA. A B16 cell line that constitutively expresses OVA (B16-OVA) was also included in the experiment. The cells were irradiated before they were used to vaccinate mice by subcutaneous injection. After the second vaccination, mice were killed and spleens were explanted for ELISPOT assay for OVA-specific CD4 and CD8 responses. With vaccinations using either B16-OVA or B16 cells transfected with pOVA, the frequency of OVA-specific CD4+ cells was at a barely detectable level (Fig. 7A). Mice vaccinated with B16 cells that were transfected with pGFP-M4-OVA showed a dramatically increased level of antigen-specific CD4+ cells, to a frequency of nearly 500 per 106 CD4+ cells. We also measured the frequency of OVA-specific CD8+ T cells. As expected, vaccination with either B16-OVA or B16 cells transfected with pOVA clearly generated measurable CD8 responses. Surprisingly, the frequency of OVA-specific CD8+ cells was also increased severalfold in mice vaccinated with pGFP-M4-OVA when compared with the other two groups (Fig. 7B).
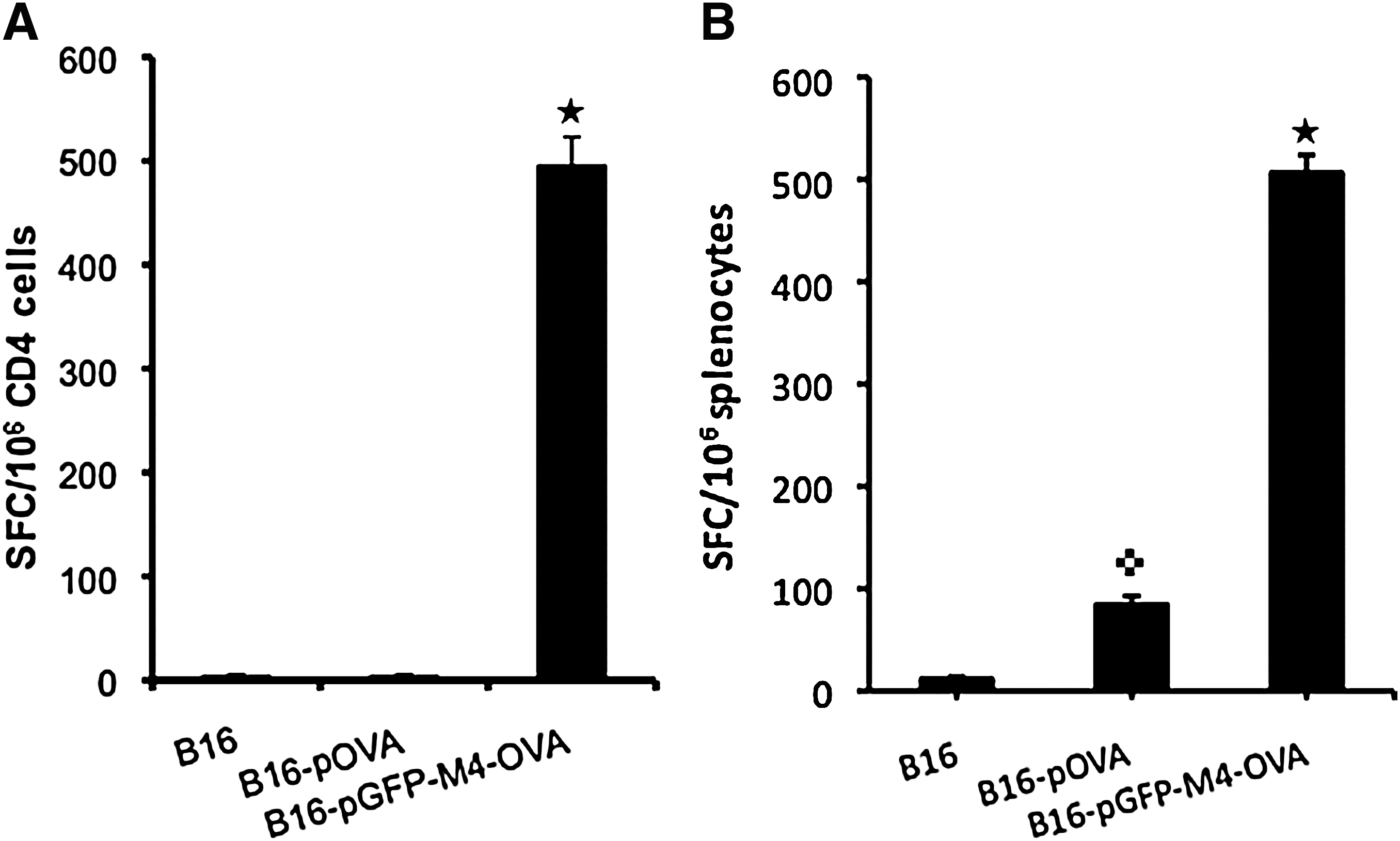
ELISPOT assays of immune responses to class I and II OVA peptides. Mice were vaccinated with the indicated constructs twice before spleens were taken for ELISPOT assays as described in Materials and Methods. (
Next, we examined whether VIPA-enhanced CD4 and CD8 immune responses could inhibit tumor growth in an established murine tumor model. For this study, we chose a therapeutic model. Mice were initially implanted with B16-OVA tumor cells. Three days later, mice were immunized once with the cell-based vaccinations as described in Fig. 7, and tumor growth was monitored. Vaccination using B16 cells without OVA antigen showed little effect on inhibiting tumor growth, as the majority of animals in the group either died or had to be killed because of large tumor burden (Fig. 8A). The antitumor effect from vaccination with pOVA-transfected B16 cells was not better than that of the B16 control group until 2 weeks after tumor implantation, when tumor size in this group was significantly smaller than that in the B16 group (p < 0.01). However, this antitumor effect seemed to occur too late to have any significant impact on animal survival (Fig. 8B). The best therapeutic result was detected in the group that was vaccinated with pGFP-M4-OVA-transfected B16 cells. The tumor size in this group was significantly smaller than in either of the other two groups at all the time points of measurement. Moreover, mice vaccinated with pGFP-M4-OVA survived significantly longer than those in the other two groups (Fig. 8B). The majority of animals in the pGFP-M4-OVA-treated group were still alive 1 month after tumor implantation, whereas all the animals in the other two groups were dead or had to be killed because of large tumor size or tumor ulceration by day 21. These results indicate that there was a strong correlation between the enhanced CD4+ and CD8+ responses mediated by pGFP-M4-OVA and the increased therapeutic outcome on tumor-bearing animals. These data also echo the notion that simultaneous induction of CD4+ and CD8+ responses is crucial for the host's defense against malignant diseases (Wang et al., 2002).
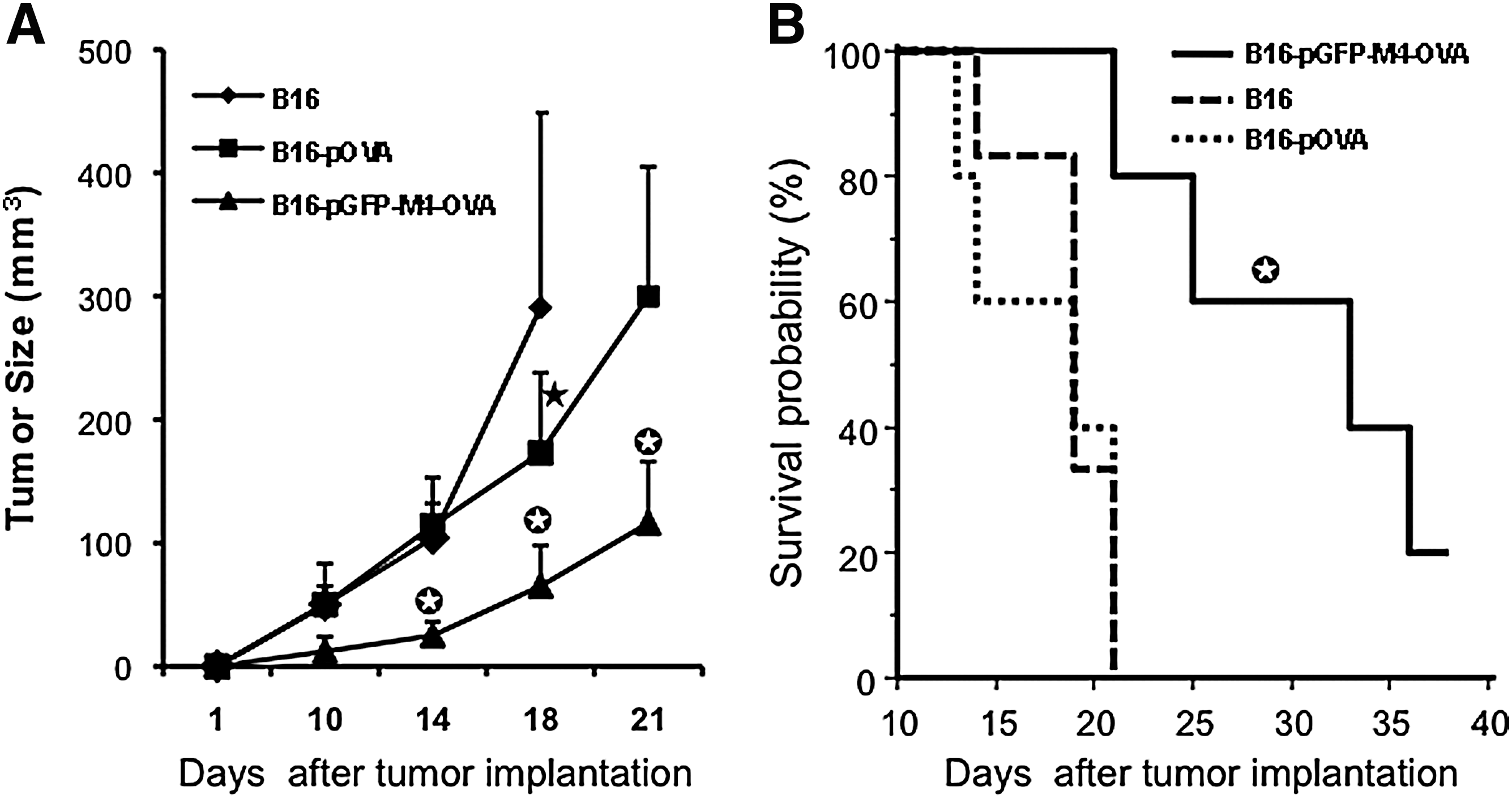
Antitumor effect of VIPA-mediated vaccination. (
Discussion
Protein aggregation is usually a result of protein misfolding. Several cellular proteins have been found to be misfolding-prone and some of them have been associated with conformational disorders such as neurological diseases (Lansbury and Lashuel, 2006). Studies have shown that certain well-defined peptides from these misfolding-prone proteins, when inserted into the C terminus of GFP, can induce aggregate formation (Garcia-Mata et al., 1999). Depending on from which proteins the polypeptides are derived, aggregate formation can locate either in the cytoplasm (Garcia-Mata et al., 1999) or in the nucleus (Fu et al., 2005). Most of the polypeptides are relatively large, in the range of 100–300 amino acids. One exception is the nonnatural 16-residue “degron” peptide, which can convert soluble GFP into an aggregation-prone toxic protein (Bence et al., 2001). To our knowledge, this is the first report that a polypeptide from a viral protein can convert soluble GFP into an aggregation-prone protein. We have named these protein sequences VIPA, that is, viral inducer of protein aggregation. We have defined the region to be as short as 13 amino acids (VIPAM4). Moreover, the N-terminal sequence of VIPA seems to contain signal peptides that dictate either cytoplasmic or nuclear localization of this aggregate formation.
Despite the demonstrated ability of VIPA to induce GFP aggregate formation in our studies, ICP10 has not been reported to form protein aggregates. This suggests that the VIPA sequence within ICP10 does not act in the same way as when fused to the C terminus of GFP. One explanation is the position effect, as demonstrated with the construct pcDNA-M4-PKF in Fig. 2. Despite the demonstrated ability to induce GFP aggregation when M4 is fused to the C terminus of GFP, M4 completely lost this ability when it was fused to the N terminus. In this context, it is interesting to note that most of the reported GFP aggregate formations occurred when the cellular polypeptides or the nonnatural degron peptide were fused to the C terminus of GFP (Garcia-Mata et al., 1999; Bence et al., 2001; Fu et al., 2005; Link et al., 2006). Another possible explanation is that either the N-terminal or C-terminal flanking region of VIPA can negate the VIPA activity. In supporting this possibility, many truncated versions of ICP10 made by Chabaud and colleagues were found to be insoluble and formed aggregates when they were maintained as purified protein in vitro (Chabaud et al., 2007).
As protein aggregates are toxic to cells, they are usually degraded by one of the two major intracellular protein degradation systems: the ubiquitin–proteasome system (UPS) and autophagy. Autophagy is a catabolic process that involves delivery of cellular components to the lysosome for degradation, and has been considered to be the major degradation process for protein aggregates (Mizushima and Klionsky, 2007). On the basis of this understanding, several groups have designed a strategy in which an antigenic protein was guided to the lysosomal degradation pathway through autophagosome formation for the purpose of enhancing class II antigen presentation. For example, Schmid and colleagues showed that targeting the influenza matrix protein-1 to autophagosomes via fusing the antigen to the autophagosome-associated protein Atg8/LC3 led to strongly enhanced MHC class II presentation to CD4+ T cell clones (Schmid et al., 2007). In this study, we chose a surrogate tumor antigen, OVA, which contains well-defined antigenic epitopes for both class I and II MHCs. When OVA is linked to VIPA (together with GFP), VIPA can efficiently guide this surrogate tumor antigen to the lysosomal degradation pathway. This has apparently resulted in dramatically enhanced CD4+ T cell responses to the antigen. Moreover, the presence of VIPA also enhanced MHC class I presentation, resulting in a significant increase in CD8+ T cell responses. The underlying mechanism of VIPA-mediated enhancement of MHC class I presentation is currently unknown. Regardless, these data suggest that VIPA can function as a molecular adjuvant to simultaneously boost MHC class I and class II antigen presentation of a surrogate tumor antigen. Experiments are currently underway to examine this strategy with endogenous tumor-associated antigens or antigens of important infectious diseases.
Studies on tumor immunology have indicated that, for eliciting effective antitumor immunity, both antigen-specific CD4+ and CD8+ cells are required (Schmid et al., 2007). Several unique strategies have been proposed to simultaneously stimulate both class I and class II antigen presentation. For example, a retrogen strategy has been proposed to present an intracellular tumor antigen, MAGE-3, to both MHC class I and MHC class II in a cognate manner. In this approach, the antigen was linked to a leader sequence at its N terminus for secretion and to a cell-binding domain at its C terminus for receptor-mediated internalization. As such, the synthesized antigen would be first released and then taken up by antigen-presenting cells via receptor-mediated internalization into the lysosomal pathway for class II MHC presentation (You et al., 2001). Immunization of mice by this strategy has been shown to efficiently induce both arms of the adaptive antitumor immune response, significantly improving the efficacy of tumor vaccines and immunotherapies (You et al., 2001). Our data show that the adjuvant effect of VIPA results in strong MHC class II presentation of an endogenously synthesized tumor antigen with simultaneous enhancement of the MHC class I response to the same antigen. As a consequence, the elicited antitumor immunity exhibits an effective antitumor effect in a challenging tumor model in which a single vaccination was administered 3 days after tumor implantation. Our data thus support the conclusion that VIPA represents an attractive candidate as a molecular adjuvant for vaccination for cancer immunotherapy and possibly also for other immunologically preventable diseases.
Footnotes
Acknowledgments
This work was partly funded by the National Institutes of Health (2R01CA106671 to X.Z.) and by the William and Ella Owens Medical Research Foundation.
Author Disclosure Statement
The authors declare that there are no financial or other interests regarding the submitted manuscript that might be construed as a conflict of interest.