
Abstract
Spinal muscular atrophy (SMA), a neurodegenerative disease, is the second most common genetic disorder and the leading genetic cause of infantile death. SMA arises from the loss of Survival Motor Neuron-1 (SMN1), leading to degeneration of lower motor neurons and, consequently, the atrophy of voluntary muscles. A duplicated copy gene called SMN2 exists in humans. SMN2 is unable to fully compensate for the loss of SMN1 because it produces very low levels of functional SMN protein due to an alternative splicing event. A C/T transition in SMN2 exon 7 results in a transcript lacking exon 7 and, therefore, creates a truncated SMN protein that cannot fully compensate for the loss of SMN1. However, SMN2 is an ideal target for therapeutic strategies that redirect this critical splicing event. Previously, we developed the first trans-splicing strategy to increase the full-length mRNA and functional SMN protein from the SMN2 gene. To improve the trans-splicing efficacy, we then developed a single-vector system that expressed a trans-splicing RNA (tsRNA) and an antisense blocking the downstream splice site. This single vector greatly enhanced trans-splicing of SMN2 transcripts in vitro and in vivo. In this report, we have added a neurotrophic factor [insulin-like growth factor (IGF)-1] to this single vector to determine whether neuroprotection and SMN induction provide greater protection in an SMA animal model. Intracerebroventricular injection of the trans-splicing/IGF vector significantly increased SMN protein in brain and spinal cord of SMAΔ7 mice and lessened the severity of disease in a more severe mouse model as evidenced by an extension of life span and increased body mass.
Introduction
Alternative pre-mRNA splicing is an important determinant of gene regulation in higher eukaryotes (Lander et al., 2001). Alternative splicing acts as a dominant mechanism in the generation of various protein isoforms; however, misregulation can also result in disease-associated mutations. Utilizing small RNA molecules to restore the accurate splicing of the defective pre-mRNA has been an expanding focus of research, including in SMA (Baughan et al., 2006, 2009; Wood et al., 2007a; Dickson et al., 2008; Khoo and Krainer, 2009). One promising therapeutic application is spliceosome-mediated RNA trans-splicing (Walsh, 2004). Several forms of trans-splicing have been reported in different species from lower to higher eukaryotes, including rodents and humans (Murphy et al., 1986; Sutton and Boothroyd, 1986; Rajkovic et al., 1990; Bruzik and Maniatis, 1992; Caudevilla et al., 1998; Finta and Zaphiropoulos, 2002; Flouriot et al., 2002; Dorn and Krauss, 2003; Labrador and Corces, 2003). The most recent study shows that trans-splicing occurs naturally in normal human cells by joining 5′ exons of the JAZF1 gene to 3′ exons of the JJAZ1/SUZ12 gene, and this chimeric RNA is translated into JAZF1-JJAZ1, a protein with antiapoptotic activity (Li et al., 2008). Functional correction using trans-splicing has been reported in several preclinical models of human disease, including cystic fibrosis, hemophilia A, X-linked immunodeficiency, and various cancers (Liu et al., 2002; Chao et al., 2003; Pergolizzi et al., 2003; Pergolizzi and Crystal, 2004; Tahara et al., 2004; Nakayama et al., 2005). The first in vivo RNA repair by trans-splicing was performed in factor VIII hemophilia A-knockout (KO) mice (Chao et al., 2003).
We developed the first trans-splicing strategy to increase the full-length mRNA and functional SMN protein from the SMN2 gene (Coady et al., 2007). The original trans-splicing RNA targeted intron 6 and replaced exon 7 (pM13) (Coady et al., 2007). The pM13 plasmid and adeno-associated virus (AAV) expressing pM13 were very effective in cell-based assays; however, the in vivo trans-splicing activity was not significant. Therefore, we improved the efficiency of trans-splicing by coexpression of an antisense RNA that blocked splicing at the boundary of endogenous SMN intron 7/exon 8. Codelivery of trans-splicing and antisense RNA to the brain of the SMAΔ7 mouse model led to a significant increase in trans-splicing and the SMN levels in the spinal cord (Coady et al., 2008). A recent study indicated that the improved version of trans-splicing extends the life span in a severe model of SMA (Coady and Lorson, 2010).
Although we have improved the efficacy of trans-splicing, increasing the level of the SMN protein per se may not be sufficient to prevent the early loss of motor neurons. Increasing the level of the SMN protein may not have any positive effect on the motor neurons that have already lost their potency and ability to survive. Neurodegenerative diseases can greatly benefit from overexpression of neurotrophic factors in motor neurons and muscles. In addition, efficient and stable expression of any molecule in the neurons will be greatly facilitated by introducing a neurotrophic factor that supports the survival of the motor neurons. Insulin-like growth factor (IGF)-1 is a pleitropic neurotrophic factor with widespread distribution of its receptor, allowing it to influence the survival of numerous populations of neurons and glial cells in both the central (CNS) and peripheral nervous systems (Dore et al., 1997). IGF-1 promotes neuronal survival during normal brain development mainly in hippocampal and olfactory systems that depend on postnatal neurogenesis (Bondy and Cheng, 2004). IGF-1 is neuroprotective in animal models of neuronal injury, including brain ischemia (Wang et al., 2000; Kawano et al., 2001) and axotomy (Kermer et al., 2000; Mathonnet et al., 2001). In cell culture models, IGF-1 prevents dopamine-induced cell death in cultured cerebellar granule cells, a model of Parkinson's disease (Offen et al., 2001). IGF-1 also protects cortical neurons from apoptosis induced by serum deprivation (Yamada et al., 2001) and motor neurons from glutamate-induced death in organotypic slice culture (Bilak and Kuncl, 2001).
IGF-1 has previously been shown to lessen the severity of disease in an animal model of amyotrophic lateral sclerosis (ALS). Delivery of AAV-expressing IGF-1 into respiratory and motor limb muscles of the ALS mouse model at the onset of clinical symptoms delayed the onset of behavioral symptoms (Kaspar et al., 2003). Injection of AAV-IGF-1 before disease onset delayed disease development by 31 days. In addition, IGF-1-treated animals showed a significant improvement in life span (Kaspar et al., 2003). Injection of an AAV2-based vector encoding human IGF-1 (CERE-130) into lumbar spinal cord parenchyma of the ALS mouse model resulted in partial rescue of lumbar spinal cord motor neurons, as well as increased animal survival (Lepore et al., 2007). In a recent study, stereotaxic injection of an IGF-1-expressing viral vector to the deep cerebellar nuclei, a region of the cerebellum with extensive brainstem and spinal cord connections, led to reduced ALS neuropathology, improved muscle strength, and significantly extended life span in ALS mice (Dodge et al., 2008).
To determine whether coexpression of the SMN trans-splicing RNA (tsRNA) and IGF-1 resulted in an enhanced level of protection, we have engineered the pMU3 trans-splicing vector to coexpress the murine IGF-1 cDNA. Our goal is to investigate the role of SMN tsRNA and IGF-1 independently to determine their individual effect and then study their role in combination to determine their potential synergistic effect in increasing the survival and quality of life in a severe SMA mouse model. Intracerebroventricular (ICV) delivery of the dual vector into the SMAΔ7 mouse model results in a significant increase of SMN in the CNS, as well as increases in body mass and life span in a severe SMA mice model.
Materials and Methods
Plasmids and cloning
pMU3 construct was described previously (Coady et al., 2008). Neuron-specific enolase (NSE) promoter was amplifed (forward outer primer, 5’-ACG CGT CGA CTA GTA GCT CTG AGC TCC TCC TCT GCT-3’) from pDRIVE01-NSE(r)-Ru5 v02 (Invitrogen, Carlsbad, CA) and linked by overlapping PCR to 357 bp of the exon 1-intron-exon 2 of pTri-EX-1.1 (Novagen) at the 3’ end to facilitate vertebrate expression of IGF-1 (reverse outer primer, 5’-CGC TCG ATA TCA GCT GTA GGA AAA AGA AGA AGA AGG-3’). The inner primers used for linking NSE promoter and sequences of pTri-EX-1.1 were as follows: forward, 5’-ATC CAA GCC ACC ATC GGG GGT TCT CGG AGT CGC TGC GCG CTG CCT TCG-3’; and reverse, 5’-CGA AGG CAG CGC GCA GCG ACT CCG AGA ACC CCC GAT GGT GGC TTG GAT-3’. The entire fragment was cloned into Sal1-EcoRV of pBlueScript (PBS). IGF-1 was amplified from a Mouse Verified Full-Length cDNA clone (Open Biosystems, Huntsville, AL) using forward primer 5’-CAC CGA TAT CCG GTC GCC ACC ATG TCG TCT TCA CAC CTC TTC T-3’ (including a Kozac sequence at the 5’ end) and reverse primer 5’- GCA CGG ATC CCT AAG CGT AAT CTG GAA CGT CGT ACT TGT GTT CTT CAA ATG TAC T-3’ [including a hemagglutinin (HA)-tag at the 3’ end]. IGF-1 fragment was cloned into EcoRV-BamH1 sites of PBS downstream of the NSE-pTri-EX-1.1 sequences. The entire fragment containing NSE and IGF-1 was amplified using forward primer 5’-ACG GAT GCA TTA GTA GCT CTG AGC TCC TCC TCT GCT-3’ and reverse primer 5’-GAG CTG TAC ACT AAG CGT AAT CTG GAA CGTC-3’. The PCR fragment was column purified (Invitrogen) and cloned into the Nsi1- BsrG1 sites of pMU3, replacing the cytomegalovirus (CMV)–enhanced green fluorescent protein (eGFP) sequence. To construct IGF-1 expression vector, the fragment containing NSE and IGF-1 was cloned into the Nsi1-BsrG1 sites of pMU1. pMU1 is the shorter version of pMU2 (Baughan et al., 2006; Coady et al., 2007), which is identical to pMU2 except that it lacks 2 kb between the SV40 polyadenylation signal and the terminal repeat.
Animal injections
All animal experiments took place in accordance with procedures approved by the Animal Care and Use Committee of the University of Missouri. SMAΔ7 (Smn–/–, SMN2+/+, SMNΔ7+/+ ) and severe (Smn–/–, SMN2+/+ ) mice with C57BL/6 genetic background were genotyped at day of birth (day 1) as described previously (Coady et al., 2008) and injected at day 2. Single ICV injections were performed at postnatal day (PND) 2 as described previously (Passini and Wolfe, 2001; Coady et al., 2008; Dickson et al., 2008; Baughan et al., 2009). Multiple injections were carried out at PND 2, 3, and 4. In brief, mice were immobilized by cryoanesthesia and injected using microliter-calibrated sterilized glass micropipets. The injection site was ∼0.25 mm lateral to the sagittal suture and 0.5–0.75 mm rostal to the neonatal coronary suture. The needle was inserted perpendicular to the skull surface using a fiber-optic light (Boyce Scientific Inc., Gray Summit, MO) to facilitate visualization of pertinent anatomical structures. The needle was removed 15 sec after the plunger movement was discontinued to prevent backflow. Mice recovered in a heating pad for 5–10 min. In vivo transfection was carried out using an injection stock solution containing 10 μg (2 μl) of either plasmid DNA (pMU3, pMU4, NSE-IGF-1), 1 μl of D-(+)-glucose 20% (w/v) (Sigma, St. Louis, MO), 2 μl of 2.5-kDa linear polyethylenimine (PEI) homopolymer (150 mM; Polysciences Inc., Warrington, PA), and 1 μl of trypan blue (4%) saline (Sigma). The negative control solution contained 2 μl of sterile PBS instead of plasmid DNA. Plasmid DNAs used for injection were prepared with endotoxin-free Maxi-Prep (QIAGEN, Valencia, CA) and were analyzed for concentration and purity by NanoDrop.
RT-PCR
Brain and spinal cord tissues were harvested either 7 or 24 hr post injection, placed immediately in liquid nitrogen, and then placed at –80°C until ready for RNA extraction. Total RNA was extracted using TRIzol Reagent (Invitrogen) according to the manufacturer's instruction. Total RNA concentration was determined and normalized before complementary DNA synthesis. RT-PCR was performed as previously described (Passini and Wolfe, 2001; Coady et al., 2008; Dickson et al., 2008; Baughan et al., 2009). The trans-spliced product was detected using exon 3 forward primer (5’-GAG AGG AGC AAA ATC TGT CCG ATC TAC-3’) and M13 reverse primer (5’-GTC ATA GCT GTT TCC TGC GAC-3’). The IGF-1 RNA was detected using IGF-1 forward primer (5’-CCG GTC GCC ACC ATG TCG TCT TCA CAC CTC TTC T-3’) and HA reverse primer (5’-GAG CTG TAC ACT AAG CGT AAT CTG GAA CGTC-3’). Mouse tubulin RT-PCR was performed using mouse tubulin forward primer (5’-TCA CTG TGC CTG AAC TTA CC-3’) and mouse tubulin reverse pimer (5’- GGA ACA TAG CCG TAA ACT GC-3’). The transcripts were amplified using the Expand High Fidelity PCR kit (Roche, AG, Basel, Switzerland) and the following conditions: 94°C for 5.00 sec; 94°C for 0.45 sec; 60°C for 1.00 sec; 68°C for 2.00 sec; repeat steps 2–4 for 10 cycles; 94°C for 0.45 sec; 60°C for 1.00 sec; 72°C for 2.00 sec; repeat steps 6–8 for 20 cycles; 72°C for 10.00 sec. Representative results are presented and have been repeated at least three times.
Western blot
Brain and spinal cord tissues were harvested either 7 or 24 hr post injection, immediately placed in liquid nitrogen, and then kept at –80°C until ready for protein extraction. Approximately 100 mg of tissue was homogenized in 500 μl of Jurkat lysis buffer [50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 20 mM NaH2(PO4), 25 mM NaF, 2 mM EDTA, 10% glycerol, 1% Triton X-100, and 1% protease inhibitor cocktail (PIC; Roche, Indianapolis, IN)]. Protein concentration was determined; equal amounts of protein were boiled in loading dye and loaded onto 12% SDS-PAGE 20 cm × 10 cm Bio-Rad Protean II xi glass plates using Protean 2000 (Bio-Rad, Hercules, CA). Resolved proteins were transferred to polyvinylidene difluoride (Immobilon) at 300 mAmp for 400 min in Towbin's buffer [24 mM Tris base, 192 mM glycine, 20% methanol (v/v)]. Blots were blocked overnight in 6% dry milk and Tris-buffered saline (TBS-T) [90 mM Tris-HCl, 10 mM KCl, 547 mM NaCl, 2% Tween (v/v) (Acros Organics), pH 7.5]. The SMN immunoblot was performed using a mouse SMN-specific monoclonal antibody (BD Biosciences, San Jose, CA) diluted 1:1,000 TBS-T in 1% dry milk. Mouse TrueBlot Ultra horseradish peroxidase (HRP) anti-mouse IgG (eBioscience Inc., San Diego, CA) was used as secondary antibody. The HA-tagged IGF-1 was detected using a polyclonal rabbit anti-HA antibody (Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:300 TBS-T in 1% dry milk. Anti- rabbit HRP was used as secondary antibody. Blots were visualized by chemiluminescence using a 1:1 mixture of Pierce West Femto reagent (Thermo Scientific, Rockford, IL). Images were captured on a Fugi Imager, LAS-3000, and quantitated by the Multi-Gauge V2.3 system. For loading control, the blots were stripped with a 1:1 mixture of 30% H2O2 and PBS for 30 min at room temperature and reprobed with a 1:3,000 dilution of rabbit anti-β-actin for SMN blots and a 1:1,000 dilution of mouse anti-α-tubulin for IGF-1 blot.
Small nuclear ribonucleoprotein (snRNP) assembly assay
Cloned mutant U1 small nuclear RNA (snRNA) (KO) lacks the Smith core site (5’-AUUUGUGG-3’) to prevent nonspecific binding. U1 snRNA plasmids [wild-type (WT) and KO] were maxi-purified, digested with EcoRI, and the U1 snRNA fragments gel-isolated (Invitrogen). The concentration of the gel-isolated fragments was determined (NanoDrop), and equal amounts of DNA were used as template for in vitro transcription. snRNA transcription reaction containing 1 μg of DNA template, 10 units of T7 RNA polymerase HC (Ambion, Austin, TX), Arca-Methyl cap (Ambion), and rNTPs supplemented with P32-labeled rUTP was incubated at 37°C for 90 min. The in vitro transcribed RNA was cleaned up using phenol/chloroform extraction, subjected to two cycles of freeze-thaw, and then resuspended in 20 μl of reconstitution buffer (20 mM HEPES-KOH, pH 7.9, 50 mM KCl, 5 mM MgCl2, 0.2 mM EDTA, 5% glycerol, 0.01% Triton X-100). Brain tissue extracts were prepared by homogenization of 100 mg of frozen tissues with ice-cold reconstitution buffer containing 0.01% NP40 as previously described (Gabanella et al., 2005, 2007). Homogenates were then passed through a 1,000-μl pipette tip multiple times and then centrifuged for 15 min at 1,000 rpm at 4°C. Supernatants were collected, and 1% PIC (Roche) was added to protein extracts. Protein concentration was measured using the Lowry method (Bio-Rad), and 50 μg of extracts was incubated with U1 snRNA (100,000 cpm) using 20 U of RNasin (Promega, Madison, WI), 1 μg of yeast tRNA, and 2.5 mM ATP. The assembly was carried out at 30°C for 30 min. Following assembly, 350 μl of DEPC-RSB600 buffer (600 mM NaCl, 20 mM Tris-HCl, pH 7.4, 2.5 mM MgCl2, and 0.01% NP40) plus 4 μl of anti-mouse Y12 monoclonal antibody (Lab Vision) were added and incubated for 30 min at 4°C with shaking. Immunoprecipitation was continued by addition of 50 μl of slurry volume Protein G Plus/Protein A Agarose (Oncogene Science, Manhasset, NY) and 1 hr of incubation at 4°C. Radioimmunoprecipitates were washed twice with DEPC-RSB600, denatured in 5× formamide loading dye, and run on 8% acrylamide TBE-urea gel. snRNP products were visualized after 2 hr of exposure on a phospho-screen (Kodak) using Fuji-Imager FLA5000 and image reader FLA5000 V2.0 software. Within experiments, the ability to assemble snRNPs is determined by the ratio of U1WT:U1KO.
Results
Design and development of the dual vector
Previous studies have led to the development of an optimized SMN trans-splicing system (Coady et al., 2008; Coady and Lorson, 2010). Here, the optimized SMN tsRNA was coexpressed with a neurotrophic factor, IGF-1, to determine whether neuroprotection provides a more suitable cellular context for therapeutics that increase SMN. The enhanced trans-splicing vector consists of (1) a tsRNA comprised of the following: an SMN intron 6 annealing sequence ∼130 bp; an optimized heterologous splice site; the SMN1 exon 7 sequence; and a specific sequence (M13 primer) immediately downstream of the native stop codon at the end of exon 7 to allow straightforward detection of the trans-spliced product by RT-PCR; and (2) a short antisense RNA that anneals to the intron 7/exon 8 splice site (Fig. 1A). In the dual vector, the Pol II-derived GFP cDNA has been replaced with the IGF-1 cDNA driven by the NSE promoter (pMU4; Fig. 1B). To analyze the effect of the neurotrophic factor independently of trans-splicing, we constructed NSE-IGF-1 plasmid that only contains IGF-1 under control of the NSE promoter (Fig. 1B). To verify IGF-1 expression, HeLa cells were transfected with NSE-IGF-1 plasmid. The ∼15-kDa HA-tagged IGF-1 protein was detected by Western blot using an anti-HA antibody 48 hr post transfection (data not shown).

Development of a single plasmid vector that contains two therapeutic elements. (
IGF-1 RNA and protein are detected in brain and spinal cord of heterozygous SMA mice injected with pMU4
To determine the capability of the dual vector to produce an IGF-1 transcript in vivo, we delivered this construct into the cerebral ventricles of a transgenic mouse that contains a single murine Smn gene and two copies of human SMN2 (Smn +/–, SMN2 +/+). Animals with this genetic background are the parental, unaffected animals used to generate a severe model of SMA (Monani et al., 2000). Neonatal mice were injected in both cerebral lateral ventricles with 10 μg pMU4 using 2.5-kDa linear PEI and glucose. After 24 hr, brain tissue was harvested and used to generate total RNA for RT-PCR. Using an HA-specific primer, the 440-bp IGF-1 RNA was detected in the brain of three injected mice (Fig. 2A), consistent with the detection of an ∼15-kDa HA-tagged IGF-1 protein in the spinal cord of three pMU4-injected animals (Fig. 2B). These results demonstrated that the pMU4 construct is capable of producing NSE-driven IGF-1 transcripts and protein in vivo.

pMU4 plasmid produces HA-tagged IGF-1 transcript and protein in the brain and spinal cord, respectively, and increases trans-splicing in a dose-dependent manner in the brain. (
The pMU4 plasmid trans -splices SMN2 pre-mRNA in the brain of injected SMAΔ7 mice
We sought to determine the levels of trans-splicing and IGF-1 expression mediated by pMU4 within the CNS. For this purpose, we chose a well characterized SMA mouse model that lacks endogenous Smn, but contains the human SMN2 transgene and an SMNΔ7 cDNA (Smn–/–, SMN2+/+, SMNΔ7+ /+ ) (Le et al., 2005). Neonatal mice were injected in both cerebral lateral ventricles with either PBS or pMU4 at 10 μg (1.08 × 1012 plasmid copies) using 2.5-kDa linear PEI and glucose. To determine whether the intensity of trans-splicing is directly correlated to the number of injections, a series of one, two, or three injections was administered. Three animals were used for each set of injections. The first set of animals was injected only once at PND 2; the second set was injected at PND 2 and 3; and the third set was injected at PND 2, 3, and 4. Brain and spinal cord tissues were harvested 7 hr after the final injection in each set for analysis in RT-PCR, Western blot, and snRNP assembly assays. RT-PCR of brain extract demonstrates that the specific 504-bp tsRNA was expressed in a dose-dependent manner (Fig. 2C). tsRNA was gel-isolated and verified by sequencing (data not shown). As expected, the trans-spliced SMN RNA was not detected in tissues derived from the untreated heterozygous control and a PBS/PEI-injected animal. The pMU4-derived IGF-1 RNA was also detected in the brain of injected animals by using IGF-1 forward and HA reverse primers; however, a plateau in IGF-1 expression appears to have been achieved (Fig. 2C). The RT-PCR results confirm the ability of the pMU4 plasmid in the production of its therapeutic elements in vivo.
The pMU4 plasmid increases the level of the SMN protein in brain and spinal cord
To determine whether the increased SMN RNA levels resulted in increased SMN protein levels, brain and spinal cord extracts were examined by Western blot. The increase in the SMN protein levels in brain (Fig. 3A) and spinal cord (Fig. 3B) of pMU4-injected SMAΔ7 mice was easily detected. The level of SMN protein in pMU4-injected SMAΔ7 mice was very similar to the SMN levels in heterozygous carrier. In general, increasing the number of injections increased SMN protein, except for the final injection in the brain (Fig. 3A). Analysis of the spinal cord demonstrated that SMN levels rose following each injection (Fig. 3B). There was a significant increase in SMN levels in the spinal cord of three-times pMU4-injected animals compared with PBS-injected littermates (P < 0.05). Potentially, the brain results could be due to the slight variations in injection sites from animal to animal, while still successfully delivering the vector to the ventricles. These results demonstrate that the tsRNA generated by pMU4 plasmid is effectively translated into SMN protein. Control vectors were also examined, including the promoterless vectors (pM13 and pMU3) and NSE-IGF-1. pM13 is the initial trans-splicing vector that contains the tsRNA but lacks the antisense Int711. As anticipated, ICV delivery of the promoterless vectors did not increase SMN levels (Fig. 4A). Although there was a modest increase in SMN levels in NSE-IGF-1-treated extracts, the precise mechanism is unclear at this point.
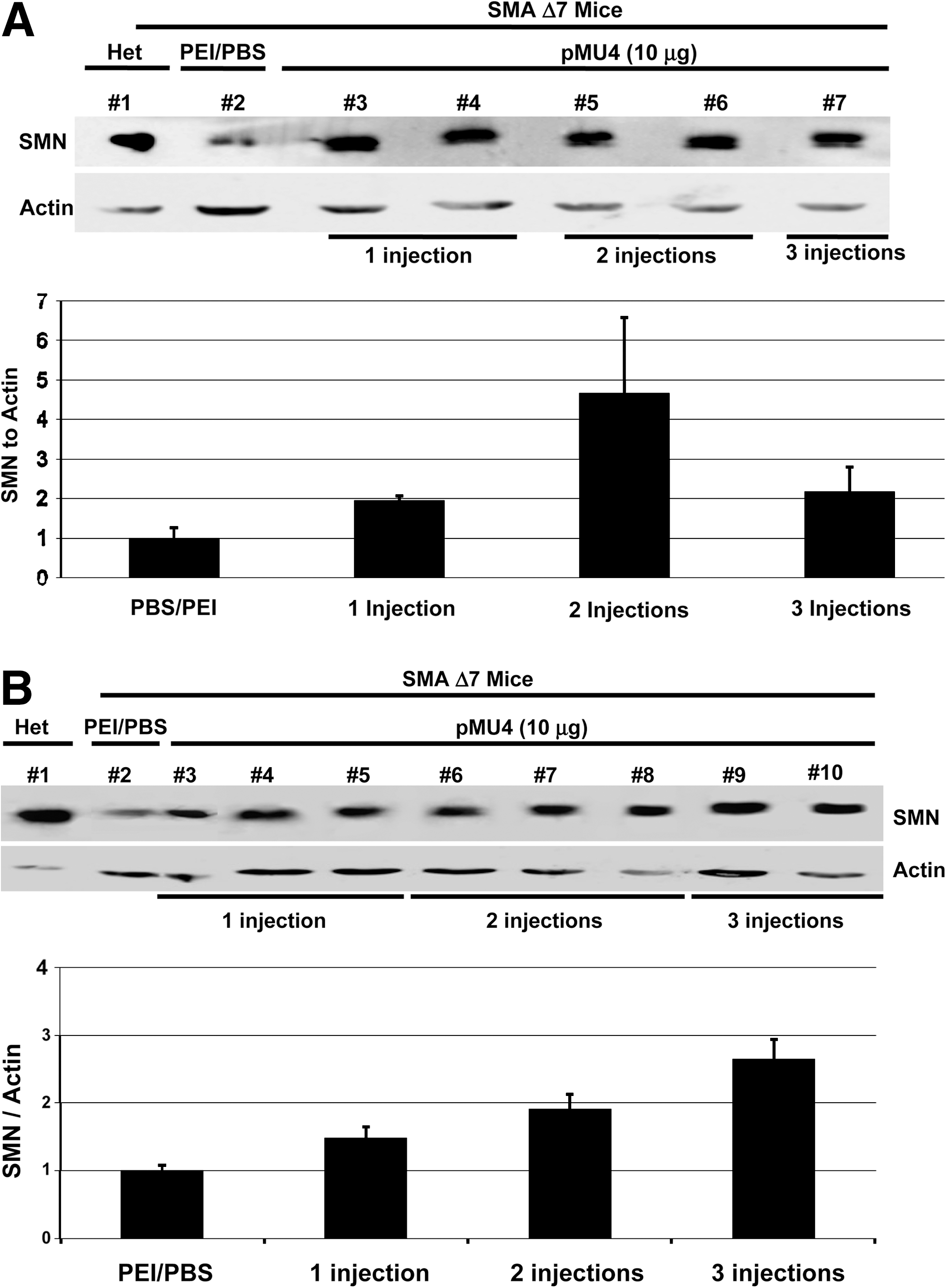
pMU4 injection increases the level of the SMN protein in an animal model of SMA (SMAΔ7). Western blot was performed with brain (
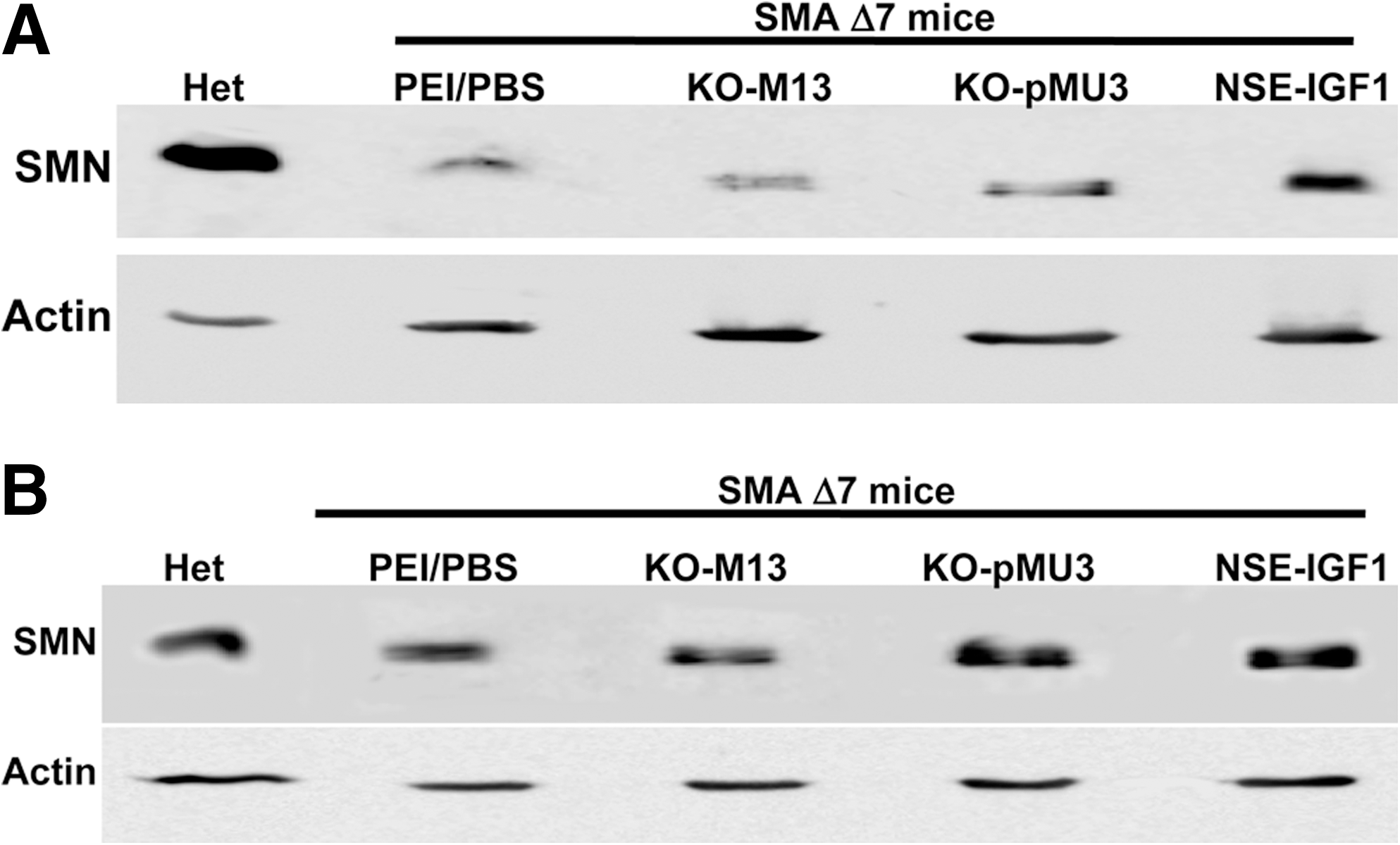
Injection of negative controls has no effect on the SMN protein levels in the SMA intermediate mouse model. Western blot was performed with brain (
SMN protein produced by pMU4 trans-splicing increases snRNP assembly in brain extracts from SMAΔ7 mice
The most comprehensively demonstrated function of SMN is its role in snRNP assembly. Although snRNP assembly activity is a well described function of the SMN protein, it remains unclear whether this is the specific defect responsible for SMA development. We next sought to determine whether the increases in SMN levels mediated by trans-splicing resulted in increased levels of snRNP assembly in vivo. To determine whether pMU4 trans-splicing generates a functional SMN protein in vivo, a portion of the brain extract analyzed by Western blot was used for snRNP assembly activity (Fig. 5). Brain extract from a PBS-injected SMAΔ7 mouse was used as negative control, and brain extract from an unaffected carrier was used as positive control. Brain extract from any of the PBS-injected SMAΔ7 mice exhibited low Sm assembly activity (Fig. 5). In contrast, all three extracts from pMU4-treated mice displayed increased levels of snRNP assembly activity compared with PBS-injected mice (Fig. 5). As expected, heterozygous carrier that contains high levels of endogenous SMN supported high levels of snRNP assembly and failed to maintain Sm assembly on the negative control snRNA that lacks the Sm assembly site (Fig. 5).
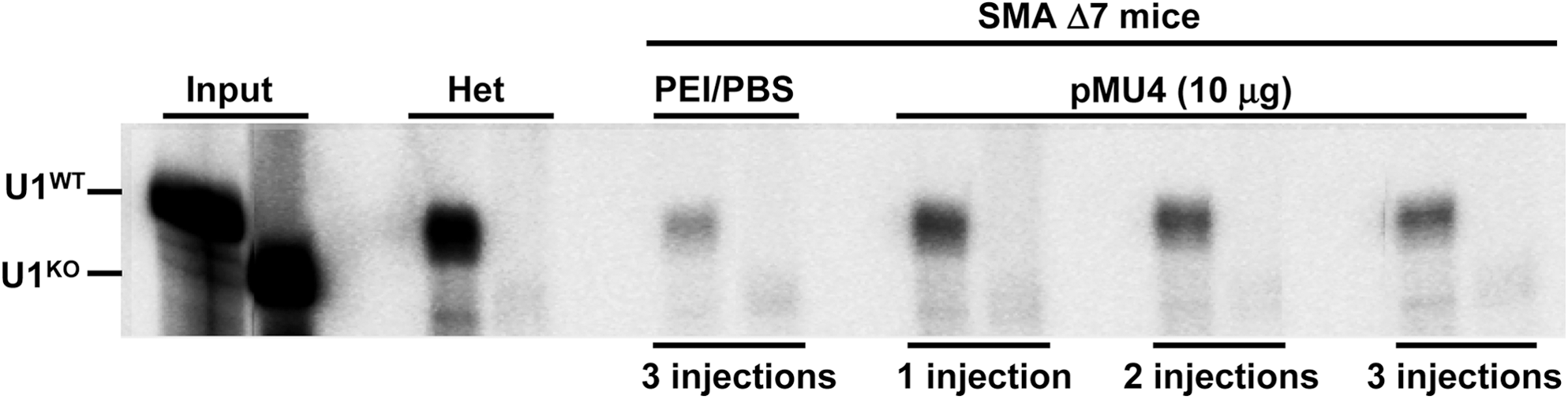
Enhanced trans-splicing by pMU4 injection enables SMAΔ7 mice to assemble U1 snRNP. Homozygous (Smn −/−, SMN2 +/+, SMNΔ7 +/+) animals were injected three times at PND 2, 3, and 4 with PEI/PBS, with pMU4 plasmid at 10 μg once at PND 2, twice at PND 2 and 3, and three times at PND 2, 3, and 4. Injection was performed using 2.5-kDa PEI and glucose into both ventricles. Brain tissues were harvested 7 hr after the last injection and used for snRNP assembly assay. Heterozygous carrier was used as positive control. U1 RNA probes (WT and KO) are shown.
Delivery of pMU4 significantly increases survival and body mass in a severe SMA model
ICV injection of the pMU4 plasmid DNA into the brain of SMAΔ7 mice confirmed the ability of this vector to (1) trans-splice, (2) produce the functional SMN protein, and (3) create IGF-1 transcripts and protein. As DNA expression is relatively transient and persists ∼48 hr following ICV delivery (data not shown), the SMAΔ7 mouse model, which lives 13–18 days (Le et al., 2005; Butchbach et al., 2010), is not an appropriate model to monitor survival and weight improvement following DNA treatment. Therefore, an alternative SMA model was selected that lacks murine Smn and contains two copies of human SMN2 (Monani et al., 2000). The average life span of the severe model is less than 5 days. To determine the impact of pMU4 on the SMA severe model, we analyzed the effect of trans-splicing and IGF-1 independently and in combination by introducing pMU3, NSE-IGF-1, and pMU4 plasmids into the brain of SMA severe mice. Neonatal mice were ICV injected with 10 μg of each plasmid (pMU3, NSE-IGF-1, and pMU4) using 2.5-kDa PEI and glucose into two ventricles. These mice were monitored every day for survival and weight gain and compared with PBS-injected SMA severe mice. A Kaplan-Meier curve indicates an extended life span for the pMU3-injected group in comparison with the PBS-injected group (Mantel-Cox, P = 0.01; Fig. 6). NSE-IGF-1 and pMU4 plasmids significantly increased the average survival of treated pups compared with PBS-injected animals (Mantel-Cox, P < 0.0001; Fig. 6). Median survival of pMU3-, NSE-IGF-1-, and pMU4-injected mice was 6, 7, and 8 days, respectively, compared with 4 days for PBS-treated animals. Even though the median survival of pMU4-injected animals extended 1 day in comparison with that of NSE-IGF-1-injected pups, their survival was not significantly different (Fig. 6). Body weight of each animal was recorded every day following plasmid delivery until the last day of its life. As the pups range in their initial weights, we normalized the weight variation between different animals by graphing the percentage of the weight gained from the day of injection until the day that maximal weight was reached (Fig. 7). The weight of pMU3-injected animals was not significantly higher than that of the PBS-injected group (Fig. 7), indicating that SMN induction per se does not have an effect on the weight of the animals. However, weights of NSE-IGF-1- and pMU4-injected mice increased significantly compared with that of the PBS-injected group (unpaired two-tailed t test, P = 0.001 and 0.0002, respectively; Fig. 7). Comparing the significant difference in survival and weight of pMU3-injected animals with that of the pMU4-injected group demonstrates the positive effects of pMU4 in improving the length and quality of life in the severe SMA model (Figs. 6 and 7). These results are consistent with the RNA and protein expression data in SMAΔ7 mice and indicate that pMU4 not only increases SMN levels in the CNS, but can also reduce the severity of the SMA phenotype in the SMA severe model. These results demonstrate that SMN expression combined with a general neuroprotectant can further lessen the severity of the SMA phenotype.
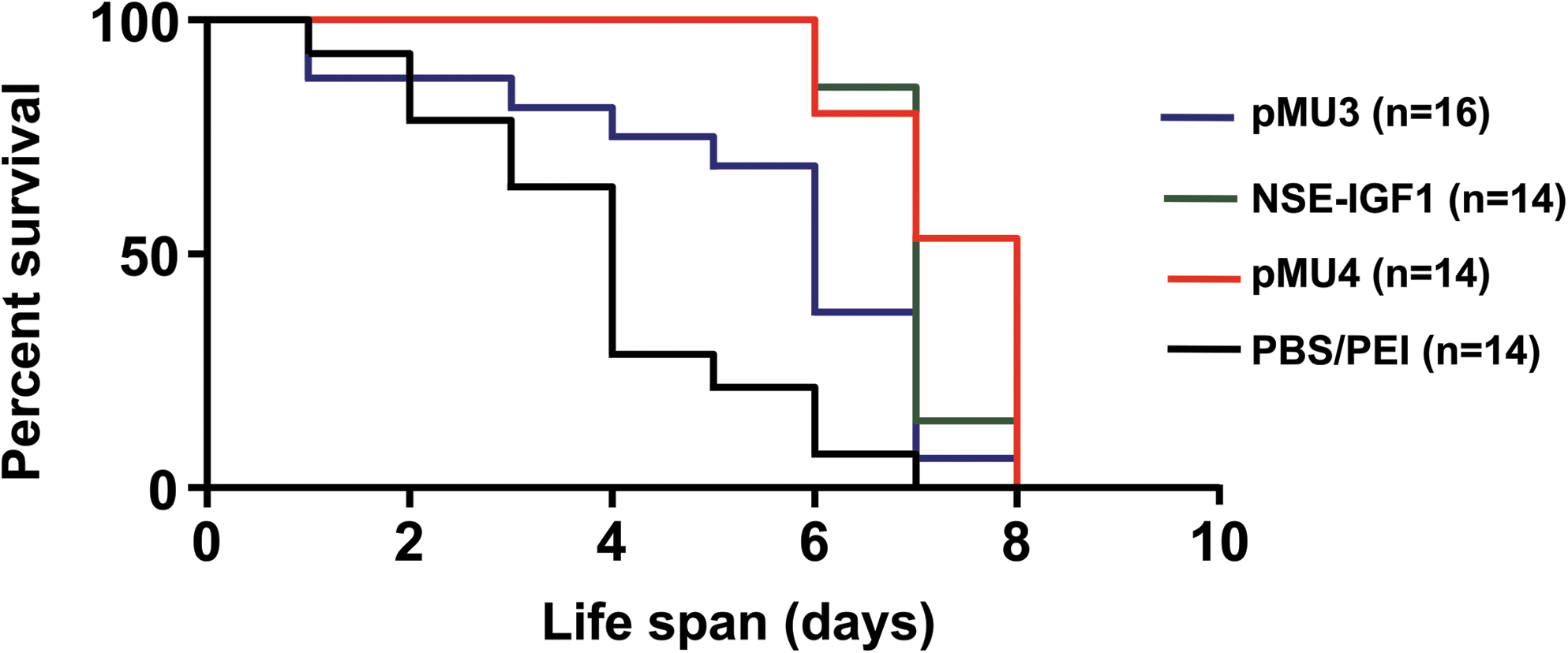
pMU4 and NSE-IGF-1 plasmid injections increase the life span in a severe SMA mouse model. Homozygous (Smn −/−, SMN2 +/+) neonatal mice were injected (PND 2) with PBS (n = 14), pMU3 (n = 16), NSE-IGF-1 (n = 14), and pMU4 (n = 14) plasmids using 10 μg with 2.5-kDa PEI and glucose into both ventricles. Survival was determined by Kaplan-Meier curves. P value was determined by the log-rank (Mantel-Cox) test. Median survival of pMU3 was 6 days in comparison with 4 days for the PBS-injected group (P = 0.01). Median survival of NSE-IGF-1 and pMU4 was 7 and 8 days, respectively, compared with 4 days for the PBS-injected group (P < 0.0001). Survival of NSE-IGF-1- and pMU4-injected mice was significantly increased in comparison with pMU3-injected mice (P = 0.01 and 0.001, respectively).
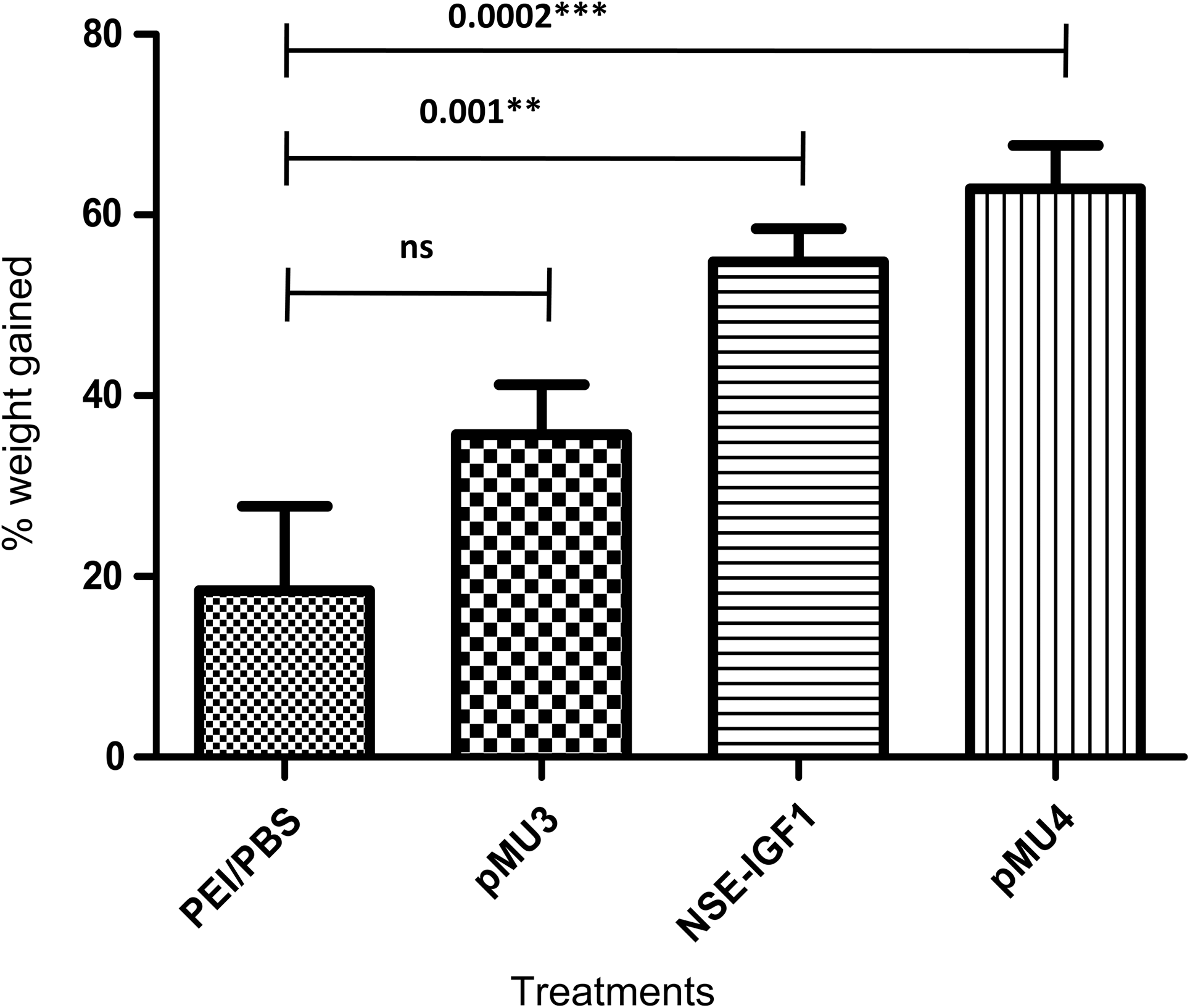
pMU4 and NSE-IGF-1 plasmid injections increase the body mass in a severe SMA mouse model. Percent weight gained post injection until maximal weight was graphed. The P value was determined by unpaired two-tailed t test. The weights of pMU3- and PBS-injected mice did not differ significantly. The weights of NSE-IGF-1- and pMU4-injected animals increased considerably compared with that of PBS-treated animals (P = 0.001 and 0.0002, respectively). The weights of pMU4- and NSE-IGF-1-treated animals were not statistically different.
Discussion
We have previously demonstrated the first SMN trans-splicing in vitro (Coady et al., 2007) and in vivo (Coady et al., 2008; Coady and Lorson, 2010). The current experiments focus on enhancing delivery and incorporating an additional transgene that could serve as a neuroprotectant. In this instance, the neuroprotectant is the IGF-1 cDNA. This clone, pMU4, was delivered via ICV injection into the brain of an SMAΔ7 mouse model and analyzed for its ability to produce tsRNA, IGF-1 transcripts, and IGF-1 protein. The capacity to produce tsRNA was increased proportionally to the number of injections, indicating that high levels of trans-splicing can be achieved by increasing the plasmid DNA input. Levels of IGF-1 transcript did not change with the number of injections, and this may be explained by the possibility that the IGF-1 mRNA expression is regulated by a negative feedback loop. Growth hormone (GH) levels have been shown to be controlled by a negative feedback loop that involves circulating IGF-1. In addition, the fluctuation in IGF status among patients with GH deficiency is partly controlled by endogenous GH secretion (Berg et al., 2009).
pMU4-mediated trans-splicing led to a substantial increase in the SMN levels in both brain and spinal cord of SMAΔ7 mice. In general, the levels of SMN increased with the frequency of injections. The SMN level in the spinal cord is directly proportional to the plasmid DNA input. However, the SMN increase in the brain tissues does not show a correlation with the frequency of injections. Results of molecular and biochemical analysis of the pMU4 vector were further confirmed by examining the life span and weight gain in the pMU4-injected SMA severe mouse model. Our results indicated that the survival of pMU4-injected severe mice expands significantly compared with either PBS- or pMU3-injected animals. However, the survival of pMU4-injected SMA mice is only slightly improved in comparison with NSE-IGF-1-injected littermates. Additionally, the amount of weight gain in NSE-IGF-1- and pMU4-injected mice is comparable and significantly higher than that in pMU3-injected mice. These comparative data suggest that the role of IGF-1 in survival and improvement of body mass was greater than that of SMN trans-splicing alone. Beneficial effects of the dual vector are achieved with only a single injection at PND 2, which is interesting given that the plasmid DNA disappears 24–48 hr post injection.
Several neurodegenerative diseases are triggered by excitotoxic cell injury mediated by excess glutamate signaling, which in turn leads to neuronal death. Glutamate inactivates Akt, a serine-kinase that plays a critical role in the pro-survival actions of IGF-1 (Dudek et al., 1997; Chalecka-Franaszek and Chuang, 1999). Glutamate-induced neurodegeneration is mediated by activation of NADPH oxidase in SH-SY5Y human neuroblastoma cells (Nikolova et al., 2005). Treatment of these cells with glutamate increases the mRNA expression level of NADPH oxidase subunits, leading to generation of reactive oxygen species that cause neurodegeneration. The involvement of NADPH oxidase in selective degeneration of motor neurons is also shown in ALS mice (Wu et al., 2006). We have recently demonstrated that the levels of Rac1 and Nox2, which are two major subunits of NADPH oxidase, were increased significantly in the heart of SMA mice at PND 9 (Shababi et al., 2010). We are currently investigating whether the increased expression of NADPH oxidase in SMA heart is correlated with the level of this enzyme in spinal cord. IGF-1 is shown to play a role in blocking glutamate-mediated death of late oligodendrocyte progenitors (OPs) by preventing Bax translocation, mitochondrial cytochrome c release, and cleavage of caspases 9 and 3 (Ness et al., 2004). Delayed delivery of IGF-1 prevents caspase 3 activation in late-stage OPs up to 16 hr following glutamate exposure and promotes maturation of oligodendrocytes previously exposed to toxic levels of glutamate (Wood et al., 2007b). Furthermore, IGF-1 in combination with glial cell-derived neurotrophic factor completely rescues motor neurons from glutamate-induced toxicity in the organotypic spinal cord model of ALS (Bilak and Kuncl, 2001; Bilak et al., 2001). The possibility that the glutamate-induced neurodegeneration plays a role in SMA pathogenesis and IGF-1 protects motor neurons from the detrimental effects of glutamate is still an open question in the SMA field.
The critical path forward for this and many gene therapy applications involves examining activity in a vector that efficiently transduces motor neurons. Recently, AAV9 has been shown to pass the blood–brain barrier and infect motor neurons in neonates following an intravenous injection (Foust et al., 2009, 2010). Recently, this group has delivered self-complementary AAV9 containing full-length SMN in the temporal vein of SMAΔ7 mice at PND 1. This treatment rescued the SMA phenotype by restoring motor function and neuromuscular physiology and extended survival up to 250 days (Foust et al., 2010). Interestingly, a therapeutic window was clearly present, because administration of the AAV9-SMN vector at PND 5 and beyond did not rescue the phenotype. This is likely due in part to the development of the blood–brain barrier; however, disease development could also impact the efficacy. An alternative study demonstrated the magnitude of benefits using self-complementary AAV versus single-stranded AAV. Single-stranded AAV8 expressing human full-length SMN was injected ICV into the SMAΔ7 mouse model and extended life to ∼50 days. Interestingly, expression of SMN through self-complementary AAV8 extends the survival up to 157 days (Passini et al., 2010). Delivery of the trans-splicing cassette and neuroprotectants will need to be examined in the self-complementary AAV9 context using intravenous injection to fully evaluate their activity in vivo, as this type of delivery route is especially attractive in future clinical trials.
Footnotes
Acknowledgments
The authors would like to thank John Marston and Tristan H. Coady. This work was funded by SMA Europe (M.S.) and FightSMA (C.L.L.) grants.
Author Disclosure Statement
There are no competing or financial interests to disclose.