
Abstract
Malignant melanoma is one of the leading causes of cancer mortality worldwide, underlining the need for effective novel therapies. In this study, the therapeutic efficacy and mechanism of systemic pro-opiomelanocortin (POMC) therapy were evaluated in mice bearing established melanoma. Injection of adenovirus encoding POMC (Ad-POMC) led to hepatic POMC overexpression and elevated adrenocorticotropin (ACTH) levels in the circulation. Systemic POMC therapy significantly attenuated the growth of established melanoma and prolonged the survival of tumor-bearing mice. Histological analysis revealed that systemic POMC therapy induced melanogenic differentiation while reducing melanoma growth. In addition, POMC therapy also elicited a significant reduction in the neovascular network of melanoma. Last, we demonstrated that POMC-derived peptides, including ACTH, α-melanocyte-stimulating hormone (α-MSH), and β-MSH, are involved in POMC-mediated melanogenic differentiation and angiogenesis inhibition. In summary, systemic POMC therapy suppresses melanoma growth via induction of melanogenic differentiation and angiogenesis blockade, thereby demonstrating its potential as a novel treatment modality for melanoma.
Introduction
Pro-opiomelanocortin (POMC) encodes a 31-kDa prohormone that is processed into various antiinflammatory neuropeptides including adrenocorticotropin (ACTH), melanotropins (α-, β-, and γ-MSH), lipotropins, and β-endorphin (β-EP) (Solomon, 1999; Catania et al., 2004). POMC peptides possess pleiotropic functions, including pigmentation, adrenocortical function, regulation of energy homeostasis, and immunity modulation, in the central and peripheral systems (Raffin-Sanson et al., 2003). Studies have shed light on the role of POMC expression during tumor progression. In malignant mesothelioma cells, expression of POMC transcripts, POMC-processing enzymes, and melanocortin-1 receptor (MC1R), the high-affinity receptor for α-MSH, constitutes an autocrine-inhibitory circuit to suppress the proliferation of tumor cells (Catania et al., 2004). Furthermore, POMC-derived peptide, α-MSH, has been reported to trigger melanoma/melanocyte differentiation, a process that involves both dendrite outgrowth and activation of melanin production (melanogenesis) (Smalley and Eisen, 2000, 2002). Other studies have also shown that treatment with α-MSH potently inhibits both the in vitro and in vivo invasion of highly invasive B16-BL6 melanoma cells (Kameyama et al., 1990; Murata et al., 1997). These findings suggest that POMC expression may inhibit the tumorigenicity of tumors and induce melanoma differentiation to intervene with tumor progression (Manna and Aggarwal, 1998; Coussens and Werb, 2002).
In our previous study, we demonstrated that prophylactic POMC gene delivery increased the release of POMC neuropeptides and attenuated tumorigenic processes including anchorage-independent growth, migration, and adhesion in melanoma cells (Liu et al., 2006). Moreover, our study also showed that POMC gene delivery significantly retarded endothelial cell activity in angiogenesis assays by the inhibition of endothelin-1 release in vitro (Lam et al., 2006) and reduced choroidal angiogenesis in laser-induced choroidal neovascularization (CNV) models (Bee et al., unpublished data). In the present study, we describe the differentiating and antiangiogenic effect of POMC gene delivery on melanoma and endothelial cells, and provide evidence concerning the influence on melanogenic differentiation and angiogenic processing. Furthermore, we also evaluated the therapeutic potential of POMC gene transfer for suppression of melanoma in a paracrine manner on tumor differentiation induction and tumor angiogenesis inhibition in established mouse melanoma models. Our results led to the hypothesis that the POMC gene offers a new approach to redifferentiation and antiangiogenic therapy in melanoma.
Materials and Methods
Cell cultures
B16-F10 melanoma cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS; PAA, Austria), 2 mM glutamine, streptomycin (100 mg/ml) (Invitrogen), and penicillin (100 U/ml) at 37°C in a 5% CO2 atmosphere. Human endothelial EA.hy926 cells (kindly provided by C.J.S. Edgell, University of North Carolina, Chapel Hill, NC) were maintained at low passage and cultured in DMEM (Invitrogen) containing 10% fetal bovine serum (FBS; PAA, Austria), 100 μM sodium hypoxanthine, 0.4 μM aminopterin, 16 μM thymidine (HAT; GIBCO-BRL, Rockville, MD), 2 mM glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C in a 5% CO2 atmosphere. ACTH, α-MSH, β-MSH, γ-MSH, and β-EP were purchased from Bachem (Torrance, CA).
Preparation of adenoviral vectors
For Ad-POMC, POMC cDNA was subcloned into pshuttle-CMV to yield the transfer vector. This was then recombined with the entire adenovirus genome in BJ5183 Escherichia coli cells and transfected into 293 cells to generate recombinant virus via homologous recombination by a calcium phosphate protocol (AdEasy XL adenoviral vector system; Stratagene, La Jolla, CA). The virus was then amplified in 293 cells, purified by two rounds of cesium chloride gradient ultracentrifugation, and dialyzed against buffer containing 10 mM Tris (pH 7.5), 1 mM MgCl2, and 10% glycerol at 4°C. The titer of the virus solution was determined by measuring its optical density at a wavelength of 260 nm and performing a plaque-forming assay in 293 cells before storage at −80°C. The control vector, enhanced green fluorescent protein (Ad-GFP), was prepared as described previously (Tai et al., 2006).
Western blot
B16-F10 cells or EA.hy926 cells were infected with adenoviral vectors. After 12 hr, cells were washed with fresh medium and continued to incubate at 37°C for an additional 48 hr. The protein extract was isolated from tissue or cells, using buffer containing 150 mM NaCl, 50 mM HEPES (pH 7), 1% Triton X-100, 10% glycerol, 1.5 mM MgCl2, 1 mM EGTA, and protease inhibitors (Roche Applied Science, Indianapolis, IN). After separation by sodium dodecyl sulfate–12.5% polyacrylamide gel electrophoresis (SDS–PAGE), protein was transferred onto a polyvinylidene difluoride membrane, using a blotting apparatus. The membrane was blocked with 5% milk in Tris-buffered saline–Tween (TBS-T) for 1 hr and then incubated with ACTH (1:500 dilution; Sigma-Aldrich, St. Louis, MO), tyrosinase (1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA), and cyclin D1 (1:500 dilution; Cell Signaling Technology, Danvers, MA) antibodies overnight at 4°C. After incubation with secondary antibody conjugated with horseradish peroxidase (HRP) (1:5000 dilution in 5% milk) for 30 min, the signals on membrane were detected with ECL-plus luminol solution (GE Healthcare Life Sciences, Piscataway, NJ) and exposed to X-ray film for autoradiography.
Transmission electron microscopy analysis
After gene transfer for 48 hr, B16-F10 cells were fixed with 2.5% glutaraldehyde in 0.1 M phosphate-buffered saline (PBS) containing 2% sucrose and then postfixed with 1% OsO4 in the same buffer. Samples were then scraped out from the plates, dehydrated in ascending ethanol concentrations, and embedded in Spurr epoxy resin (Agar Scientific, Stansted, Essex, UK). Ultrathin sections were stained with uranyl acetate and lead citrate and observed under a Jeol-1230 electron microscope.
Melanin assay
For determination of melanin content as previously described (Liu et al., 2006), tumor tissue and cells were dissolved in 1 N NaOH for 1 hr at 60°C. The absorbance was measured by reading optical densities in a microplate reader (Dynex Technologies, Chantilly, VA) at an absorption wavelength of 470 nm.
Flat colony formation assay
B16-F10 cells in logarithmic phase were digested with 0.25% tryptase to produce a single-cell suspension. Five hundred cells were dispersed in a culture dish (diameter, 60 mm); culture medium was added, and the cells were cultured for 2 weeks. When clones were visible, the supernatant was removed, and the clones were fixed in methanol for 15 min. The samples were stained with 0.4% crystal violet for 20 min, rinsed in water, and air dried, and the number of clones that contained more than 50 cells was calculated.
Cell cycle analysis
B16-F10 cells were infected with adenoviral vector at a multiplicity of infection (MOI) of 500 or 1000. After 12 hr, cells were supplemented with fresh medium and allowed to continue incubation at 37°C for an additional 48 hr. The harvested cells were washed twice with PBS before fixation with ice-cold 70% ethanol and storage overnight at −20°C. Cell aliquots were again washed twice with PBS before incubation with RNase A (10 μg/ml) and propidium iodide (50 μg/ml) for 60 min at 37°C. DNA content of 10,000 events was analyzed with a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) equipped with CellQuest software.
Cell proliferation: MTT assay
EA.hy926 cells were cultured in a 96-well plate at a density of 4 × 104 cells/ml. Cells were supplemented with fresh medium containing 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT; 0.456 mg/ml) and incubated for 2–4 hr at 37°C. The formazan in viable cells was dissolved with 100 μl of dimethyl sulfoxide and determined by reading optical densities in a microplate reader (Dynex Technologies) at an absorption wavelength of 570 nm.
Cell migration assay
The migration assay was performed in a Boyden chamber as previously described (Lam et al., 2006). EA.hy926 cells were seeded in triplicate in the upper compartment of the chamber (1 × 105 cells in 400 μl). The lower compartment was filled with 200 μl of DMEM containing basic fibroblast growth factor (bFGF, 100 ng/ml; R&D Systems, Minneapolis, MN) as the chemoattractant. A polycarbonate filter (pore size, 8 μm; Nucleopore-Costar, Cambridge, MA) coated with 0.005% gelatin to allow cell adhesion separated the compartments. After incubation for 6 hr in a humidified 5% CO2 atmosphere at 37°C, cells on the upper side of the filter were removed, and those that had migrated to the lower side were fixed in absolute ethanol, stained with 10% Giemsa solution (Merck, Darmstadt, Germany), and counted in five different high-power fields to determine the mean ± SEM per filter.
Tube formation assay
The tube formation assay was performed as previously described (Lam et al., 2006). Briefly, Matrigel (BD Biosciences) was diluted with cold serum-free medium to 10 mg/ml. The diluted Matrigel solution was added into a 24-well plate (200 μl per well) and allowed to form a gel at 37°C for 30 min. EAhy926 cells (1 × 105 cells/ml) were incubated with Matrigel and then subsequently added to each well to incubate for 6–8 hr at 37°C in 5% CO2. Under these conditions, endothelial cells form delicate networks of tubes that are detectable within 2–3 hr and are fully developed after 8–12 hr. After incubation, the endothelial tubes were fixed with 3% paraformaldehyde and counted under four different high-power fields.
Bioluminescence imaging
After the animals were anesthetized with a cocktail of ketamine–xylazine (4:1) in PBS, mice were injected intraperitoneally with 100 μl of
ACTH radioimmunoassay
The radioimmunoassay (RIA) was performed as previously described (Liu et al., 2006). ACTH concentrations were determined with RIA kits (Nichols Institute Diagnostics, San Juan Capistrano, CA) with a linear range of measurement between 5 and 1000 pg/ml for ACTH and a detection threshold of 5 pg/ml. A standard curve was constructed for each assay.
Primary melanoma models and gene delivery
To induce the primary melanoma, B16-F10 cells were subcutaneously injected into C57BL/6 mice (5 × 105 cells in 0.1 ml of PBS; n = 13–15) to monitor tumor growth. After tumors had grown to at least 100 mm3 (∼7–14 days after implantation), mice were administered adenoviral vector at the indicated doses in 0.1 ml of PBS via the tail vein. Subsequently, tumor volumes were measured with a dial-caliper according to the following formula: width2 × length × 0.52. For tumor-suppressing studies, experiments were terminated when the tumor burden exceeded 10% of the animal's normal body weight. To evaluate the life-prolonging effect of therapy, identical experiments were performed in a large group of tumor-bearing mice (n = 13–15) and the survival rate in various groups of mice was compared by Kaplan–Meier analysis.
Immunohistochemical analysis
Paraffin-embedded 5-μm-thick sections were cut from all blocks and stained with hematoxylin and eosin to examine the overall tissue. To identify tumor vessels, CD31 antibody was used to stain and count vessel formations in tumor tissue. Tissue sections were dewaxed and treated in proteinase K (Dako, Glostrup, Denmark) for 8 min. Sections were then washed in TBS, pH 7.5, and treated with 3% H2O2 in methanol for 10 min to block endogenous peroxidase. After another wash in TBS, the sections were incubated for 20 min in blocking solution (10% goat serum), followed by an overnight incubation in primary antibody to CD31 (1:100; BD Biosciences) and Ki-67 (1:100; Dako) for 1 hr at room temperature. The primary antibody was detected with secondary antibody: biotinylated goat anti-rat (1:200; Dako) for 30 min at room temperature. The biotinylated complexes were detected with streptavidin–HRP (1:400; Dako) and visualized with 3,3′-diaminobenzidine (Dako). The negative controls included substitution of a primary antibody with a corresponding serum isotype.
Statistical analysis
Differences between the groups were statistically evaluated by unpaired Student t test. The results are presented as means ± SEM. All p values were two-tailed, and a p value less than 0.05 was considered statistically significant.
Results
POMC gene delivery induces melanogenic differentiation in melanoma cells and inhibits angiogenic activity in endothelial cells
The E1,E3-defective recombinant adenovirus encoding POMC (Ad-POMC) was generated for gene delivery studies. To evaluate the efficacy of POMC processing, the levels of POMC-derived peptides and ACTH in the cell lysates of B16-F10 or EA.hy926 cells were determined by Western blot. Ad-POMC-infected B16-F10 or EA.hy926 cells expressed significantly higher levels of ACTH compared with cells from control groups (Figs. 1A and 2A).
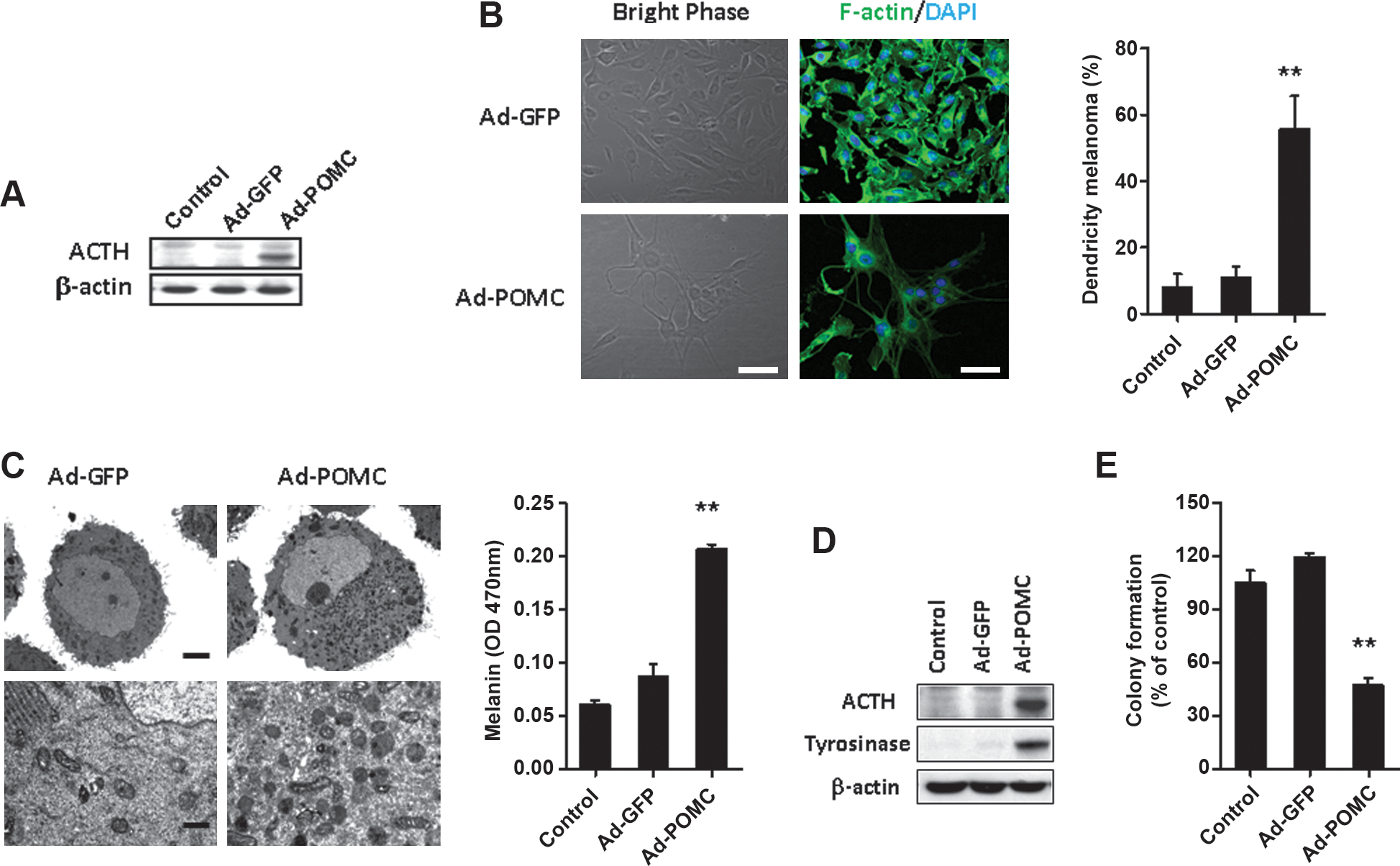
Overexpression of POMC-derived peptides and induced melanogenic differentiation by adenovirus-mediated POMC gene delivery in B16-F10 melanoma cells. Shown is the effect of POMC gene delivery on the levels of ACTH in cell lysates of B16-F10 cells. After infection with adenoviral vectors for 48 hr, cell lysates were harvested from B16-F10 cells and analyzed for the levels of ACTH by Western blot analysis (

The effect of POMC gene delivery on angiogenic activity in EA.hy926 endothelial cells. Shown is the effect of POMC gene delivery on the levels of ACTH in cell lysate of EA.hy926 endothelial cells. After infection with adenoviral vectors for 48 hr, cell lysates were harvested from endothelial cells and analyzed for the levels of ACTH by Western blot analysis (
Induction of melanoma differentiation in B16-F10 cells is characterized by the stimulation of melanogenesis resulting from an increase in long dendrite formation that transfers mature pigmented melanosomes to the neighboring keratinocytes (Wu et al., 2002). It is also characterized by melanin synthesis (Hill et al., 1989) and cell cycle arrest (Strasberg Rieber and Rieber, 1995). By phase-contrast and F-actin staining, it was observed that POMC gene delivery significantly increased the proportion of cells with dendrite differentiation (Fig. 1B; p < 0.01), a morphological feature of differentiated melanocytes. This was correlated with an increment in melanin content (Fig. 1C; p < 0.01), elevated expression of tyrosinase (Fig. 1D), and cell cycle arrest on G1 phase (Table 1). We subsequently investigated the colony-forming capability of B16-F10 cells after POMC gene delivery. It was found that the POMC-transduced melanoma cells showed a significantly reduced ability to form colonies compared with cells of control groups (p < 0.01; Fig. 1E). These results indicate that POMC gene delivery significantly induces redifferentiation by POMC-derived peptides and reduces the malignant features of melanoma cells.
After infection with adenovirus vectors for 48 hr, the effect of POMC gene delivery on cell cycle progression in B16-F10 melanoma cells was analyzed by flow cytometery.
p < 0.05 versus Ad-GFP or control.
p < 0.01 versus Ad-GFP or control.
To evaluate whether POMC gene delivery altered the angiogenic activities of endothelial cells, various angiogenesis assays using endothelial cells including proliferation, migration, and tube formation were performed. After POMC gene delivery, despite a lack of effect on the proliferation of endothelial cells (Fig. 2B), endothelial cells exhibited significantly decreased ability to induce chemotaxis (Fig. 2C) and to form tubelike structures in Matrigel (Fig. 2D). Given the pivotal roles of migration and tube formation during angiogenesis, POMC gene delivery may also perturb the vascular network in melanoma.
Intravenous injection of adenovirus encoding POMC leads to systemic production of POMC neuropeptides and attenuates melanoma progression with associated prolonged survival of tumor-bearing mice
To evaluate the tropism of the transgene after intravenous injection of adenoviral vector, we investigated luciferase transgene expression in mice after injection of adenovirus encoding luciferase (Ad-luc) by bioluminescence analysis. Three days after injection, luciferase expression was localized mainly in the livers of mice and remained detectable for at least 28 days (Fig. 3A). To evaluate the duration of liver-based POMC expression, the plasma ACTH concentration were determined by RIA at various time intervals after adenovirus injection. It was found that the circulating ACTH level rose within 24 hr of injection and reached the maximal level between days 7 and 14, yet remained higher than that of control groups for at least 28 days (Fig. 3B). Thus, intravenous injection of Ad-POMC resulted in liver-based POMC expression and elevated ACTH levels in circulation during experimental period.
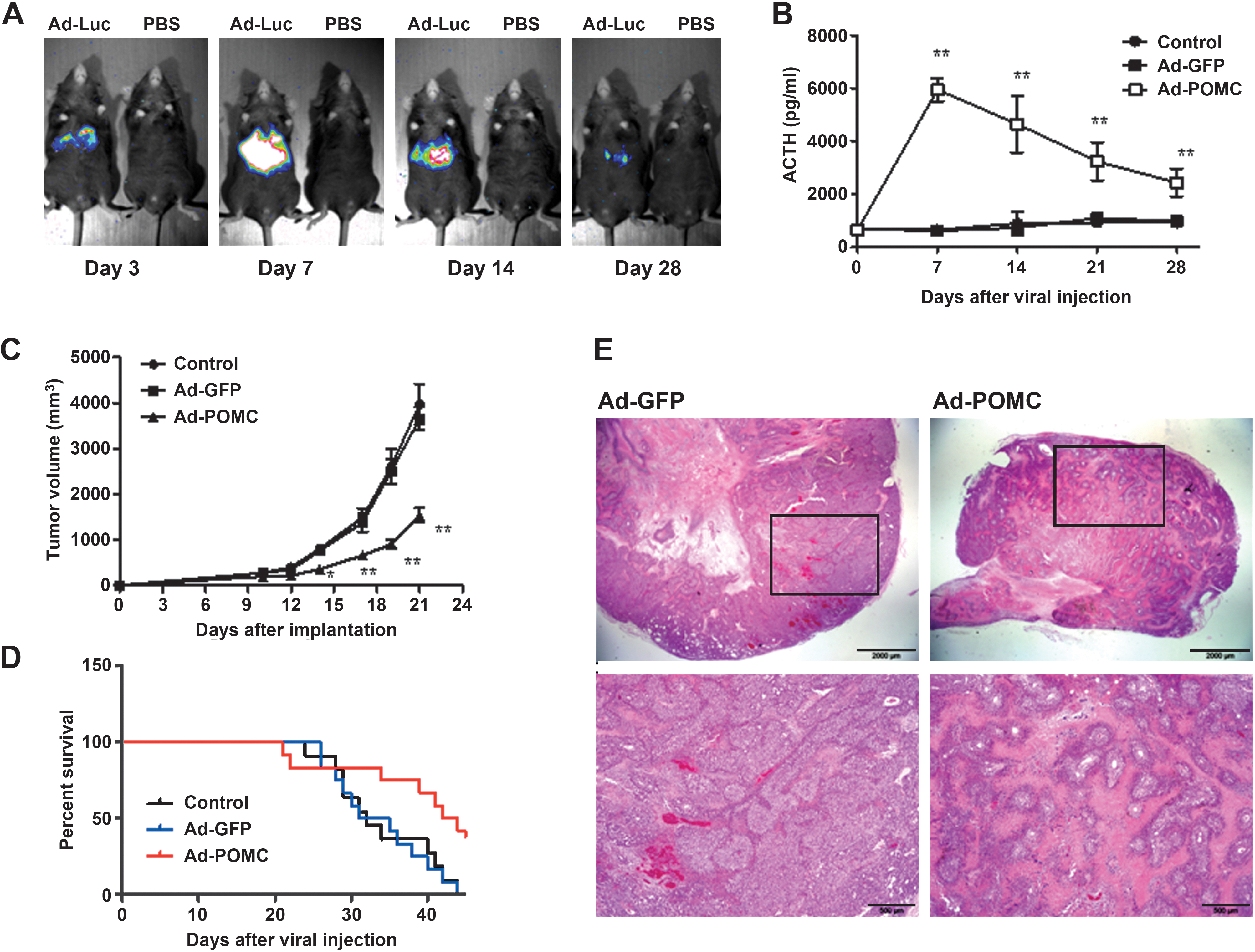
Systemic POMC expression and the effect on growth of established melanoma after intravenous Ad-POMC injection in mice. (
To evaluate the therapeutic potential of systemic POMC delivery, mice bearing established melanoma were administered adenoviral vector and then monitored for tumor progression. Mice treated with Ad-POMC showed significantly retarded melanoma growth compared with those in control animals (the tumor size for Ad-POMC-, control-, and Ad-GFP-treated mice was 1537 ± 547, 3969 ± 1466, and 3648 ± 926 mm3, respectively, on day 21; p < 0.01; Fig. 3C). Moreover, the Ad-POMC-treated mice survived significantly longer than those in the control groups (average survival time for Ad-POMC-, control-, and Ad-GFP-treated mice was 43, 32, and 33 days, respectively; p = 0.038; Fig. 3D). Furthermore, histological analysis showed that only small nodules were found within the Ad-POMC-treated tumor tissues compared with the larger nodules in the Ad-GFP-treated group (Fig. 3E). There was no significant difference in body weight between Ad-POMC-treated mice and the control groups. In addition, biochemical examination revealed no dramatic difference in biochemical parameters between Ad-POMC and control groups (Supplementary Table S1; supplementary data are available online at
We subsequently investigated the dose dependency of systemic POMC delivery for melanoma. By injecting various doses of adenoviral vector, it was found that a dose of 1 × 108 plaque-forming units (PFU) was sufficient to evoke a significant increment in plasma ACTH levels and to attenuate melanoma progression (Table 2). We also evaluated whether POMC gene therapy augments the efficacy of cisplatin chemotherapy in melanoma. It was shown that a combination of cisplatin and POMC gene delivery significantly prolonged the life span of tumor-bearing mice compared with either treatment alone (average survival time for mice treated with Ad-GFP, Ad-POMC, cisplatin, and Ad-POMC combined with cisplatin was 35, 41, 39, and 51 days, respectively; Supplementary Fig. S1).
B16-F10 cells were implanted into C57BL/6 mice on day 0. After tumors reached the size of ∼100 mm3 (on day 10), mice were treated by intravenous injection of adenoviral vectors (1 × 107, 1 × 108, 1 × 109 PFU per mouse) and monitored for ACTH level and tumor growth on day 21.
p < 0.05 versus Ad-GFP (1 × 109 PFU).
p < 0.01 versus Ad-GFP (1 × 109 PFU).
Systemic POMC gene delivery induces melanogenic differentiation and inhibits blood vessel formation in melanoma tissue
Histological analysis of melanoma tissues was performed in mice receiving adenovirus gene delivery. Ad-POMC-treated melanoma tissues were smaller and darker than melanomas of the control groups. Histological analysis further revealed dark deposits surrounding tumor nodules in Ad-POMC- but not Ad-GFP- or control-treated melanomas (Fig. 4A). By measuring melanin content and protein levels of tyrosinase in melanoma tissues, it was found that there was higher melanin content in Ad-POMC-treated melanoma than in Ad-GFP-treated melanoma (p < 0.01; Fig. 4B and C). Subsequently, we studied the correlation between melanogenesis and proliferation events by Ki-67 staining and measuring cyclin D1 expression. It was found that Ad-POMC-treated melanoma exhibited significantly decreased numbers of Ki-67-positive cells and expression levels of cyclin D1 compared with Ad-GFP-treated melanoma (p < 0.05; Fig. 4D and E).
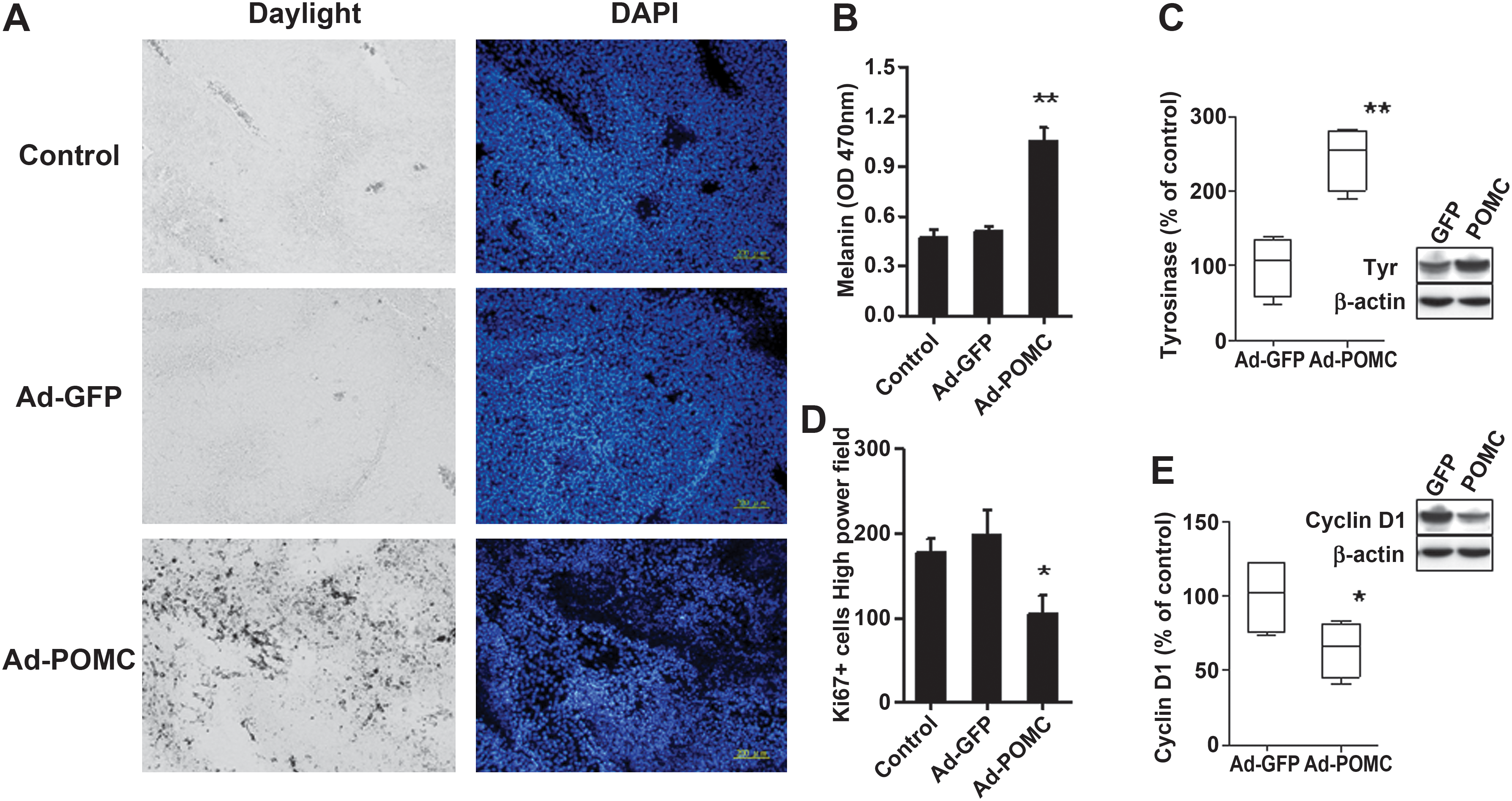
Effect of systemic POMC expression on melanogenic differentiation in melanoma tissues. After injection of adenoviral vector for 21 days, the melanogenic and proliferation extent in melanoma tissues was examined. (
We subsequently investigated whether POMC gene delivery also perturbed the tumor angiogenesis of melanoma. Histological analysis showed that the number of blood vessels analyzed by CD31 immunostaining in Ad-POMC-treated melanoma was significantly decreased compared with tumors of control groups (p < 0.05; Fig. 5A and B). This finding was strengthened by immunohistochemical studies, which revealed a significant reduction in vessel number as well as vessel size in Ad-POMC-treated melanoma compared with control groups by α-smooth muscle actin (α-SMA) (Fig. 5C) or von Willebrand factor (vWF) immunostaining (Supplementary Fig. S2). Together, these results indicate that systemic POMC expression suppressed melanoma growth by reducing cell proliferation, inducing melanoma differentiation, and perturbing the neovascularization in melanoma.

Effect of systemic POMC expression on tumor angiogenesis in B16-F10 melanoma tissues. After injection of adenoviral vectors for 21 days, the blood vessel network in melanoma tissues was examined by CD31 (
POMC-derived peptides mimicking POMC gene delivery induce features of melanoma differentiation and inhibit angiogenic processing
To identify the neuropeptide(s) that contributed to the differentiation-inducing and antiangiogenic effect of POMC gene delivery, B16-F10 and EA.hy926 cells were treated with various POMC peptides (ACTH; α-, β-, and γ-MSH; and β-EP) to recapitulate the effect of POMC gene delivery. For inducing melanogenic differentiation, phase-contrast microscopy analysis showed that application of ACTH, α-MSH, and β-MSH significantly increased the percentages of dendritic formation in B16-F10 cells (Fig. 6A). In contrast, γ-MSH and β-EP treatment had marginal effect. Similarly, treatment with ACTH, α-MSH, and β-MSH induced a significant increase in melanin content in B16-F10 cells whereas γ-MSH and β-EP had no such effect (Fig. 6B). Moreover, by evaluating the effect of POMC-derived peptides on tumorigenicity of melanoma, it was found that an exogenous supply of ACTH, α-MSH, or β-MSH significantly reduced colony formation, whereas γ-MSH and β-EP had no such effect (Fig. 6C).
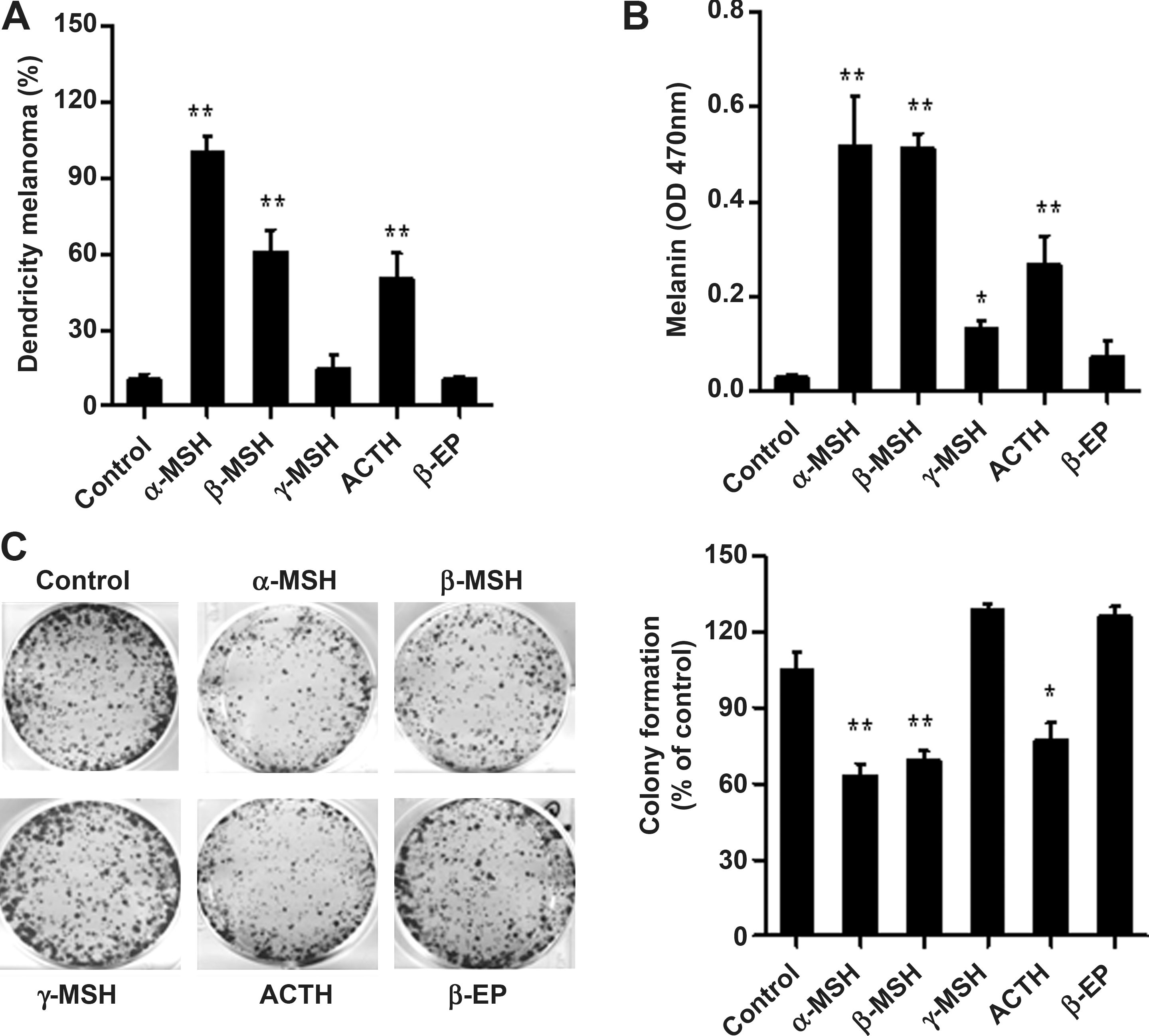
Effect of POMC-derived peptides on induction of melanogenic differentiation and tumorigenicity in B16-F10 melanoma cells. After POMC-derived neuropeptide (10 nM ACTH, 10 nM α-, β-, γ-MSH, and 10 nM β-EP) treatment, B16-F10 melanoma cells were cultured for 48 hr. Morphological changes (
To address which POMC peptide(s) contribute to the antiangiogenic effect of POMC gene delivery, we treated endothelial cells treated with various POMC peptides and then evaluated their influence on angiogenic processes. It was shown that treatment with ACTH, α-MSH, or β-MSH significantly reduced tube formation and migration of endothelial cells (Fig. 7A and B). On the other hand, γ-MSH and β-EP treatment had no effect on these angiogenic processes. Therefore, the production of ACTH, α-MSH, and β-MSH but not γ-MSH and β-EP participated in POMC-mediated melanogenic differentiation and inhibition of tumor angiogenesis in melanoma.
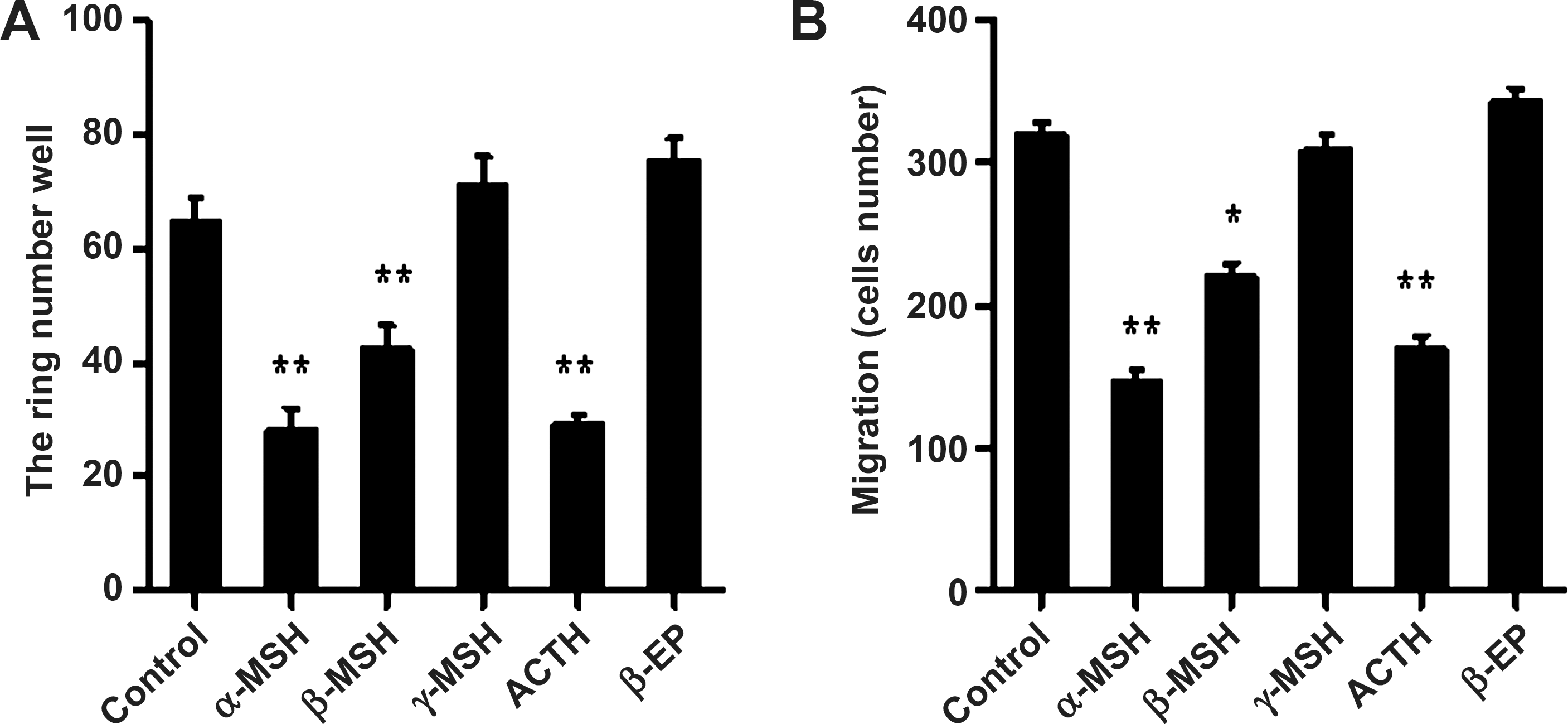
Effect of POMC-derived peptides on inhibition of angiogenic processing in EA.hy926 endothelial cells. After POMC-derived neuropeptides (10 nM ACTH, 10 nM α-, β-, γ-MSH, and 10 nM β-EP) treatment, the representative profile of migration (
Discussion
The novel findings of the present study are as follows. First, we demonstrated the feasibility of using peripheral organs such as the liver as a reservoir for sustained production of POMC prohormone, expression and processing of which normally take place primarily in the central nervous system. Second, we validated the antineoplastic potential of systemic POMC therapy for the control of distant melanoma in vivo. Third, we demonstrated that POMC-derived peptides such as ACTH, α-MSH, and β-MSH are involved in POMC-mediated melanoma suppression through melanogenic differentiation induction and angiogenesis inhibition (Fig. 8).

Schematic mechanism of POMC gene therapy for melanoma suppression.
This study demonstrates the potential of systemic POMC therapy in suppressing melanoma, which also enhances the survival of tumor-bearing mice. In fact, in addition to mouse B16-F10 melanoma, POMC therapy similarly reduced the tumor burden in human A375 xenograft melanoma in immune-deficient mice (data not shown), suggesting that this approach may be extrapolated to human melanoma. To our knowledge, this study provides the first proof-of-principle evidence for the antineoplastic function of POMC hormone. Because mice receiving POMC therapy showed no obvious changes in body weight and feeding behavior, systemic POMC expression seemed tolerable in the animals. We have demonstrated the feasibility of using a similar strategy to prevent capsaicin-induced neurogenic inflammation in rat airways (Liu et al., 2009a). However, because of its inability to completely eradicate the melanoma, POMC therapy may be better used in conjunction with current cancer therapies such as chemotherapy or radiotherapy. Indeed, our studies suggested that combining POMC gene therapy with cisplatin further augmented the survival-prolonging effect of the therapy. Because prophylactic POMC overexpression inhibited the lung metastasis of B16-F10 melanoma in mice, it remains to be determined whether POMC therapy could be applied to the treatment of metastatic diseases or other kinds of cancer. Furthermore, the adverse effects of POMC-based therapies should be carefully characterized before advocating its clinical application.
Our previous studies showed that intratumor POMC expression by adenovirus gene delivery suppressed growth and metastasis of B16-F10 melanoma via autocrine production of POMC neuropeptides (Liu et al., 2006). We also previously showed the intramuscular route to be an effective route for systemic gene expression (Liu et al., 2009b). Similarly, in the current study, we compared the various delivery routes for efficacy in melanoma suppression to determine the optimal delivery route for systemic POMC gene therapy. In this study, systemic POMC expression produced POMC peptides in the liver, which were released into the circulation and reached tumor tissues in a paracrine manner. To our surprise, the liver-based, paracrine POMC expression elicited better therapeutic efficacy in reducing tumor burden than the tumor-based, autocrine approach (p < 0.05; Supplementary Fig. S3). This could be attributed to the higher transduction efficiency of adenoviral vectors in the liver compared with melanoma or muscle tissues. This was reflected by the stronger bioluminescence in mice receiving luciferase vector via the intravenous route.
One important finding of this study is that systemic POMC delivery induced melanogenic differentiation in distant melanoma tissues. The low degree of differentiation shown by melanoma cells (similar to that of cells in early stages of melanocytic development) may be related to their aggressiveness (Elder et al., 2005). The appropriate therapy could induce cell changes to result in a loss in proliferative capacity and an increase in differentiation (Leszczyniecka et al., 2001). It has been reported that adult human metastatic melanoma cells expressing a differentiated phenotype can be reprogrammed to modify this phenotype, a highly promising development (Kulesa et al., 2006). In the present work, we provide evidence that POMC gene delivery induces differentiation of B16-F10 melanoma cells and reverts the cells from the proliferating to the differentiated state. The differentiation pathway, characterized by growth inhibition, reorganization of the cytoskeletal network, and dendrite outgrowth, has been reported to modulate cell growth and differentiation and found to suppress tumor cell invasion in vitro and metastasis in vivo (Jacob et al., 1998; Pereda et al., 1999).
Angiogenesis plays a critical role in tumor development as well as metastasis in many types of cancer including melanoma (Marcoval et al., 1997; Vacca et al., 2000; Hlatky et al., 2002; Streit and Detmar, 2003; Folkman, 2006). The present study showed that systemic POMC therapy retarded vascular formation in melanoma, which was likely due to inhibition of the angiogenic process through a direct effect on endothelial cell function. Furthermore, systemic POMC gene delivery may not only affect tumor vasculature, but also disrupt normal endothelium as depicted in our previous study showing that POMC gene transfer possibly retards the migration and tube formation of endothelial cells by inhibition of endothelin-1 release (Lam et al., 2006). Together, systemic POMC expression or exposure to POMC peptides could perturb the angiogenic activity of endothelium during melanoma growth. Future work is required to characterize the peptide(s) and receptors that contribute to the POMC-induced angiogenesis blockade.
In summary, the present study provides evidence and mechanistic insights supporting systemic POMC delivery, which may facilitate a novel therapy for melanoma control. The potential of POMC-based therapy for metastatic melanoma and other types of cancer deserves future investigation. Moreover, delineation of antineoplastic POMC peptide(s) and their signaling pathway(s) is pivotal to optimize POMC therapy. This study not only sheds light on the novel function of POMC prohormone, but may also assist the development of therapeutic strategies to combat cancer and inflammatory diseases.
Footnotes
Acknowledgments
This work was supported by grants from the National Science Council, Taiwan (NSC 98-2320-B-110-004-MY3), Kaohsiung Veterans General Hospital, Taiwan (VGHKS-98-007 and VGHKS-99-036), the National Sun Yat-Sen University-Kaohsiung Medical University Joint Research Center, and the Asia-Pacific Ocean Research Center of National Sun Yat-Sen University.
Author Disclosure Statement
The authors who have taken part in this study declare that they do not have anything to disclose regarding funding from industry or conflict of interest with respect to this manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.