
Abstract
Polypurine reverse-Hoogsteen hairpins (PPRHs) are double-stranded DNA molecules formed by two polypurine stretches linked by a pentathymidine loop, with intramolecular reverse-Hoogsteen bonds that allow a hairpin structure. PPRHs bind to polypyrimidine targets by Watson–Crick bonds maintaining simultaneously a hairpin structure due to intramolecular Hoogsteen bonds. Previously, we described the ability of Template-PPRHs to decrease mRNA levels because these PPRHs target the template DNA strand interfering with the transcription process. Now, we designed Coding-PPRHs, a new type of PPRHs that directly target the pre-mRNA. The dihydrofolate reductase (dhfr) gene was selected as a target in breast cancer therapy. These PPRHs caused a high degree of cytotoxicity and a decrease in DHFR mRNA and protein levels, but by a different mechanism of action than Template-PPRHs. Coding-PPRHs interfere with the splicing process by competing with U2 auxiliary factor 65 for binding to the polypyrimidine target sequence, leading to a lower amount of mature mRNA. These new PPRHs showed high specificity as no off-target effects were found. The application of these molecules as therapeutic tools was tested in breast cancer cells resistant to methotrexate, obtaining a noticeable cytotoxicity even though the dhfr locus was amplified. Coding-PPRHs can be considered as new molecules to decrease gene expression at the mRNA level and an alternative to other antisense molecules.
Introduction
Hoogsteen bonds are hydrogen bonds between nucleic acids, with a different binding code than Watson–Crick bonds. The main benefit of this kind of linkage is to allow the formation of triplex structures, in which one DNA strand can simultaneously form Watson–Crick bonds with one strand and Hoogsteen bonds with the other (Praseuth et al., 1999). The only requirement for the usage of Hoogsteen structures is the presence of polypyrimidine/polypurine stretches, as this type of binding can only happen when these sequences are present. There are two major groups of Hoogsteen geometries: parallel and antiparallel. Parallel triplexes follow the d(T#A•T) and d(C#G•C) code in which the Watson–Crick polypurine strand is in the same orientation as the Hoogsteen polypyrimidine strand, whereas antiparallel triplexes need the formation of the triads d(A#A•T) and d(G#G•C), and the orientation between the Watson-Crick polypurine strand and the Hoogsteen polypurine strand is antiparallel. The main benefit of using antiparallel Hoogsteen bonds is that they can be formed at physiological pH, unlike parallel Hoogsteen bonds.
Hoogsteen bonds play an important role in the mechanism of action of triplex-forming oligonucleotides and circular oligonucleotides, which have been used with great success but have found to have some stability and specificity problems (Chan and Glazer, 1997; Giovannangeli and Heléne, 1997; Ryan and Kool, 1998; Casey and Glazer, 2001). As an attempt to solve these problems, a molecule that binds to its target sequence by Watson–Crick bonds instead of by Hoogsteen bonds has been designed. PPRHs are non-modified DNA molecules formed by two polypurine stretches linked by a five-thymidine loop (Aviñó et al., 2002; Coma et al., 2005). The hairpin structure is maintained by intramolecular reverse-Hoogsteen bonds between the two polypurine stretches, which are in antiparallel orientation. These molecules bind to their polypyrimidine target sequence by Watson–Crick bonds, thus forming a triplex structure.
In a previous study, we described a type of PPRHs that had the ability to bind to a target sequence located in the template DNA strand, Template-PPRHs (de Almagro et al., 2009). The formation of the triplex between these PPRHs and their double-stranded (ds) DNA target sequence interfered with the transcription process, thus producing an mRNA decrease. In vitro binding studies revealed that Template-PPRHs were able to bind to their dsDNA polypyrimidine target sequence in a sequence-specific manner. These PPRHs showed a remarkable stability, almost 5 days, without containing modified nucleotides. In comparison with other molecules that decrease mRNA levels, Template-PPRHs showed activity greater than that of aODNs and similar to that produced by siRNAs. The difficulty in designing PPRHs resides in finding pure polypyrimidine stretches, but this problem was minimized by placing adenines in front of purine interruptions.
In this study, a new type of PPRHs has been developed, Coding-PPRHs. These PPRHs directly target the pre-mRNA and the coding strand, thus allowing the application of the PPRH technology to genes whose polypyrimidine sequences are located in the coding DNA strand. We studied the activity of PPRHs targeting mRNA and their mechanism of action. The Coding-PPRHs' binding ability were analyzed, as well as their effects on mRNA, protein, enzymatic activity, and cell viability, using the dihydrofolate reductase (dhfr) gene as a model. DHFR is involved in the synthesis of glycine, hypoxanthine, and thymidylate; thus its inhibition blocks DNA synthesis and cell growth. The therapeutic application of these PPRHs in breast cancer cells resistant to methotrexate was also studied.
Materials and Methods
Design and usage of PPRHs
The PPRHs used in this study were made up of two antiparallel polypurine domains, bound by intramolecular reverse-Hoogsteen bonds and linked by a pentathymidine loop. The Triplex-Forming Oligonucleotide Target Sequence Search software (M.D. Anderson Cancer Center, Houston, TX) (
Preparation of polypurine/polypyrimidine duplexes
The duplexes to be targeted by the hairpins were formed by mixing 25 μg of each single-stranded (ss) polypurine and polypyrimidine oligodeoxynucleotide with 150 mM NaCl and incubated at 90°C for 5 minutes as described by de Almagro et al. (2009).
Oligodeoxynucleotide labeling
One hundred nanograms of ss or ds oligodeoxynucleotides or RNA was 5’-end-labeled with [γ-32P]ATP as described by de Almagro et al. (2009).
DNA–PPRH and RNA–PPRH binding analysis
Triplex formation was analyzed by incubating radiolabeled ss or ds DNA or RNA targets (2 nM strand concentration; 20,000 cpm) in the presence or absence of unlabeled PPRHs (250 nM strand concentration) in a buffer containing 10 mM MgCl2, 100 mM NaCl, and 50 mM HEPES, pH 7.2. Binding reactions (20 μl) were incubated for 45 min at 37°C before electrophoresis, which was performed on a nondenaturing 12% polyacrylamide gel containing 10 mM MgCl2, 5% glycerol, and 50 mM HEPES, pH 7.2. Gels were electrophoresed for 3–4 hr at 190 V at 4°C, dried, and analyzed on a Storm 840 Phosphorimager (Molecular Dynamics, Sunnyvale, CA). Quantification was performed using ImageQuant version 5.2 software (Molecular Dynamics). Binding specificity was tested by addition of 2 μg of tRNA (Sigma-Aldrich) to the binding reaction.
Plasmid construction
The Chinese hamster dhfr minigene construct pDCHIP has been described previously (Ciudad et al., 1988). It contains the six exons of the gene, intron-1, about 900 bp of the 5′ flank, and the first of the two major polyadenylation sites in exon-6. Plasmid pDCHIP-Hp was constructed by cloning the Coding-PPRH target sequence (Fig. 1b or Table 1), present in the intron-3 of the human dhfr gene, into the unique PstI site in intron-1 of pDCHIP. This 31-nucleotide sequence contained a NotI site close to its 5′ end, to select the plasmids containing the insert. The number of inserts cloned was measured by polymerase chain reaction (PCR) product size, and a plasmid containing just one insert was selected.
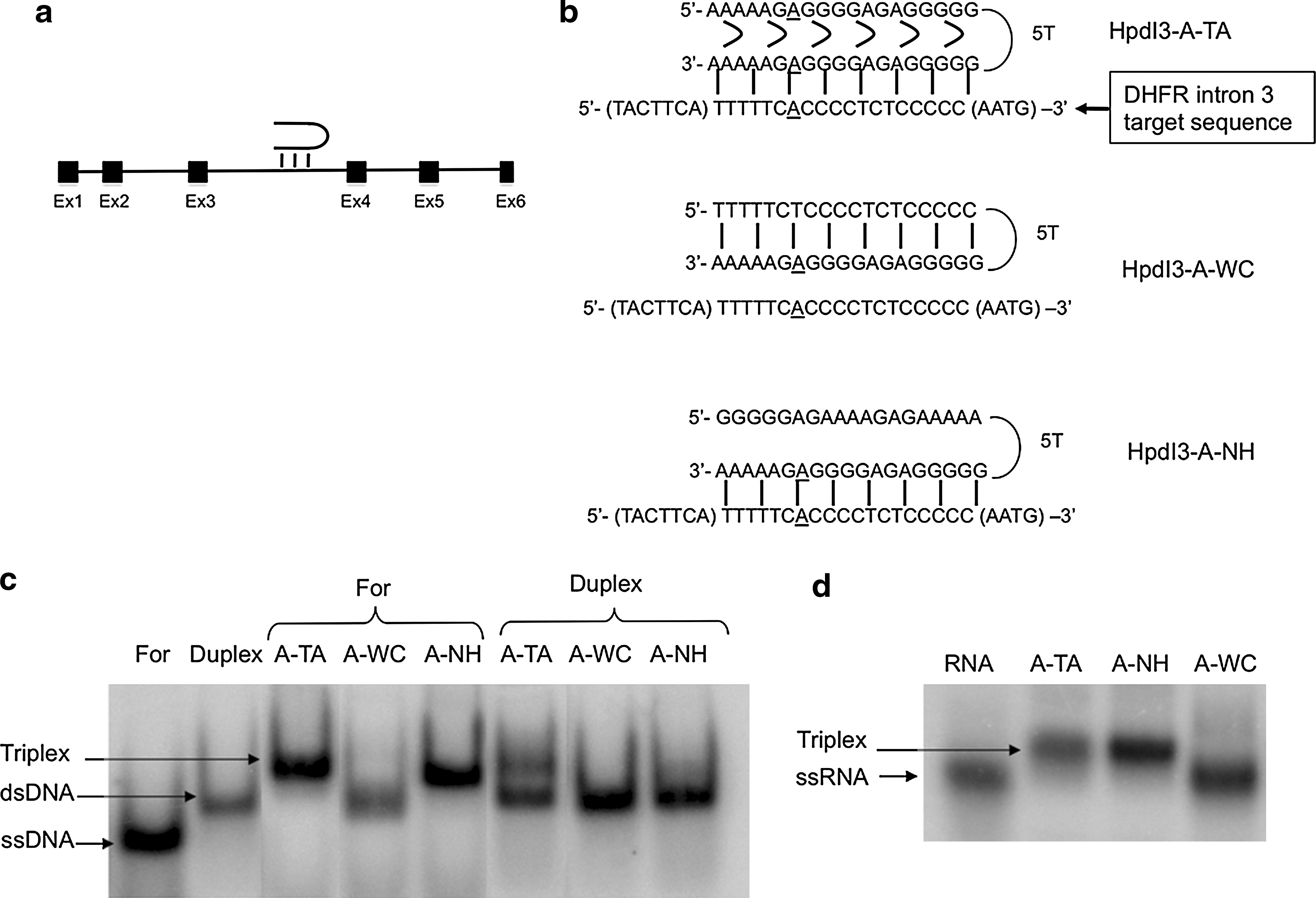
Coding-PPRH binding to its target sequence in the dhfr gene. (
Bold letters indicate mismatches between the target sequence and the PPRH. The sequences in parentheses are not PPRH target sequences.
Cell culture
SKBR3 breast cancer cells were grown in Ham's F-12 medium containing 7% fetal bovine serum (GIBCO, Invitrogen, Barcelona, Spain) and incubated at 37°C in a humidified 5% CO2 atmosphere. MCF7-R breast cancer cells resistant to 10−6 M methotrexate were previously generated in our laboratory (Selga et al., 2009) and bear amplification of the dhfr locus. This cell line was grown in Ham's F-12 medium lacking the final products of DHFR activity (glycine, hypoxanthine, and thymidine [–GHT medium]) (Chasin, 1985) containing 7% dialyzed fetal bovine serum (GIBCO, Invitrogen). pDCHIP and pDCHIP-Hp plasmids were stably transfected into dhfr − DG44 Chinese hamster ovary cells to obtain the DCHM1 and DCHM1-Hp cell lines, respectively. In brief, 500,000 dhfr − DG44 cells were transfected with 1 μg of plasmid pDCHIP or pDCHIP-Hp or with unspecific calf DNA. Five hours after the calcium phosphate transfection the medium was changed, and 24 hr later the selective medium –GHT was added. Transfectants were selected for survival in –GHT medium. These cell lines were used for specificity studies.
Transfection
SKBR3, MCF7-R, DCHM1, and DCHM1-Hp cells were plated in 35-mm-diameter dishes. PPRHs were mixed with N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate (DOTAP) (Roche, Mannheim, Germany) at the appropriate oligonucleotide–DOTAP molar ratio (1:10–1:100) for 15 min at room temperature before lipofecting the cells.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay
SKBR3, MCF7-R, DCHM1, and DCHM1-Hp cells (10,000) were plated in 35-mm-diameter dishes in –GHT medium. After 7 days, 500 μg of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide and 5.6 mg of succinate (both from Sigma-Aldrich) were added to the culture medium and allowed to react for 3 hr at 37°C before addition of the solubilization reagent (0.57% acetic acid and 10% sodium dodecyl sulfate in dimethyl sulfoxide). Cell viability was measured at 570 nm in a WPA S2100 Diode Array spectrophotometer (Biochrom Ltd., Cambridge, UK).
mRNA analysis
At 72 hr after PPRH transfection, total RNA from 30,000 SKBR3 or MCF7-R cells was extracted using Ultraspec RNA (Biotech Laboratories, Houston) following the manufacturer's specifications. Reverse transcription and real-time PCR were performed as described by de Almagro et al. (2009).
The primer sequences for Sybr were as follows: Dhfr-Ex3 forward, 5′-GAAGACCTGGTTCTCCATTCC-3′; Dhfr-Int3 reverse, 5′-GCAGCTTCATCAATAGCTCCT-3′; Dhfr-Ex5 reverse, 5′-GGCAGAAGTTTATATTTCTCCAAA-3′; Dhfr-Ex4 reverse, 5′-TGCCACCAACTATCCAGACC-3′; Tert-forward, 5′-AGGAGCTGACGTGGAAGATG-3′ Tert-reverse, 5′-GCTCGACGACGTACACACTC-3′; Surv-forward, 5′-AGCCAGATGACGACCCCATAG-3′; Surv-reverse, 5′-CACAGGAAGGCTGGTGGCAC-3′; Aprt-forward, 5′-GCAGCTGGTTGAGCAGCGGAT-3′, and Aprt-reverse, 5′-AGAGTGGGGCCTGGCAGCTTC-3′. S100A4, DHFR, UGT1A10, and adenine phosphoribosyltransferase (APRT) mRNA Taqman probes were used (Applied Biosystems, Barcelona). A 5% polyacrylamide gel was used to analyze the size of the PCR products.
Western analysis
MCF7-R cells were used to detect DHFR protein. MCF7-R cells (30,000) were plated in 35-mm-diameter dishes and treated with PPRHs. At 72 hr after transfection, total protein extracts were obtained, and western blot analysis to detect the levels of DHFR protein was performed as described by de Almagro et al. (2009).
DHFR activity assay
At 72 hr after PPRH transfection, DHFR activity was determined by incorporation of tritiated deoxyuridine to cellular DNA (Ciudad et al., 1988) with modifications (Noe et al., 1998).
In vitro transcription
Linear DNA templates for in vitro transcription assays were generated by PCR using pDCHIP and pDCHIP-Hp as templates and the following primers: T7Dex1 (5′-TAATACGACTCACTATAGCCAAACTTGGGGGAAGCA-3′) and Dex3U1Tail (5′-ATACTTA CCTGCGATTCTTCTCAGGAATGGAG-3′). PCR products were purified in a 5% polyacrylamide gel.
The in vitro transcription reactions were performed with the RiboScribe T7 Probe Synthesis Kit (Epicentre Biotechnologies, Madison, WI). Reactions contained 0.5 μg of template DNA, 1 × Transcription Buffer, 0.5 mM each ATP, CTP, and GTP, 15 μM unlabeled UTP, 30 μCi of [α-32P]UTP (3,000 Ci/mmol, Perkin Elmer, Madrid), 10 mM dithiothreitol, 40 units of RNase inhibitor (Promega Biotech Ibérica, Madrid), and 750 μM Cap analog, in a final volume of 20 μl. The reaction was incubated for 3 hr at 37°C and then stopped by addition of formamide loading buffer (80% [v/v] formamide, 20% [v/v] 5 × Tris-borate-EDTA, bromophenol blue, and xylene cyanol). The samples were heated at 85–95°C for 5 min, placed on ice, and resolved in 7 M urea/5% polyacrylamide gels using Tris-borate-EDTA as the electrophoresis buffer. The electrophoresis was carried out at 350 V and 55°C. Following electrophoresis, gels were subjected to autoradiography, and the transcription products were gel-extracted, precipitated with ethanol, and resuspended in diethylpyrocarbonate-water.
In vitro splicing
PPRHs were incubated for 45 min at 37°C with DCHIP or DCHIP-Hp transcripts (20,000 cpm or 1 μg) in the presence of 4 units of RNase inhibitor and 5.8 mM magnesium acetate. After PPRH binding, splicing reaction was started by adding 1 mM ATP, 5 mM creatinine phosphate, 0.5 mM dithiothreitol, 80 mM potassium acetate, and HeLa nuclear extract specific for splicing (CilBiotech, Mons, Belgium) to the reaction mixture, in a final volume of 25 μl. The splicing reaction was carried out at 30°C for 2 hr. The reaction was stopped by addition of 100 mM Tris (pH 7.5), 10 mM EDTA, 1% sodium dodecyl sulfate, 150 mM NaCl, and 300 mM sodium acetate (AppliChem, Ecogen, Barcelona). Nucleic acids were extracted with phenol/chloroform/isoamyl alcohol (25:24:1 v/v/v) and precipitated from the aqueous phase by adding 1 volume of isopropanol (AppliChem, Ecogen). The pellets were then dissolved in formamide loading buffer and heated for 5 min at 85–95°C, before electrophoresis in 7 M urea/5% polyacrylamide gels at 55°C and 350 V. The gels were dried and analyzed on a Storm 840 Phosphorimager, and the transcripts were quantified using ImageQuant version 5.2 software. Results were normalized by the signal of the whole lane after background correction.
Electrophoretic mobility shift assay
An ssDNA probe corresponding to HpdI3-A-TA target sequence 5′-TACTTCATTTTTCACCCCTCTCCCCCAATG-3′ was end-labeled with T4 polynucleotide kinase (New England Biolabs, Beverly, MA) and [γ-32P]ATP (3,000 Ci/mmol, Perkin Elmer) and used in the gel shift assays. A DNA probe was used because U2 auxiliary factor (U2AF) 65 is able to bind either ssDNA or ssRNA polypyrimidine sequences as reported by Singh et al. (2000). One microgram of HeLa nuclear extract specific for splicing (CilBiotech) was used. In competition experiments, the different PPRHs used were added to the reaction mixture in excess to the radiolabeled probe ranging from 10- to 200-fold. DNA binding assays were performed as described (Noe et al., 2001) and were analyzed on a Storm 840 Phosphorimager. tRNA was used as a nonspecific competitor in a 1:1 nonspecific/specific ratio. In the supershift assays, 1 μg of rabbit polyclonal antibody U2AF65 (Santa Cruz, Heidelberg, Germany) was added to the reaction mixture in combination with 0.5 μg or 1 μg of nuclear extract and incubated overnight at 4°C.
Statistical analysis
Data are presented as mean ± SEM values. Statistical analysis was performed using Student's t test using SPSS (Chicago, IL) version 13.0 software for Mac OS X (Apple Computer, Cupertino, CA). Results were considered significant if p < 0.05 (*), p < 0.01 (**), or p < 0.005 (***).
Results
Design of Coding-PPRHs
We searched for polypyrimidine stretches in the coding DNA strand of the human dhfr gene. One polypyrimidine stretch was found in intron-3, which contained a purine interruption (Fig. 1a and b). Two PPRHs were designed against this polypyrimidine target—one with an adenine matching the adenine interruption (HpdI3-A-TA) of the target sequence and another with a guanine (HpdI3-A-TG)—because we had determined that placing either a T or C in the PPRH sequence was less effective (de Almagro et al., 2009). To study the importance of the hairpin structure and Hoogsteen bonds on PPRH activity, we used a polypurine structure that had the same target sequence as HpdI3-A-TA, but without the ability to form a hairpin structure due to its lack of intramolecular Hoogsteen bonds (HpdI3-A-NH). This PPRH would act as an aODN with a hanging tail. As negative controls we used a PPRH with a scrambled sequence (HpdI3-Sc) and a PPRH with intramolecular Watson–Crick bonds instead of Hoogsteen bonds, thus displaying a structure able neither to form triplexes nor to bind DNA (HpdI3-A-WC). To test cell responsiveness, HATNL-24, a phosphorothioate aODN targeting dhfr translational start (5′-gatGcAGtttAGcGAAccAAccaT-3′ [where lowercase letters indicate phosphorothioate bonds]) (Rodriguez et al., 1999), and the dhfr Template-PPRH HpdI3-B directed against intron-3 of the dhfr template DNA strand (de Almagro et al., 2009) were used. BLAST analyses were performed to assess PPRH specificity. The PPRH sequences are listed in Table 1, and their structures are depicted in Fig. 1b.
Binding of Coding-PPRHs to ssDNA, dsDNA, and RNA
The ability of Coding-PPRHs to bind to their target sequence was studied by electrophoretic mobility shift assays. Three probes were used: the For sequence, containing the polypyrimidine ssDNA target; the Duplex probe, which corresponds to the polypyrimidine/polypurine dsDNA target sequence; and the RNA probe corresponding to the polypyrimidine target sequence. Figure 1b shows the different PPRH sequences and their corresponding binding structures. In Fig. 1c and d, the mobility of the For, the Duplex, and the RNA probes can be observed. The mobility shift due to PPRH binding to ssDNA (For) indicated that HpdI3-A-TA and HpdI3-A-NH (no Hoogsteen bonds) were able to bind to its target sequence. However, only HpdI3-A-TA was able to bind to its dsDNA target sequence (Duplex) and to form a triplex. To assure that PPRHs were also able to bind to its corresponding RNA target sequence, they were incubated in the presence of the radiolabeled RNA probe (Fig. 1d). HpdI3-A-TA and HpdI3-A-NH bound to its RNA target sequence, whereas HpdI3-A-WC was unable to bind to it. Binding specificity was tested by addition of 2 μg of tRNA to the binding reactions. It is interesting to note that ssDNA probe incubated with HpdI3-A-WC suffered a shift in mobility corresponding to a duplex structure. If that shift were due to the formation of a duplex with the polypurine sequence of the Hp-A-WC after displacement of the polypyrimidine strand, it would form a structure similar to that of Hp-A-NH, which produced higher retardation in mobility. However, when testing Hp-A-WC with its real target, ssRNA, no retardation was observed.
Coding-PPRH cytotoxicity
We tested the cytotoxic effect of HpdI3-A-TA and HpdI3-A-TG in SKBR3 breast cancer cells. In Fig. 2a, it is shown that HpdI3-A-TA produced 67% cytotoxicity, whereas the cell death caused by HpdI3-A-TG was 55%; thus HpdI3-A-TA was selected for the following assays. The specificity of HpdI3-A-TA was studied using a series of different controls that did not cause a significant change in cell viability (Fig. 2b). On the other hand, HATNL-24 (at its optimal conditions, 1:10 aODN:DOTAP molar ratio) and HpdI3-A-NH caused a significant drop of cell survival, as they acted as aODNs. Additionally, an HpdI3-A-TA dose–response curve was performed (Fig. 2c). The highest cytotoxicity was 86% at 100 nM HpdI3-A-TA using a 1:100 PPRH:DOTAP molar ratio (10 μM DOTAP), similar to that produced by the Template-PPRH HpdI3-B. The minimum time that HpdI3-A-TA needed to cause cell death was analyzed by removing the PPRH at different intervals (Fig. 2d). At 48 hr, almost the maximum activity was achieved.
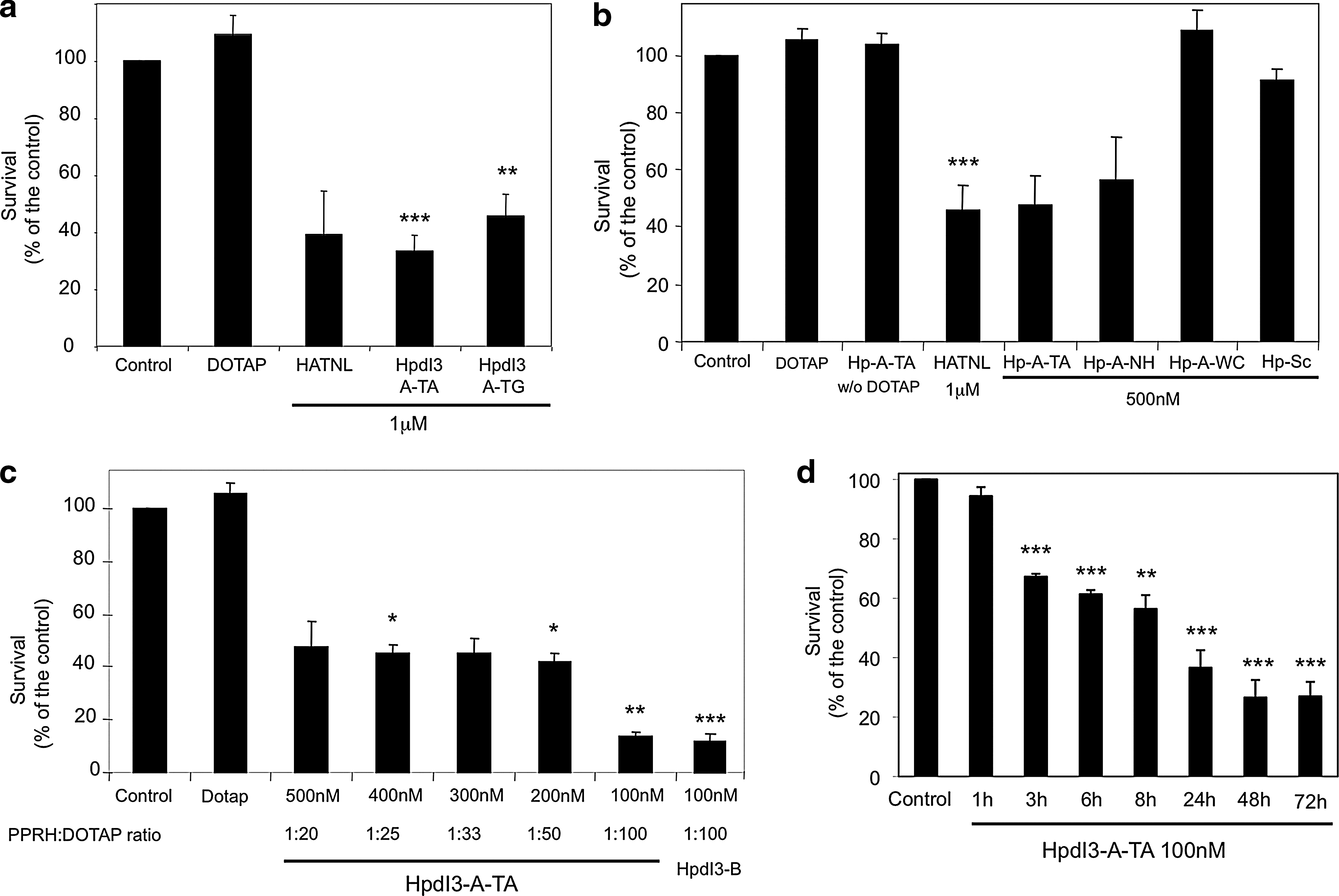
Cytotoxicity caused by Coding-PPRHs. (
Effects of Coding-PPRHs on DHFR activity
The 6-[3H]deoxyuridine assay was used to determine DHFR activity in SKBR3 cells incubated with Coding-PPRHs (against DHFR mRNA) for 72 hr. HpdI3-A-TA (100 nM):DOTAP (10 μM) (1:100 ratio) showed the greatest inhibition of DHFR activity (60%) (Fig. 3a), in agreement with the cytotoxicity results. Lower PPRH:DOTAP molar ratios caused less DHFR inhibition, despite the increase in PPRH concentration. HpdI3-A-NH also inhibited DHFR activity but in a lesser extent than HpdI3-A-TA (100 nM), whereas DOTAP (10 μM) alone or HpdI3-A-WC did not produce any decrease in DHFR activity.

DHFR activity and mRNA levels in SKBR3 cells treated with Coding-PPRHs. (
Effects of Coding-PPRHs on DHFR mRNA levels
SKBR3 cells were incubated with Coding-PPRHs for 72 hr. Surprisingly, HpdI3-A-TA (100 nM) produced a decrease of only 20% in DHFR mRNA levels, whereas a reduction close to 50% was caused by the aODN HATNL-24 and the dhfr Template-PPRH HpdI3-B (Fig. 3b). HpdI3-A-NH also caused a very slight decrease in DHFR mRNA levels (17%). The different controls did not produce a drop in DHFR mRNA levels.
Effects of Coding-PPRHs on DHFR pre-mRNA levels
Taking into account that the PPRH target sequence was included in intron-3, we decided to study if there was any change in the pre-mRNA levels. Three different species were analyzed by reverse transcription–real-time PCR: Exon3–Intron3, corresponding to pre-mRNA levels; Exon3–Exon4, corresponding to the splicing of intron-3; and Exon3–Exon5, to explore exon-4 skipping (Fig. 3c). As shown in Fig. 3d, when SKBR3 cells were incubated for 72 hr with HpdI3-A-TA, an increase of more than 200% was observed in the pre-mRNA levels, whereas the amounts of the Exon3–Exon4 and Exon3–Exon5 products were reduced by 22% and 42%, respectively. The relative abundance of each RNA species was 3.8% for pre-mRNA, 50% for Exon3–Exon4, and 46.2% for Exon3–Exon5. The negative control HpdI3-WC did not produce a significant drop in DHFR mRNA levels. No loss of exon-4 was observed (data not shown).
Effects of Coding-PPRHs on DHFR pre-mRNA splicing
The increase in the pre-mRNA DHFR levels, followed by a decrease of the spliced form, suggested a possible alteration in the splicing process of intron-3 in the presence of Coding-PPRHs. To study this possibility, we generated the plasmids pDCHIP and pDCHIP-Hp. The insertion of the human dhfr HpdI3-A-TA target sequence in the intron sequence present in pDCHIP to generate pDCHIP-Hp did not affect the splicing of intron-1 and the functionality of the DHFR protein (data not shown), as DCHM1 and DCHM1-Hp cells grew normally in –GHT medium. No other HpdI3-A-TA target sequence was found in the Chinese hamster ovary dhfr gene.
Next, we proceeded to perform in vitro splicing experiments with probes generated by in vitro transcription from pDCHIP and pDCHIP-Hp plasmids. Representative images of the different species after DHFR pre-mRNA splicing from the DCHIP and DCHIP-Hp probes are shown in Fig. 4a and c. The upper band corresponded to the pre-mRNA probe, the second band to the spliced product, and the lower band to free exon-1. Figure 4a and c also include the effect of Coding-PPRHs on the in vitro splicing reaction from the DCHIP or DCHIP-Hp probes, respectively. No decrease in splicing was observed when DCHIP was incubated with Coding-PPRHs (Fig. 4b). In contrast, when the DCHIP-Hp probe was incubated with HpdI3-A-TA, the decrease in splicing was dose dependent, reaching a 51% decrease at 400 nM (Fig. 4d). HpdI3-A-NH also caused a decrease in splicing (29%) but to a lesser degree than HpdI3-A-TA. The negative controls HpdI3-A-WC and HpdI3-Sc did not affect splicing.

Effect of Coding-PPRHs on in vitro splicing. (
Effects of Coding-PPRHs on U2AF65 binding
The effects of PPRHs on protein binding to HpdI3-A-TA target sequence were studied by electrophoretic mobility shift assays. A radiolabeled probe corresponding to the HpdI3-A-TA target sequence in dhfr intron-3 was incubated with HeLa nuclear extracts specific for splicing, as shown in Fig. 5a. A DNA probe was used because U2AF65 is able to bind to either ssDNA or ssRNA polypyrimidine sequences (Singh et al., 2000). A shifted band was observed due to protein binding to the probe (gel shift). When HpdI3-A-TA was added as competitor, a dose-dependent decrease in the gel-shifted band was observed, as well as the appearance of a lower band due to the binding of the PPRH to the probe and an upper band due to PPRH–probe–protein binding. The upper band seemed to be mainly due to albumin binding (data not shown). To test the involvement of U2AF65, an important splicing factor that recognizes intronic polypyrimidine sequences and recruits the splicing machinery, in the molecular mechanism of Coding-PPRHs splicing blockage, a supershift assay using U2AF65 antibody was performed (Fig. 5b). When using 1 μg of nuclear extract in the presence of U2AF65 antibody, a supershifted band and a decrease of 40% in the intensity of the shifted band were observed, whereas with 0.5 μg of nuclear extract, a major decrease of 88% in the gel-shifted band was observed, although the supershifted band was not visually detectable.

PPRH competition for U2AF65 binding. A radiolabeled probe corresponding to the HpdI3-A-TA target sequence in dhfr intron-3 was incubated with 1 μg of HeLa nuclear extract (NE) and tRNA as unspecific competitor. The first lane in each panel corresponds to the probe in the absence of NE. (
Specificity
The specificity of HpdI3-A-TA was assessed by BLAST analysis, by determining the mRNA levels of unrelated genes, and by cell survival analysis after its transfection in cells lacking the HpdI3-A-TA target sequence. The mRNA levels of the Telomerase, Survivin, UGT1A10, and S100A4 genes did not change significantly in SKBR3 cells incubated for 72 hr with HpdI3-A-TA, as shown in Table 2.
SKBR3 cells were transfected with HpdI3-A-TA, and a reverse transcription–real-time PCR was performed to determine the mRNA levels of unrelated genes. Data are mean ± SEM values of at least four experiments.
DCHM1 or DCHM1-Hp cells were incubated with Coding-PPRHs in DHFR-selective medium (–GHT). Interestingly, after 1 week of incubation (Fig. 6b) only DCHM1-Hp (cells containing the HpdI3-A-TA target sequence) suffered a decrease in cell survival of 40% when treated with HpdI3-A-TA and of 21% when incubated with HpdI3-A-NH. Negative controls HpdI3-A-WC and HpdI3-Sc produced a relative cell death that was not statistically significant in both transfected cell lines.
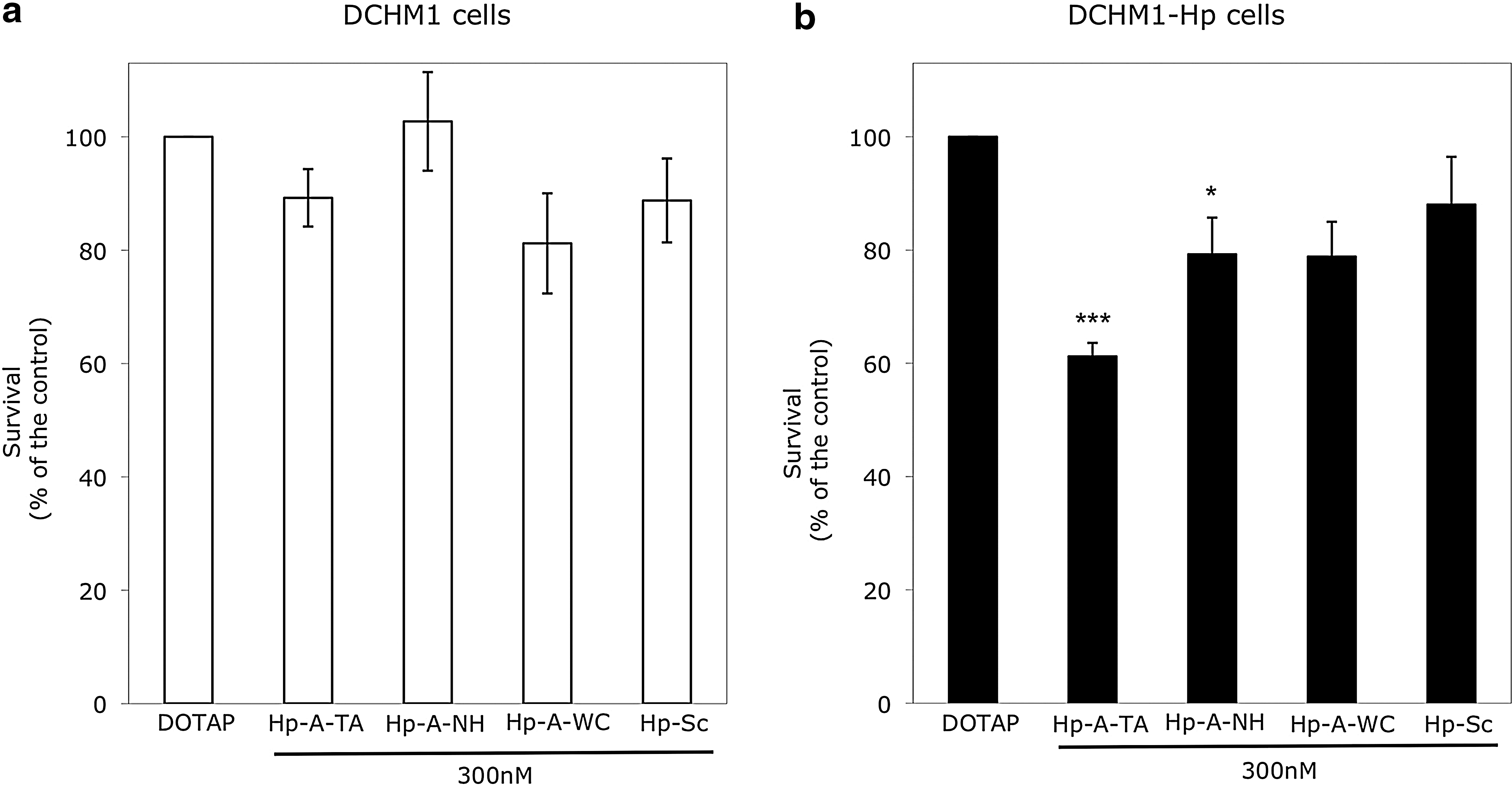
Coding-PPRH specificity. Dhfr
− DG44 cells (30,000) stably transfected with (
Effects of Coding-PPRHs on methotrexate-resistant cells
The effectiveness of Coding-PPRHs in MCF7 breast cancer cells resistant to the chemotherapeutic drug methotrexate at 10−6 M was tested. These cells have the dhfr locus amplified (gene copy number 58) and overexpress DHFR mRNA (mRNA change 33-fold) (Selga et al., 2009). HpdI3-A-TA presented a higher cytotoxicity than HpdI3-A-TG and HpdI3-A-NH (58%, 30%, and 45%, respectively) in these cells (Fig. 7a). The aODN HATNL-24 (at its optimal conditions, 1:10 aODN:DOTAP molar ratio) scarcely showed any effect. DOTAP (10 μM) and HpdI3-Sc caused a certain degree of cell death, unlike HpdI3-A-TA without DOTAP and HpdI3-A-WC. Template HpdI3-B showed a greater effect than HpdI3-A-TA (71%). As previously noted in SKBR3 cells, MCF7-R cells did not present a large DHFR mRNA decrease after HpdI3-A-TA transfection (Fig. 7b). However, at the protein level, a 40% decrease in DHFR level was observed upon incubation of MCF7-R cells with HpdI3-A-TA (Fig. 7c). HpdI3-B (Template-PPRH) and HpdI3-A-NH also caused a decrease in DHFR protein levels, whereas DOTAP and HpdI3-A-WC did not cause a significant effect.
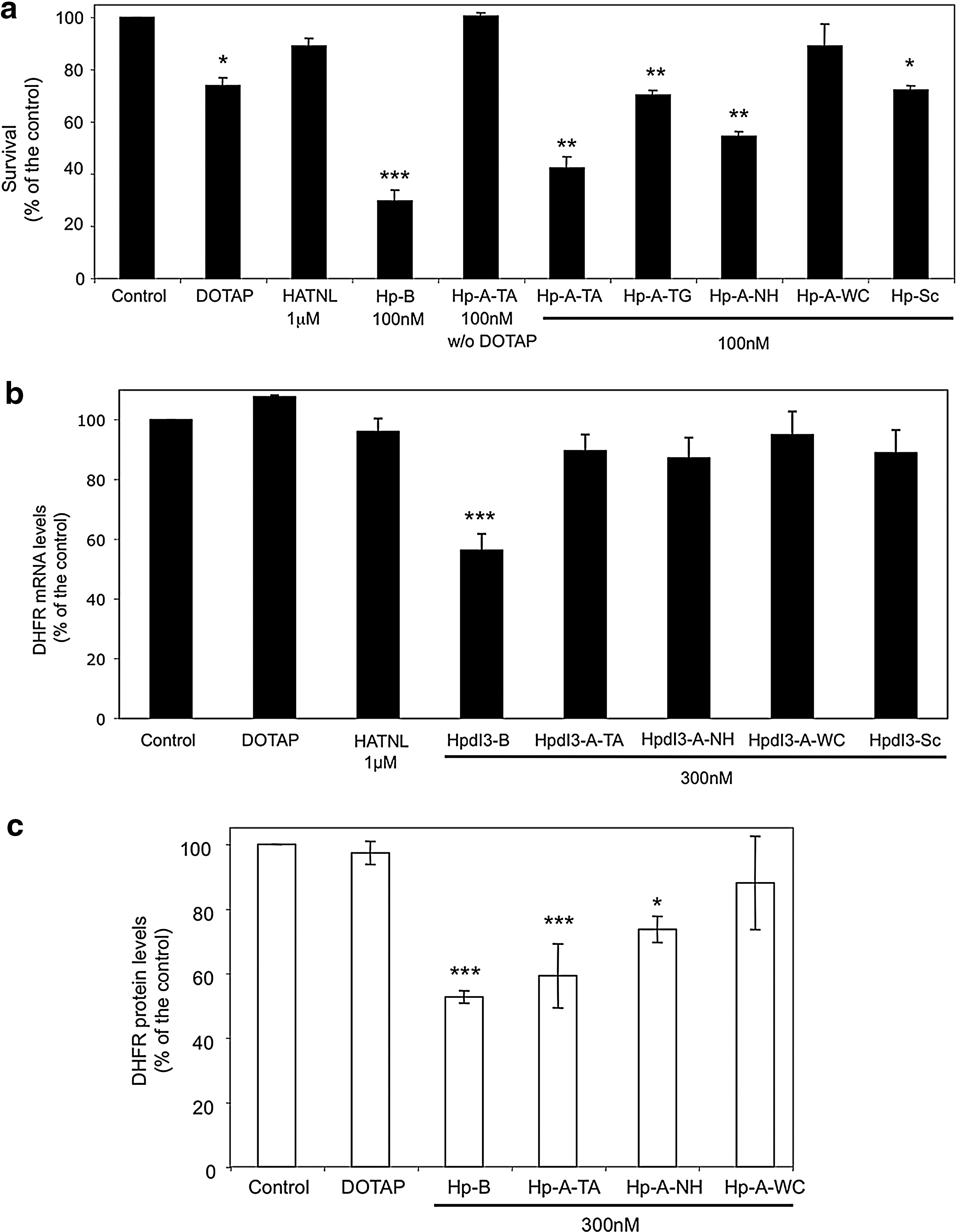
Effects of Coding-PPRHs in MCF7 cells resistant to 10−6 M methotrexate. (
Discussion
The ability to selectively modulate the activity of genes is a long-standing goal in molecular medicine (Jain et al., 2008). Template-PPRHs were previously described in our laboratory (de Almagro et al., 2009) as molecules targeting the template DNA strand, thus producing a decrease in gene transcription. In the present study we developed a new usage for PPRHs as Coding-PPRHs that target pre-mRNA. RNA targeting has recently emerged as a potential alternative to more conventional approaches in gene therapy (Wood et al., 2007; Madsen et al., 2008). aODNs that bind to pre-mRNA and sterically alter RNA processing have been successfully used in cell culture to demonstrate the utility of this approach (Suwanmanee et al., 2002; Gebski et al., 2003; Bruno et al., 2004; Du et al., 2007). Moreover, we can also find molecules that target mRNA and produce its degradation, like siRNAs.
Template-PPRHs were designed to bind the template DNA strand. However, targeting a PPRH towards an mRNA sequence implies that it is also directed towards the DNA coding strand. Thus, Coding-PPRHs could theoretically bind to both mRNA and DNA. As shown in the binding assays, HpdI3-A-TA is able to bind both ss and ds sequences, although it preferably binds to the ss species. Pre-mRNA accumulation followed by a decrease of the spliced forms of RNA suggests that the predominant PPRH mechanism of action is related to splicing interference. The possibility of a slight decrease in transcription due to the binding of the PPRH to the coding strand cannot be ruled out.
Coding-PPRHs against the human dhfr gene showed a noticeable activity in decreasing DHFR expression. At the suitable PPRH:DOTAP molar ratio (1:100), 100 nM HpdI3-A-TA caused more than 90% cell death, similar to that caused by Template HpdI3-B (targeting the template DNA strand). HpdI3-A-TA was more active than the aODN HATNL-24, when used at their optimal conditions. The time of action of Coding-PPRHs was very fast because 48 hr was enough to exert their action.
The treatment of the cells with HpdI3-A-TA resulted in a decrease in levels of both DHFR protein and enzymatic activity, but interestingly, when determining DHFR mRNA levels, there was only a small decrease, lower than expected. Because the mRNA analysis method to measure DHFR mRNA was based on exon-1 detection, changes in mRNA levels around intron-3 may have been overlooked. When DHFR hnRNA levels were measured after HpdI3-A-TA transfection, a surprising increase was observed, in contrast with the decrease in the species that had spliced intron-3. These results suggested that HpdI3-A-TA could provoke interference in the splicing process, as it caused an accumulation of pre-mRNA, followed by an mRNA decrease after splicing. The small decrease observed at first instance when measuring DHFR mRNA levels using the exon-1 probe could be explained considering that it corresponded to the pool of all the DHFR pre-mRNA and mRNA species present in the cell at that moment, thus representing the mean between the increase of the pre-mRNA levels and the decrease of the spliced forms. Despite the noticeable pre-mRNA rise, there is a resulting slight decrease of the DHFR mRNA pool value; this could be due to the presence of higher levels of mature mRNAs than of pre-mRNAs because of their longer half-life (relative abundance of pre-mRNA was 3.8% and 96.2% mature mRNA). Because Coding-PPRHs interfered with splicing, it raised the possibility for exon-4 skipping, but no loss of this exon was observed. The involvement of splicing in the mechanism of action of Coding-PPRHs was studied by in vitro splicing reactions. Indeed, when an intron containing a Coding-PPRH target sequence was incubated with HpdI3-A-TA, a noticeable decrease of 50% in the spliced form was observed.
The target sequence of Coding-PPRHs consists of a polypyrimidine stretch, and the importance of polypyrimidine stretches in intron splicing is well known. Regions of 15–30 contiguous purine or pyrimidine tracts are greatly overrepresented in all eukaryotic species, ranging from yeast to human (Bucher and Yagil, 1991; Behe, 1995), and are preferably found in promoters and introns and with less frequency in exons. The polypyrimidine tract is one of the important cis-acting sequence elements directing intron removal in pre-mRNA splicing, but it is not the only polypyrimidine stretch that plays an important role in splicing. Substitutions, as well as progressive deletions in the 3′-splice site polypyrimidine tract, were found to inhibit spliceosome formation and splicing (Maschhoff and Padgett, 1992; Coolidge et al., 1997). Despite the important role of the polypyrimidine tract in splicing, there appears to be great flexibility in the specific sequence of a given tract including the introduction of purines (Coolidge et al., 1997).
In mammals, U2AF binds to the polypyrimidine tract (Zamore et al., 1992), and splicing is regulated by having multiple proteins compete for binding to that pyrimidine tract, particularly sex-lethal protein and polypyrimidine tract binding protein (Valcarcel et al., 1993; Lin and Patton, 1995; Singh et al., 1995). U2AF recognizes consensus 3′-splice site sequences in the pre-mRNA and coordinates the initial states of spliceosome assembly. Mammalian pyrimidine tracts vary in length and sequence composition, which are frequently interrupted with purines (Senapathy et al., 1990; Sickmier et al., 2006). U2AF is formed by two subunits: U2AF65, which binds to the polypyrimidine target sequence, and U2AF35, which interacts with other proteins to form the spliceosome. Because formation of the U2AF complex commits the pre-mRNA to be spliced (Michaud and Reed, 1991), U2AF/pre-mRNA interactions present a key target for regulation. Coding-PPRHs could act as an additional competitor for U2AF binding sites or just constitute a steric blockage for splicing, avoiding spliceosome formation. According to our results, coding-PPRHs would preferably prevent U2AF rather than polypyrimidine tract binding binding to their target sequences, as polypyrimidine tract binding is involved in alternative splicing (Cote et al., 2001; Le Guiner et al., 2001; Shen et al., 2004; Izquierdo et al., 2005; Stern et al., 2009), and no modification of the exon pattern was observed.
There have been previous studies reporting the use of oligonucleotides to regulate splicing of specific genes with successful results. In those approaches, aODNs were implemented to interfere with translation and pre-mRNA splicing, changing levels of alternatively spliced genes, or skipping an exon in order to restore a disrupted reading frame (Aartsma-Rus and van Ommen, 2007; Marshall et al., 2007). Antisense RNAs have also been studied. HeLa or K562 cells that stably expressed thalassemic genes, when treated with U7 and U1 small nuclear RNAs modified to contain sequences antisense to the aberrant splice sites, restored correct splicing and correct expression of β-globin (Vacek et al., 2003).
One of the major problems of all molecules targeting nucleic acid sequences is the specificity of recognition of their target sequence. To assess Coding-PPRH specificity, aside from BLAST analysis, different negative controls (Hp-Sc and Hp-A-WC) and changes in mRNA levels of unrelated genes were studied. Moreover, incubation of HpdI3-A-TA in a cell line without the target sequence (DG44 plus pDCHIP) did not show any change in cell viability. On the other hand, cells containing the PPRH target sequence inserted in intron-1 (DG44 plus pDCHIP-Hp) suffered a decrease in cell survival upon incubation with Coding-PPRH.
Possible therapeutic applications of Coding-PPRHs were tested by incubating HpdI3-A-TA with methotrexate-resistant MCF7 cells. These cells that survived 10−6 M methotrexate died when incubated with HpdI3-A-TA. These results could have an important repercussion as drug resistance caused by gene overexpression could be overcome using PPRHs.
Coding-PPRHs extend the application of PPRHs, as the use of Template-PPRHs was limited to the presence of polypyrimidine sequences in the template DNA strand. Coding-PPRHs allow the use of this technology for genes where the polypyrimidine sequence is located in the coding DNA strand. Both types of PPRHs showed very similar activity.
In previous studies we compared the action of Template-PPRHs with that of an siRNA directed towards the dhfr gene (de Almagro et al., 2009). Using the same concentration in both cases (100 nM) HpdI3-B achieved the same cytotoxicity as the siRNA, but with the advantages of (1) remarkable stability (half-life of 5 days compared with the 2 days for modified aODNs), (2) easier handling (because they are DNA molecules instead of RNA), and (3) ease of synthesis and inexpensive cost. For these reasons, PPRHs represent an alternative to the use of siRNAs.
Coding-PPRHs showed a noticeable ability to specifically decrease gene expression. Thus, they could have potential applications for diseases coursing with deregulated gene expression.
Footnotes
Acknowledgments
This work was supported by grants SAF08-43 from the “Plan Nacional de Investigación Científica” (Spain) and RD06/0020/0046 from the ISCIII-RETIC. Our group holds the Quality mention from the “Generalitat de Catalunya” SGR09-118. M.C.deA. is the recipient of a predoctoral fellowship from the “Generalitat de Catalunya” (FI), and N.M holds an APIF fellowship from the University of Barcelona.
Author Disclosure Statement
The authors declare that they have no competing interests.