
Abstract
Rheumatoid arthritis is a proinflammatory autoimmune disease attributed to failure of both CD4+CD25+ regulatory T (Tr) and CD8+CD28− suppressor T (Ts) cells to control autoreactive CD4+CD28+ Th1 (Th1) and autoantibody-producing B cells. Here we show a single intramuscular injection of our novel targeted DNA vaccine encoding Pseudomonas exotoxin A and costimulatory molecule B7-2 without autoantigens in a collagen-induced arthritis model simultaneously increased Tr and Ts cells and selectively decreased autoreactive Th1 cells. The vaccine induced a shift from Th1 to Th2 and Th3 cellular and cytokine profiles and a decrease in CD4+/CD8+ cell ratios. Importantly, the vaccine showed potent antirheumatic activity by clinical and other examinations such as X-ray, histopathology, and anti-type II collagen IgG levels and was comparable to methotrexate, the current “gold standard” treatment. As an effective stimulator of both Tr and Ts cells and a specific suppressor of autoreactive Th1 cells, this vaccine is a promising therapeutic approach for rheumatoid arthritis.
Introduction
The ultimate goal for treating RA should be to selectively block abnormal autoimmune responses such as autoreactive Th1 cell- and B cell-mediated inflammatory processes. Ideally, a therapeutic DNA vaccine targeting the relevant RA autoantigens to restore impaired Tr and Ts regulatory/suppressor mechanisms in RA without generalized immunosuppression may be highly beneficial (Lider et al., 1989). To date a few preventive or therapeutic DNA vaccines target only one or several autoantigens; but some nonspecific autoantigens have been effective, at least partly, in some animal models of autoimmune diseases including RA (Robinson et al., 2003; Sercarz, 2003; Steinman, 2004; Holmgren and Czerkinsky, 2005; Larche and Wraith, 2005; Song et al., 2009).
The crucial role of the B7-2/CD28 costimulatory signaling pathway has been implicated in the development, activation, differentiation, and function of Tr and Ts cells (Cortesini et al., 2001; O'Garra and Vieira, 2004; Paust et al., 2004; Lyddane et al., 2006; May et al., 2007; Prod'homme et al., 2007; Sakaguchi and Powrie, 2007) in the immunopathogenesis of RA mediated by autoreactive CD4+CD28+ Th1 and B cells (Tada et al., 1999; Cortesini et al., 2001; Feldmann, 2001; Marrack et al., 2001; Fu and Storb, 2002; O'Garra and Vieira, 2004), prompting some to use antibody-mediated blockade of CTLA-4 or of both the B7.1 and B7.2 costimulatory molecules to treat RA (Knoerzer et al., 1995; Webb et al., 1996; Steinman, 2004). Here, we designed a new class of DNA vaccine for RA, selectively targeting B7-2/CD28 (not B7-1/CTLA-4) expressing a truncated Pseudomonas exotoxin A (PEA) fused to a costimulatory B7-2 molecule. We further compared the antirheumatic activity of this targeted DNA vaccine, which does not express any autoantigens, with methotrexate (MTX), the current “gold standard” treatment for RA (Song et al., 2010) in a CIA model. To our knowledge, this paper constitutes the first treatment and preventive strategy against CIA with a targeted DNA vaccine directed toward the B7-2/CD28 costimulatory signaling system.
Materials and Methods
Development of the vaccine
The prokaryotic expression vector pRSETA/B7-2-L-PE40KDEL, previously constructed in our laboratory (Xi et al., 2006), contains the functional domains Ib, II, and III of the 1083-bp PE40 (C-terminal 40-kDa segment of the Pseudomonas exotoxin A protein) but has a deletion of the cell-binding domain Ia; in addition, the REDLK sequence at the C terminus of PE40 was replaced with KDEL in the 3′ linker to the fused 675-bp human B7-2 extracellular domain. The full-length B7-2-PE40KDEL cDNA was amplified from pRSETA/B7-2-L-PE40KDEL and cloned into the pcDNA3.1/Zeo(+) expression vector (Invitrogen, Leek, The Netherlands), using the following primers: sense, 5′-CGG
Characterization and expression analysis of the vaccine in stable cells
pcDNA3.1/B7-2-PE40KDEL was transfected into the CHO-K1RPE.40 toxin-resistant cell line, a generous gift from T.J. Moehring and J.F. Sucic (Sucic et al., 1998), and selected with Zeocin. Total RNA was extracted from stably transfected CHO-K1RPE.40 cells (TRIzol reagent; Invitrogen), and B7-2-PE40KDEL cDNA was synthesized (RNA LA PCR kit; TaKaRa Bio, Otsu, Shiga, Japan). The primers, 5′-CGGGGTACCATGGCTGCTCCTCTGAAGATTCAAG-3′ and 5′-GCTCTAGATTACTTCAGGTCCTCGCGCGGCGGTTTG-3′, amplified a 1900-bp fragment. Human β-actin cDNA (650 bp) was amplified as the control.
To investigate the expression of pcDNA3.1/B7-2-PE40KDEL from CHO-K1RPE.40 cells, the supernatants obtained from 1 × 106 stable cells cultured for 24 hr were concentrated with an Amicon Ultra-4 filter (Millipore, Cork, Ireland) and analyzed by standard Western blot and quantified by ELISA.
Selective cytotoxicity in vitro of the vaccine
Jurkat cells expressing high levels of CD28 and Hut28 cells expressing low levels of CD28 (2 × 105/ml) were cocultured on 96-well plates with stably transfected CHO-K1RPE.40 cells (2 × 105/ml) for 48 hr. CellTiter 96 AQueous One Solution cell proliferation assay kit (Promega, Madison, WI) was used to detect viable cells in the cocultures.
Expression analysis in vivo of the vaccine in CIA
On days 2, 4, 7, 14, 28, and 56 after intramuscular injection of 150 μg of the vaccine, rats were killed and the uninjected-side quadriceps was collected from each. Total RNA was immediately isolated with TRIzol and 1 μg of RNA was transcribed by reverse transcription-polymerase chain reaction (RT-PCR), using B7-2-PE40KDEL gene sense (5′-CGGGGTACCATGGCTGCTCCTCTGAAGATTCAAG-3′) and antisense (5′-GCTCTAGATTACTTCAGGTCCTCGCGCGGCGGTTTG-3′) primers, or a pair of rat β-actin cDNA gene sense (5′-AGGCATCCTGACCCTGAAGTAC-3′) and antisense (5′-TCTTCATGAGGTAGTCTGTCAG-3′) primers. Thirty cycles (94°C for 1 min, 55°C for 1 min, and 72°C for 3 min) of amplification were performed, using a OneStep RT-PCR kit (TaKaRa Bio).
Induction of CIA in the rat model
Inbred female Wistar rats (RT1u) at 4–6 weeks old were obtained from the Animal Breeding Center of the Academy of Military Medical Sciences of Beijing (Beijing, China) and maintained under specific pathogen-free conditions. The experiments were performed under the supervision and guidelines of the Academy of Military Medical Sciences Animal Welfare Committee. CIA was induced as described previously (Song et al., 2009, 2010).
Flow cytometric analysis of Tr, Ts, Th1/Th2, and CD4+/CD8+ cells
Peripheral blood samples were prepared from various experimental group rats on day 28 after CIA induction. CD4+CD25+ Tr, CD8+CD28− Ts, and CD4+/CD8+ cells were detected with various monoclonal antibodies. Th1/Th2 cell discrimination was performed by intracellular cytokine staining, using a BD Cytofix/Cytoperm Plus fixation/permeabilization kit (BD Biosciences, San Jose, CA). Phycoerythrin (PE)-conjugated anti-rat interleukin (IL)-4, fluorescein isothiocyanate (FITC)-conjugated anti-rat interferon (IFN)-γ, and PE–cyanine 5 (Cy5)-conjugated anti-rat CD4 monoclonal antibodies (Song et al., 2009, 2010) were used.
Cytokine assays for Th1, Th2, and Th3 cytokines
On day 28 after CIA induction, serum was obtained and stored at −20°C until use. Rat IL-2, IL-4, IFN-γ, tumor necrosis factor (TNF)-α, IL-10, and transforming growth factor (TGF)-β were quantitated with commercial enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN), and IL-10 was quantified with an IL-10 immunoassay system (BioSource Division, Invitrogen). The lower limits of detection of the assays were 7 pg/ml (Song et al., 2009, 2010).
Vaccine administration protocols for CIA
After rats were anesthetized with sodium pentobarbital, the vaccine, solubilized in 150 μl of 0.9% NaCl, was injected into the gastrocnemius muscle of the left hind leg. About 20 sec after injection, transcutaneous electric pulses were applied with two stainless steel plates, 1.0 cm in diameter. The conditions were as previously described for mouse and rat skeletal muscle (Mir et al., 1999; Ruiz et al., 1999). In both preventive and treatment groups 1–3, rats received a single intramuscular injection of 150, 200, or 300 μg, respectively, of the vaccine on day 0 of CIA induction immunization (preventive groups 1–3), or on day 12 after the onset of arthritis (treatment groups 1–3). The negative control group rats received only a single 200 μg dose of empty vector pcDNA3.1/Zeo(+), and the standard positive control group rats were administered intraperitoneally a low (0.75 mg/kg) dose of MTX (Shanghai Pharmaceutical, Shanghai, China) diluted in phosphate-buffered saline (PBS) weekly for 4 weeks. At least 10 rats were included in each group.
Clinical examinations for antirheumatic efficacy
Disease progression in all experimental rats included in prophylactic, treatment, and negative and positive groups was monitored and assessed daily over a 28-day period, using a macroscopic scoring system (range, 0–4 for each paw), as previously described (Song et al., 2009, 2010).
Determination of serum antibody against type II collagen
Anti-type II collagen (CII) antibody titers were determined in serum of blood obtained by retro-orbital bleeding on day 28 after CIA induction. The levels of serum antibodies to CII were measured by ELISA (Song et al., 2009, 2010).
Determination of serum antibody against Pseudomonas exotoxin A
Sera were obtained on day 56 after CIA induction and measured by ELISA (Pasten and FitzGerald, 1991).
Histopathology examination
Histopathological examination of the hind ankle joints of arthritic rats was performed on day 28 after CIA induction (Song et al., 2009, 2010).
Statistical analysis
Data were analyzed with the SPSS version 11.5 software program (SPSS, Chicago, IL). Dichotomous variables, that is, the incidence of arthritis in various groups of mice, were analyzed by χ2 test. Nonparametric data, that is, arthritis scores and the values with a skewed distribution, antibody levels, and stimulation indices, were evaluated by Mann–Whitney test. The level of significance was set at α = 0.05. The difference in cytokine levels and the frequency of various T cell subclasses were compared by Student t test.
Results
Development of the targeted DNA vaccine
We reported on a recombinant human B7-2-L-PE40KDEL fusion exotoxin, a new B7:CD28/CTLA-4 blocker that could specifically kill CD28+ T cells but not CD28− T cells (Xi et al., 2006), suggesting it could be a promising novel treatment approach to RA driven by proinflammatory CD4+CD28+ Th1 cells. In addition, accumulated evidence indicated that the secreted PE40 exotoxin selectively kills targeted cells after internalization and release into the cytosol but does not affect PE40 exotoxin-expressing cells lacking the targeted antigens (Chen et al., 1997). This previous work provided the theoretical and material basis for our current development of a new class of targeted DNA vaccine, pcDNA3.1/B7-2-PE40KDEL (Fig. 1A). We confirmed expression of the vaccine construct by amplifying the expected 1919-bp transcript from RPE.40 cells, a mutant CHO-K1 strain resistant to Pseudomonas exotoxin A, which were stably transduced with pcDNA3.1/B7-2-PE40KDEL (Fig. 1B). The 90-kDa B7-2-PE40KDEL fusion exotoxin secreted by these stably transfected cells could be specifically detected with anti-human B7-2 monoclonal antibody (mAb) and rabbit anti-PEA antiserum in a Western blot (Fig. 1C). The fusion protein was apparently larger than that expressed in Escherichia coli cytosol (70 kDa), suggesting that it was posttranslationally modified, potentially at some of the eight predicted N-glycosylation sites (Asn-X-Ser or Asn-X-Thr) within the B7-2 moiety of the B7-2-PE40KDEL fusion protein. In addition, the stable cell line produced high amounts of the fusion protein: equivalent to more than 0.23 μg of PEA per liter was produced in 24 hr from 1 × 106 stably transfected CHO-K1RPE.40-B7-2-PE40KDEL cells ml–1 (data not shown).
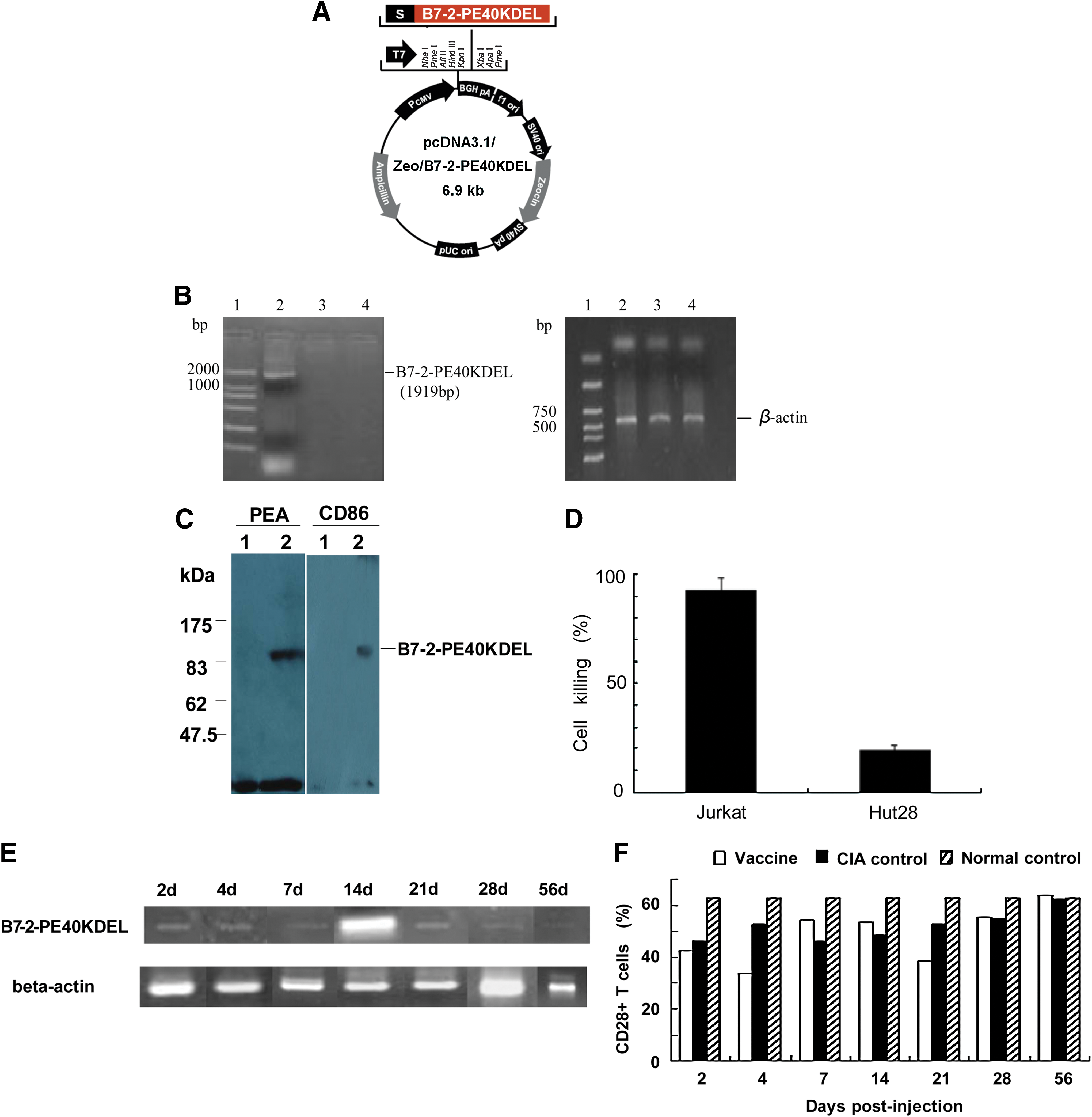
(
We observed a high level of selective killing by stably transfected CHO-K1RPE.40-B7-2-PE40KDEL cells of cocultured CD28-positive Jurkat T cells (96.9%), but not of cocultured CD28-negative Hut28 T cells (0.0%), whereas CHO-K1RPE.40 cells alone (control) had no effect on either T cell line (Fig. 1D). Therefore, B7-2-PE40KDEL fusion protein produced by the DNA vaccine can selectively kill CD28-positive but not CD28-negative T cells in vitro.
The full-length B7-2-PE40KDEL fusion transcript could readily be detected in vivo by PCR from all CIA rat muscle samples on day 2 after a single intramuscular injection of 150 μg of the vaccine (Fig. 1E). On day 14, expression of the B7-2-PE40KDEL fusion gene peaked, and then gradually decreased thereafter. In addition, in vivo expression of the B7-2-PE40KDEL fusion gene over days 21–28 was equal to that on days 2–7 after vaccination. By day 56, B7-2-PE40KDEL fusion gene expression was low or absent. Meanwhile, CD28+ T cells were reduced 31% on day 2 and significantly decreased by 47% on day 4 compared with the pcDNA3.1 negative control group (p < 0.05), corresponding to in vivo expression of the B7-2-PE40KDEL fusion gene. Interestingly, CD28+ T cells recovered transiently (16%), but their level was still lower than that of the CIA negative control group over days 7–14. After B7-2-PE40KDEL fusion gene expression peaked on day 14, CD28+ T cells were suppressed again on day 21 (p < 0.05), similar to the reduction observed on day 4. Thereafter, CD28+ T cells gradually recovered to levels similar to the CIA negative control group when the B7-2-PE40KDEL fusion gene was weakly expressed on day 56 (Fig. 1F). These results indicate that our novel targeted DNA vaccine was successfully expressed and functional in vivo.
The vaccine can simultaneously increase both CD4 +CD25 + Tr and CD8 +CD28 − Ts cells and selectively decrease autoreactive CD4 +CD28 + Th1 cells in CIA
We analyzed the effect of the vaccine on the three T cell major subsets, that is, Tr, Ts, and autoreactive CD28+ Th1 cells, known to be involved in RA immunopathogenesis. Indeed, all three T cell subsets were markedly lower and the CD4+/CD8+ T cell ratios were markedly higher in established CIA rats compared with normal rats (Fig. 2A–D; p < 0.05). These findings, except for the CD28+ T cells, are highly consistent with the deduced and theoretical immunopathogenesis of RA. Surprisingly, the CD28+ T cell level in the established CIA rats was markedly lower than in normal rats. In established CIA rats, a single muscle injection of the vaccine at 150, 200, or 300 μg/rat, either in the treatment (day 16) or preventive groups (day 28) on the fourth week after CIA induction, simultaneously and dramatically increased both Tr and Ts cells over that of CIA negative control rats (Fig. 2A and B), although not to the levels of normal rats. In addition, the vaccine had a clear-cut dose-dependent effect on Ts cells in the treatment but not preventive groups. No dose-dependent effects on Tr cells were seen in either the treatment or preventive groups.
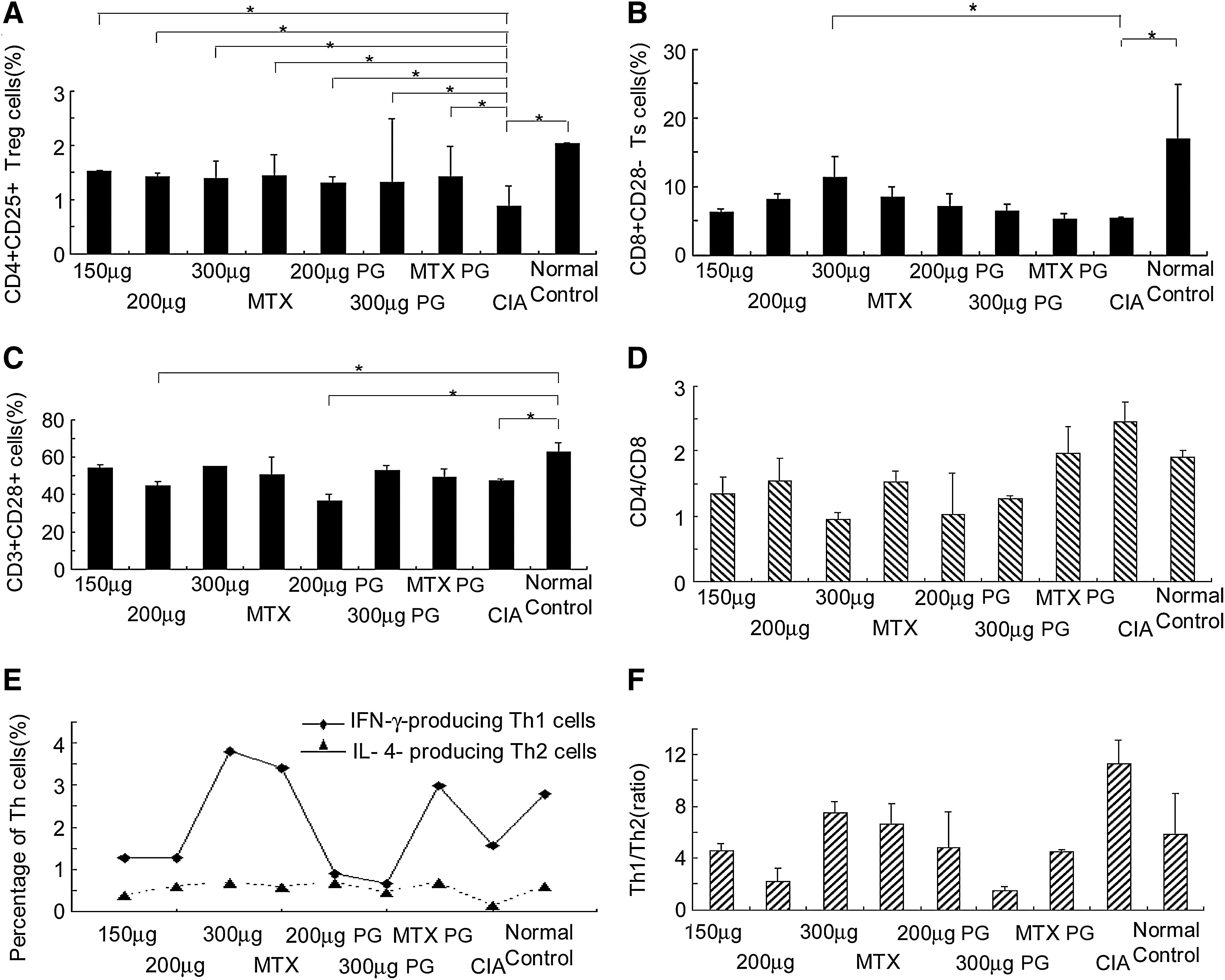
(
As mentioned previously, the vaccine significantly decreased CD28+ T cells in CIA rats at two separate time points on days 4 and 21 after a single vaccine injection (Fig. 1F). However, we only observed obviously decreased CD28+ T cells in the treatment and preventive groups at the 200 μg/rat dose in the fourth week after CIA induction (Fig. 2C). The reason for this result is that the examined time point (day 16 for the treatment groups and day 28 for the preventive groups) was exactly the time for partial recovery of CD28+ T cells. Indeed, a slight increase in CD28+ T cells was observed at the 150 or 300 μg/rat vaccine dose in the treatment, preventive, and MTX treatment groups relative to the CIA control group. In contrast, only the 200 μg/rat vaccine dose was effective in killing CD28+ T cells not only in the treatment but also the preventive groups compared with the CIA negative control group (Fig. 2C; p < 0.05). Importantly, the CIA groups that received the 200 μg/rat vaccine dose were better protected against CIA than the other dose groups (see below). In addition, a markedly higher CD4+/CD8+ cell ratio was found in the blood of established CIA rats compared with normal rats; this increased ratio was significantly reversed in both the treatment and preventive groups (Fig. 2D). These results clearly demonstrated that the vaccine can efficiently correct the impaired T cell regulatory/suppressor mechanisms in the treatment and prevention of RA immunopathogenesis.
The vaccine can induce a Th1-to-Th2 cell shift, downregulate Th1 cytokines, and upregulate Th2 and Th3 cytokines in CIA
CIA is considered a well-established model of Th1-mediated autoimmune arthritis (Seo et al., 2004; Song et al., 2009). Our previous study and the present data confirmed that established CIA rats have markedly higher Th1/Th2 cell ratios compared with normal rats; however, established CIA rats also had lower IFN-γ-producing Th1 cells and cytokine IL-4-producing Th2 cells than normal rats (Fig. 2E; p < 0.05). Notably, all doses of the vaccine induced a significant shift of Th1 to Th2 cells in both treatment and preventive groups compared with the CIA negative control group (Fig. 2F; p < 0.05). Likewise, both MTX positive control treatment and preventive groups had significantly higher Th1 cells and lower Th2 cells than the controls. The immediate cause of the Th1-to-Th2 cell shift by the vaccine and MTX is likely due to the increase in Th2 cells and not the decrease in Th1 cells (data not shown). Although the Th1/Th2 ratio in the 300-μg/rat group was close to the baseline of normal rats, there were significantly more Th1 cells than in the other treatment groups, possibly caused by the severe infections occurring in the inflamed paw and ankle joints of the rats in this group.
The imbalance between proinflammatory Th1 cytokines and antiinflammatory Th2 and/or Th3 cytokines is a critical factor in the immunopathogenesis of RA. Th1 cytokines IL-2, IFN-γ, and TNF-α were markedly higher whereas Th2 cytokine IL-10 and Th3 cytokine TGF-β in established CIA rats were significantly lower than in normal rats (Fig. 3A–E; p < 0.05). However, serum IL-4 was too low to detect in all rats. All these changes are typical of RA immunopathogenesis. The vaccine administered to either the treatment or prevention groups dramatically reduced IFN-γ and TNF-α, but not IL-2, compared with CIA negative control rats (Fig. 3A–C; p < 0.05). Serum IL-2 also was partially decreased, but not significantly, in both the treatment and preventive groups. In contrast, IL-10, but not IL-4, significantly increased on treatment with the 200 and 300 μg/rat vaccine doses and MTX (Fig. 3D). Both the vaccine and MTX also elevated serum TGF-β, but not significantly (Fig. 3E). Although IL-4 was detected in the treatment and preventive groups, the levels were low. These results suggest that the vaccine effectively treated or prevented the systemic inflammatory response and enhanced broad antiinflammatory activity.
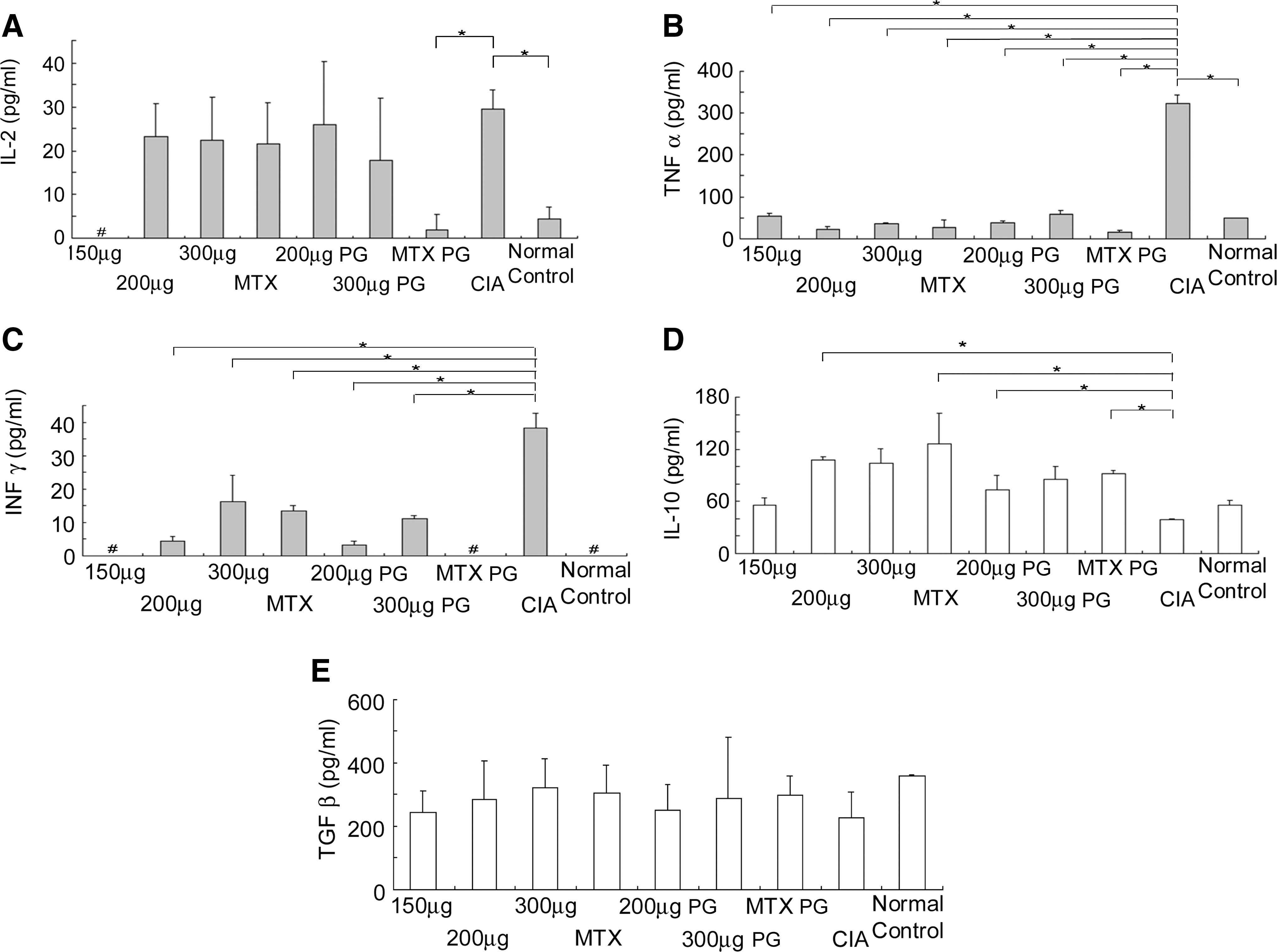
(
The vaccine has potent antirheumatic activity in both the treatment and prevention of CIA
We compared both therapeutic and preventive efficacy of the vaccine with MTX, which is a widely accepted treatment for RA. In the preventive groups, single vaccine doses of 200 and 300 μg/rat were started on day 0 of CIA induction. Mild arthritis developed in rats in the first week after preventive immunization with all vaccine doses (Fig. 4A). Higher expression of the vaccine in vivo corresponded with significant suppression of footpad swelling, arthritic incidence and score, and the onset of CIA disease up to 24 days compared with CIA rats and the CIA negative control group (p < 0.05). The suppressive effect on CIA development of the 200 μg/rat vaccine dose was more potent than the 300 μg/rat dose, as observed up to 10 weeks. Notably, the potency of the 200 μg/rat vaccine dose against CIA development in rats was comparable to that of MTX.

(
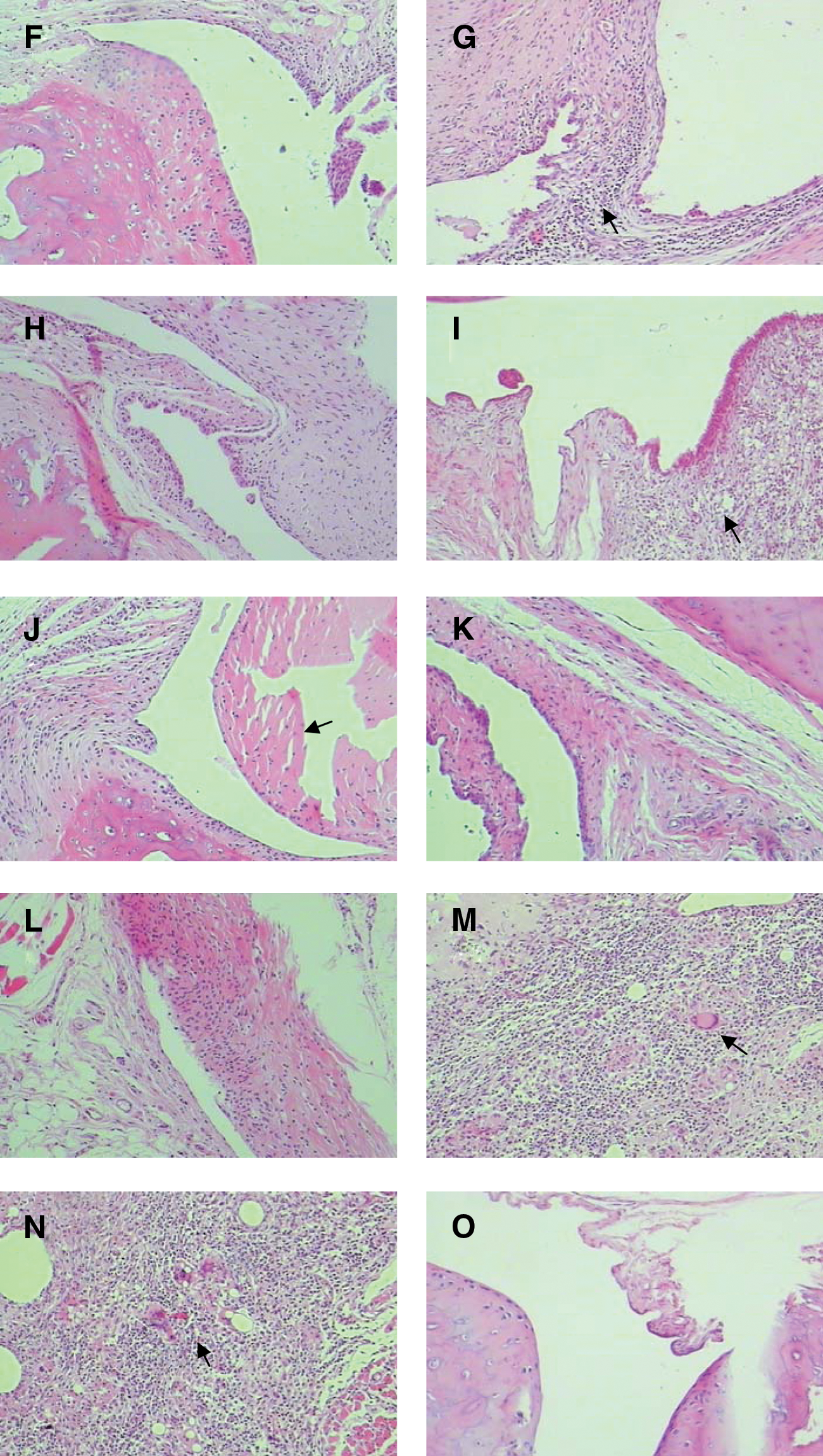
In treatment groups, a single muscle injection of vaccine at 150, 200, and 300 μg/rat was given on day 12 after the establishment of CIA. Dose-dependent therapeutic effects were observed on day 5 after treatment (Fig. 4B). In particular, the 200 and 300 μg/rat vaccine doses significantly reduced the severity of CIA and deferred the onset of CIA up to 10 weeks (p < 0.05). More importantly, a single vaccine dose of 200 or 300 μg/rat was as effective as MTX administered weekly intraperitoneally at a low (0.75 mg/kg) dose for 4 weeks. Vaccine at the 150 μg/rat dose could also partially reduce the severity and development of CIA in rats compared with CIA negative controls. In contrast, pcDNA3.1 naked vector at 200 μg/rat had no effect. It should be noted that immunizations with a dose of 300 μg/rat in the treatment group resulted in injection site ulcerations that did not easily heal.
The serum level of anti-CII IgG antibodies is considered the most reliable marker for arthritic severity in patients with RA and in the established CIA model (Williams et al., 1998; Kim et al., 2000). Similar to MTX treatment, various doses of the vaccine in both treatment and preventive groups tended to reduce serum anti-CII IgG antibody levels; however, only the 200 μg/rat dose in the preventive groups was significantly different from CIA controls (Fig. 4C and D). We further examined the development of anti-PEA IgG antibody in the animal model to predict potential complications of the in vivo use of immunotoxins in humans. Serum levels of anti-PEA IgG antibody detected by ELISA were low not only in the treatment groups but also in the preventive groups, but a dose-dependent induction effect was observed (Fig. 4E). Nevertheless, the presence of low-level anti-PEA IgG antibody did not preclude clinical therapeutic efficacy of the vaccine.
Furthermore, the hind ankle joints of vaccinated arthritic rats were histopathologically examined either on day 16 after treatment or on the fourth week after preventive vaccination. All CIA rats and CIA negative control group rats exhibited typical arthritic lesions, with prominent swelling of the ankle joints, severely inflamed synovium, inflammatory cell infiltration, erosion and degeneration of the cartilage cap, and pannus formation (Fig. 4H and I). Compared with normal rats (Fig. 4J), synovium hyperplasia and cartilage and bone destruction were also observed in both CIA rats and the CIA negative control group (Fig. 4H and I). This damage rapidly expanded the synovial pannus. In contrast, these histopathological changes were significantly ameliorated by the vaccine in both treatment groups (Fig. 4D–F) and preventive groups (Fig. 4A and B). Similar to MTX treatment, vaccine at 200 μg/rat in the treatment groups abrogated pannus formation, cartilage destruction, and bone erosion, while only showing mild inflammation of synovial tissue (Fig. 4G). In the preventive groups, the vaccine at 200 μg/rat effectively inhibited the development of arthritic lesions, only showing mild inflammation of synovium, and seemed to be superior to MTX (Fig. 4C). These histopathological results were consistent with the observed clinical therapeutic efficacy and were corroborated by radiographic X-ray examination of the hind ankle and paw joints in both treatment and preventive groups (data not shown).
Discussion
Despite many advances in DNA vaccine studies, clinical RA has been not successfully treated by either a specific antigen or a targeted DNA vaccine. Unlike infectious diseases, in which the pathogenesis generally results from only a particular microbe or a related antigen, multiple and as yet unknown autoantigens appear to be involved in RA (Sercarz, 2003; Holmgren and Czerkinsky, 2005; Larche and Wraith, 2005). Moreover, both intra- and intermolecular epitope spreading probably play a key role in the pathogenesis of RA (Robinson et al., 2003; Sercarz, 2003). However, expression of numerous autoantigens by a vaccine cocktail may have the potential to exacerbate rather than ameliorate RA (Sercarz, 2003). These are major challenges to developing a single specific DNA vaccine to simultaneously tolerize against multiple immune responses in RA. We provide here the proof-of-principle for a novel therapeutic approach to autoreactive CD4+CD28+ Th1-mediated RA through selective blockade or inhibition of B7-2/CD28. We successfully developed a new class of targeted DNA vaccine expressing a truncated exotoxin (PEA) and a costimulatory molecule, B7-2, without autoantigens. This vaccine could selectively inhibit the critical B7-2/CD28 costimulatory signals, and importantly, displayed potent antirheumatic activity to both prevent and treat CIA.
We chose truncated PEA and B7-2 for inclusion in our targeted DNA vaccine for several reasons. First, PEA has been shown to be highly cytotoxic against targeted cells (Pasten and FitzGerald, 1991; Xi et al., 2006), yet does not kill PEA-expressing cells themselves (Chen et al., 1997). Second, CD28 expression is essential for cellular and humoral immunity against type II collagen (CII), and CD28 costimulation is required for CIA development (Tada et al., 1999). CD28-deficient mice never develop arthritis and show markedly decreased IgG and IgM anti-CII antibody levels and low IFN-γ production by lymph node cells in response to CII (Tada et al., 1999). Studies have shown that the selective inhibition of CD28 can decrease the activation of alloreactive and autoreactive T cells, but not the activation of T cells stimulated by exogenous antigens. In addition, because agonistic anti-CD28 antibodies promote Th1 differentiation in vitro, inhibition of CD28 is likely to modify the Th1/Th2 balance. The selective blockade of CD28 was also reported to be more immunosuppressive than blocking B7 with CTLA-4/Ig in a murine model of graft-versus-host disease (Vanhove et al., 2003). Thus, CD28 blockade may allow selective inhibition of pathologic T cells in autoimmunity without inhibiting other protective T cell responses (Tada et al., 1999; Vanhove et al., 2003; Lyddane et al., 2006). Last, Ts cells do not express CD28, which is constitutively expressed on all Th1 cells, and Tr-mediated suppression is not dependent on CD28 (Filaci et al., 2004; Davila et al., 2005; Lyddane et al., 2006; Xi et al., 2006). By contrast, CTLA-4 is preferentially and highly expressed on both Tr and Ts cells and may play an essential role in the suppressor function of Tr cells, although this idea is still controversial (Khoury, 2003; Shevach, 2008). Theoretically, inactivation of B7-1/CTLA-4 with CTLA-4/Ig might prevent the development of both autoimmune and transplantation tolerance and may ultimately oppose the effect of B7 blockade (Vanhove et al., 2003).
Our findings here provide direct evidence that a single intramuscular injection of the vaccine can potently inhibit rheumatic symptoms in both therapeutic and preventive CIA models. The CIA animal model is most commonly used to study the effectiveness of both anti-T cell and antiinflammatory therapies for human RA (Liu et al., 2003; Seo et al., 2004). The significant clinical therapeutic and preventive efficacy of the vaccine was convincingly verified in a series of clinical examinations and detection of reduced serum anti-CII IgG levels. The latter is important because anti-CII IgG levels have been demonstrated to reflect inflammatory activity with the potential to destroy cartilage in the early stages of RA (Williams et al., 1998; Kim et al., 2000). Furthermore, the clinical therapeutic and preventive efficacy of the vaccine was closely associated with B7-2-PE40KDEL fusion exotoxin expression in vivo and the reduction of CD4+CD28+ Th1 cells. Significantly, the clinical efficacy of the vaccine was comparable to that of MTX, the current clinical treatment of choice for RA (Song et al., 2010). More importantly, in addition to selectively decreasing autoreactive CD4+CD28+ Th1 cells, the vaccine also increased both Tr and Ts cells simultaneously and caused polarization of Th1/Th2/Th3 cells/cytokines in the CIA model. Such specific and pronounced immunologic effects strongly suggest that the vaccine may serve as either an effective stimulator of the two regulatory T subsets or as a specific suppressor of autoreactive CD4+CD28+ Th1 cells simultaneously. With these unique biological activities, this targeted DNA vaccine could also provide a new class of therapeutics for the treatment of other autoreactive Th1-mediated autoimmune diseases.
In this study, the vaccine could induce long-term stable high expression of B7-2-PEA fusion protein in vivo. Interestingly, the 200 μg/rat dose was more effective than the higher (300 μg/rat) dose as well as the lower (150 μg/rat) dose in the CIA model in both therapeutic and preventive settings, suggesting that the therapeutic window for the vaccine may be somewhat narrow, not unlike many widely used immunosuppressive agents such as tacrolimus (FK506) and cyclosporine A (CsA) (Song et al., 2009). As is well known, the therapeutic or pharmaceutical window is an important index for estimating the drug dosage that can treat disease effectively while staying within the range of safety. Therefore, a vaccine with a small pharmaceutical window must be administered with care and control, for example, by frequently measuring the blood concentration of B7-2-PE40. Concerning whether the vaccine is toxic, much work remains to be done in future studies; in particular because a phase 1 trial and further clinical development of superagonist anti-CD28 mAb TGN1412, targeting the B7/CD28 axis, probably the first antihuman CD28 mAb to be tested clinically, had to be stopped because of the massive release of inflammatory cytokines triggered by TGN1412 in humans (Suntharalingam et al., 2006).
Induction of anti-immunotoxin neutralizing antibodies is always a crucial barrier to the use of immunotoxins (Pasten and FitzGerald, 1991; Wolf and Elsasser-Beile, 2009). Both bacterial and plant toxins are highly immunogenic foreign proteins, and high levels of neutralizing antibodies generally develop about 10 days after exposure in the absence of immunosuppression (Pasten and FitzGerald, 1991; Wolf and Elsasser-Beile, 2009). Strikingly, serum levels of anti-PEA IgG antibody produced by our vaccine were low and did not affect the clinical therapeutic efficacy. The reason is that DNA vaccines have generally been considered weakly immunogenic. Moreover, the vaccine treatment most likely eliminated autoreactive CD4+CD28+ Th1 cells and inhibited antibody-producing B cells by increasing both Tr and Ts cells. Thus, anti-PEA IgG antibodies would not be readily produced with high long-term stable expression of B7-2-PE40KDEL fusion protein in vivo.
In summary, we have developed a new class of targeted DNA vaccine that effectively stimulates both Tr and Ts subsets while specifically suppressing autoreactive CD4+CD28+ Th1 cells. This new vaccine has potent antirheumatic activity to both treat and prevent CIA, with effects comparable to MTX. Our results support the concept that selective blockade of B7-2/CD28 costimulation may represent a powerful strategy for the treatment of RA, and this vaccine could be a promising new approach for the prevention of autoimmune diseases driven by proinflammatory CD4+CD28+ Th1 cells.
Footnotes
Acknowledgments
The authors thank Drs. T.J. Moehring and J.F. Sucic (Department of Microbiology and Molecular Genetics, University of Vermont, Burlington, VT) for providing us with the toxin-resistant cell line CHO-K1 RPE.40. This study was supported by grants from the National Natural Sciences Foundation (39900187) and by the National Key Technologies R&D Program (2009ZX09103-624) of China. X.H. designed and performed research, and analyzed and interpreted data; L.F., L.N., S.X., Y.F., L.Y., Z.X., L.J., and S.Y. performed research, and analyzed and interpreted data; X.Y. designed research, analyzed and interpreted data, and wrote the manuscript.
Author Disclosure Statement
The authors declare no competing financial interests.