
Abstract
This review describes the historical emergence of the concept of bone marrow mesenchymal stem cells (MSCs), summarizing data on Wolf and Trentin's hematopoietic inductive microenvironment; Dexter's hematopoiesis-supportive stromal cells; Friedenstein's osteogenic cells; and Pittenger's trilineal osteoblastic, chondrocytic, and adipocytic precursors; to finally introduce the specific bone marrow mesenchymal stem cells with differentiation potential to four lineages (mesenchymal and vascular smooth muscle lineages), and stromal and immunomodulatory capacities. Two points are the object of detailed discussion. The first point envisions the stem cell attributes (multipotentiality, self-renewal, tissue regeneration, population heterogeneity, plasticity, and lineage priming) compared with that of the paradigmatic hematopoietic stem cell. In the second point, we discuss the possible existence of bone marrow cells with greater differentiation potential, eventually pluripotential cells. The latter point raises the issues of cell fusion, reprogramming, or selection under nonstandardized conditions of rare populations of neuroectodermal origin, or of cells that had undergone mesenchymal-to-epithelial transition. In the last section, we review data on MSC senescence and possible malignant transformation secondary to extensive culture, gene transfer of telomerase, or mutations such as leading to Ewing's sarcoma. The set of data leads to the conclusion that bone marrow MSCs constitute a specific adult tissue stem cell population. The multiple characteristics of this stem cell type account for the versatility of the mechanisms of injured tissue repair. Although MSC administration may be extremely useful in a number of clinical applications, their transplantation is not without risks that must not be overlooked when developing cell therapy protocols.
The Hematopoietic Supportive Stromal Cell
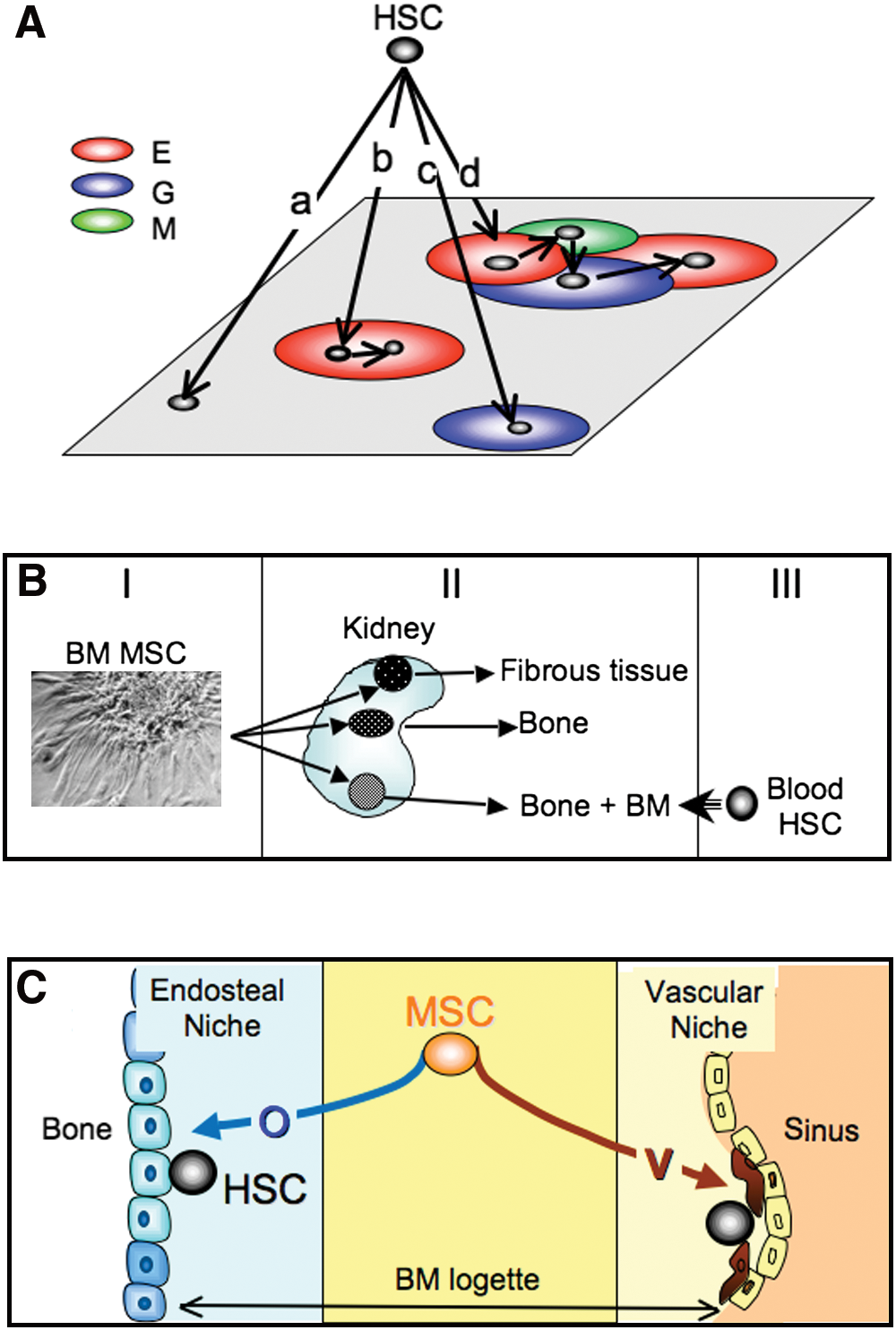
Historical overview. (
In 1977, Dexter and colleagues described bone marrow long-term cultures (Dexter et al., 1977). In this system, the maintenance over time of mouse HSCs was strictly dependent on the generation of an adherent layer of stromal cells. Various investigators adapted the system to human bone marrow (Gartner and Kaplan, 1980). In the following years, systems were made more analytical because various populations of hematopoietic precursors (including HSCs) were seeded on preformed layers of stromal cells. Stromal cells were isolated according to their membrane antigen expression pattern (Simmons and Torok-Storb, 1991). Alternatively, continuous lines generated from bone marrow of mice or humans served as feeders (review in Zipori, 1989).
These systems demonstrated the requirement of stromal cells for HSC maintenance. The model of the stem cell niche illustrated this requirement (Schofield, 1978). In this model the HSC must be in physical contact with the stromal cell, or with its associated extracellular matrix (ECM), to maintain the adequate balance (“stemness”) between self-renewal and commitment to the various blood lineages (Moore and Lemischka, 2006).
The model of the niche has been generalized to almost all types of stem cells (Spradling et al., 2001). In the Drosophila ovariole, the physical association between a “hub” stromal cell and a germinal stem cell could be visualized. On division, the daughter cell that lost its contact with the hub cell became committed to the germinal lineage, whereas the daughter cell that kept its contact remained a stem cell. Several publications have described two HSC niche models (reviews in Kopp et al., 2005; Adams and Scadden, 2006). In the osteoblastic niche model the stromal component is close to the endosteal lining or may be a subtype of osteoblasts. In the vascular niche model the stromal component is a cell constituting the vascular sinus.
A large number of regulatory pathways are operative in the molecular control of HSC by stromal cells (reviews in Charbord, 2001, 2004). Adhesive pathways are essential, be it homophilic adhesion such as cadherin/cadherin, heterophilic adhesion such as very late antigen-4/vascular cell adhesion molecule-1 (VLA4/VCAM1), or cell–ECM adhesion such as integrins to fibronectins or collagens. Cytokine or morphogen pathways are also crucial, stromal cells expressing a wide range of early-acting cytokines or morphogens such as hedgehog, notch, or Wnt families members. A point to be emphasized is the overlap of, and redundancy in, the pathways, accounting for fine-tuning and robustness, features characteristic of network systems (Kitano, 2002).
A critical issue for this review is that of the differentiation program and origin of the stromal cells. At the time when long-term marrow cultures were developed two major contending hypotheses were raised.
The most challenging was that stromal cells were the progeny of HSCs, as suggested by simultaneous growth of HSCs and stromal cells in long-term marrow cultures. However, identification of the stromal/hematopoietic precursor has proven difficult. One study showed that rigorously cloned stromal cells generated, after infection with entire simian virus 40 (SV40) virus, both CD45+/CD34+ round cells and vimentin+/smooth muscle (SM)-actin+ elongated cells (Singer et al., 1987). However, in the absence of colony-forming hematopoietic progenitors, any conclusion about the hematopoietic nature of the round cells remained uncertain. Another study claimed to have purified to homogeneity a population of CD34+/HLA-DR+/CD38– cells with stromal and hematopoietic potential (Huang and Terstappen, 1994). However, data were subsequently retracted because colonies proved to be generated by more than one clonogenic cell. Is the hypothesis of a common stromal/hematopoietic precursor completely disqualified? There are still a few reports that support it (Ogawa et al., 2006; Menendez et al., 2009). In particular, mouse multipotent adult progenitor cells (MAPCs) generated by Catherine Verfaillie's team (Jiang et al., 2002) were shown to give rise to HSCs with multilineage hematopoietic engraftment of immunodeficient mice (Serafini et al., 2007). MAPCs have been difficult to reproduce in other laboratories. However, taken together the data leave open the possibility that there exists within stromal layers a very minor bipotential population that could be selected under specific culture conditions.
The mesenchymal origin of the stromal cells had been advocated early on. The capacity of the mesenchyme to synthesize and assemble the ECM accounts for the inclusion as mesenchymal cells of connective tissue-forming cells such as dermal and interstitial tissue fibroblasts, osteoblasts, chondrocytes, adipocytes, and smooth muscle cells. These cells are, for the most part, but not exclusively (as discussed below), of mesodermal origin. Many stromal lines were described as preadipocytic cells filled with fat-laden vesicles (Zipori, 1989). We have described the vascular smooth muscle differentiation of human and murine stromal cells/lines acquiring with time in culture vascular smooth muscle cytoskeletal markers, up to the most specific alternatively spliced isoforms of caldesmon (h-caldesmon) and of smooth muscle myosin heavy chains (SM1 and SM2) (Galmiche et al., 1993; Remy-Martin et al., 1999). Other reports have shown the presence of osteoblasts within stromal layers (Long et al., 1990). Collectively, these data allow the definition of stromal cells as mesenchymal cells, which does not specify the stem cell type of origin.
The Tripotential Adipocytic, Osteoblastic, and Chondrocytic Precursor Cell
Definitive evidence that bone marrow includes cells that can generate connective tissue-forming cells was originally provided by the pioneering work of Alexander Friedenstein, summarized in Fig. 1B (review in Friedenstein et al., 1970). In the 1960s, this investigator made a number of experiments pointing to the existence within the bone marrow of various species of a precursor for osteoblasts and fibrous tissue. One to 2 weeks after seeding bone marrow cells at low density in liquid cultures containing serum, he observed discrete colonies consisting of plastic-adherent, nonphagocytic, elongated cells of fibroblastic appearance. The clonogenic cell at the origin of each colony was called colony-forming unit-fibroblasts (CFU-f ). CFU-fs were shown, by suicide technique and cell cycle analysis, to be quiescent in vivo and at culture inception. Each colony seeded under the renal capsule of semisyngeneic animals gave rise, a few weeks after transplantation, to fibrous tissue, to bone and to bone containing bone marrow. Using chimeric animals, Friedenstein further showed that marrow hematopoietic cells within the bony spaces were of recipient origin, in contrast to bone cells or fibrous tissue, which were from the donor. These latter data suggested that some of the transplanted colonies constituted an adequate microenvironment for HSC homing and subsequent hematopoiesis, reinforcing therefore the hypotheses of the hematopoietic-inductive microenvironment and HSC niche.
In the 1980s and 1990s, other work extended these observations and made clear that the cells identified by Friedenstein were multipotent and could give rise to osteoblasts, chondrocytes, and adipocytes (review in Prockop, 1997). In 1999, Mark Pittenger showed that trilineal adipocytic, osteoblastic, and chondrocytic clones were present in human bone marrow and provided a membrane antigen analysis of the cells indicating that few endothelial or hematopoietic specific markers were expressed (Pittenger et al., 1999). Later studies asserted that the MSC population was clearly distinct from that of endothelial or hematopoietic cells (Delorme et al., 2008).
Since their initial description, these multipotent bone marrow cells have been given different names. Friedenstein used the term osteogenic stem cells, whereas Maureen Owen called them marrow stromal stem cells to emphasize that they generate stromal cells in long-term cultures (Owen et al., 1987). Arnold Caplan was the first to introduce the term mesenchymal stem cells (Caplan, 1991), which has become popular in the current literature. However, Paolo Bianco and Pamela Robey have suggested the alternative term skeletal stem cells to underscore their potential to give rise to the cellular components of the skeleton (Bianco et al., 2008), and James Dennis has suggested the name mesenchymal progenitor cells, arguing that these cells were not bona fide stem cells (Dennis et al., 1999). Following a similar line of reasoning, the International Society for Cellular Therapy has proposed the term multipotent mesenchymal stromal cells (Horwitz et al., 2005).
Taken together, the data indicate that mesenchymal precursors are present in the bone marrow of multiple species. These cells can be extensively amplified in vitro, which allows their use in cell therapy applications.
The Mesenchymal Stem Cell
The minimal criteria to qualify adult tissue stem cells are that they constitute an immature population of heterogeneous, self-renewing cells able to regenerate, after injury, their tissue of origin (they are multipotential if the tissue consists of several cell types). An additional criterion is the flexibility of the stem cell attributes, for example, that they can shift from quiescence to a proliferative state or that differentiation can be reversed at least up to a certain stage (Loeffler and Potten, 1997; Loeffler and Roeder, 2002).
May the Trilineal Mesenchymal Precursor Be Qualified as a Stem Cell?
Multipotentiality is one of the hallmarks of these cells. Clones can be differentiated under appropriate conditions not only into adipocytes, chondrocytes, and osteoblasts, but also into vascular smooth muscle cells (Kashiwakura et al., 2003; Kobayashi et al., 2004; Jeon et al., 2006; Kim et al., 2008; Delorme et al., 2009; Kurpinski et al., 2010). Moreover, these cells may differentiate into tenocytes as inferred from in vivo studies (Hoffmann et al., 2006), although evidence in vitro at the clonal level is at present lacking.
In 2007, Paolo Bianco's team showed that clonogenic human CD146+/CD90+ CFU-fs were able to self-renew (Sacchetti et al., 2007). One colony of such phenotype was implanted subcutaneously into an immunodeficient mouse. After 8 weeks the implant was retrieved and the CD146+/CD90+ minor population was sorted and cultured at cloning density. Two colonies of the same phenotype were obtained. Because it is generally agreed that the ability of a stem cell population to regenerate cells in vivo with characteristics identical to the initially explanted fraction practically reflects its self-renewal capacity, these data indicate that MSCs are able to self-renew. More extended self-renewal capacity, requiring sequential passages to secondary and even tertiary recipients, has yet to be evidenced. However, there are no data supporting the notion that the in vivo serial repopulation assay is either feasible or entirely applicable in the case of MSCs, and it might not therefore represent the most appropriate in vivo model for determining their functional potential. Indeed, MSCs need to be cultured extensively before transplantation to attain sufficient numbers to regenerate a bone or cartilage defect. In addition, the turnover rate of the mesenchymal tissue is much lower than that in hematopoiesis, which complicates the experimental setting (Bianco and Gehron Robey, 2000). Self-renewing mouse MSCs appear to be of smaller size and to express a series of surface antigens not expressed by other culture-amplified cells (Colter et al., 2001). Sca-1/Ly-6A, another membrane murine MSC marker, has proven essential for maintenance of the self-renewal capacity (Bonyadi et al., 2003).
Numerous in vivo experiments in animals and a few clinical trials in humans have shown that MSCs are able to regenerate bone and cartilage. Particularly demonstrative were experiments in the mouse model of osteogenesis imperfecta (OI). One to 4 months after systemic injection, into normal irradiated mice, of MSCs expressing a human minigene for collagen I, cells with the characteristic collagen were found in bone, bone marrow, and cartilage, and infusion of wild-type MSCs into OI mice improved bone mineralization and collagen synthesis (Pereira et al., 1995, 1998; Li et al., 2007). MSCs also contribute to the formation of vessels in part because of their capacity to differentiate into vascular smooth muscle cells (Au et al., 2008; Dufourcq et al., 2008).
MSCs constitute a heterogeneous population of immature cells as suggested by the multimodal expression (in flow cytometry studies) of membrane antigens specific for nondifferentiated cells, such as CD146 or CD200 (Delorme et al., 2008). However, heterogeneous expression of certain markers can also be observed in cells from clones, which may be due to gene or network noise (Kaern et al., 2005). This is why heterogeneity is optimally assessed in functional studies, which have revealed great clonal heterogeneity in terms of proliferative capacity and differentiation potential (Pittenger et al., 1999; Muraglia et al., 2000). Moreover, the generation of bonelike tissue in vitro does not appear to reflect the capacity to form bone in vivo, and only a fraction of MSC clones are able to generate bone in vivo (Bianco et al., 2008).
Flexibility of MSC attributes is exemplified by their differentiation plasticity. It is well known that mesenchymal cells can shift from one differentiation pathway to another under modified external conditions. Chondrocytes in the growth plate undergo hypertrophy before turning into osteoblasts. Rocky Tuan's team has shown that cloned osteoblasts can turn into chondrocytes or adipocytes (Song and Tuan, 2004; Song et al., 2006), and we have shown that cloned MSCs differentiated into vascular smooth muscle cells can turn into adipocytes, osteoblasts, or chondrocytes (Delorme et al., 2009). Lineage conversion may proceed via a dedifferentiation process (Song et al., 2006). To some extent, plasticity and self-renewal are two opposing attributes because the potential for a cell among the progeny of a stem cell to reacquire its full differentiation potential negates the need for an indefinite self-renewal capacity to maintain the stem cell pool. Indeed, pedigree hierarchical models of stem cell differentiation, in which stem cells are preordained entities differentiating into discrete populations of progenitors and differentiated cells, are not compatible with plasticity (Loeffler and Roeder, 2004). Other models that account for the capacity of dedifferentiation may apply, such as the phase space model (Kirkland, 2004; Morad et al., 2008) or a model based on the concept of noise-driven stem cell differentiation (Krinner et al., 2010).
Lineage priming, another property of stem cells shared by MSCs (Delorme et al., 2009), is a molecular model for stem cell differentiation in which self-renewing stem cells, not induced to differentiate, express a subset of genes associated with the differentiation pathways to which they can commit (Hu et al., 1997). Of major interest is the expression of key transcription factors for differentiation, such as, for HSCs, PU.1 and GATA1 for myeloid and erythroid differentiation, respectively (Huang et al., 2007; Huang, 2009). Analysis of individual HSCs has revealed variable expression of these transcription factors from one cell to another, which might reflect the high level of noise characteristic of stem cells. Induction of differentiation is marked by the decline of one factor at the expense of the other (e.g., downregulation of PU.1 and upregulation of GATA1 as HSCs undergo erythroid differentiation). Sui Huang has posited that a similar process occurs in MSCs (e.g., RUNX2 [runt-related transcription factor-2] downregulation and PPARG [peroxisome proliferator-activated receptor-γ] upregulation in the adipogenic condition), which remains to be tested at the single cell level (Huang, 2009).
Taken together, the data indicate that MSCs are bona fide stem cells. However, HSCs and MSCs may constitute two extremes among adult tissue stem cells: in HSCs self-renewal appears to be the cardinal property whereas in MSCs plasticity would be the predominant feature (Zipori, 2005).
A Unifying Concept: MSCs as Multipotential Stem Cells with Stromal and Immunomodulatory Capacities
That MSCs support hematopoiesis and are implicated in the regulation of the immune system indicates properties beyond those implied by stemness.
As already alluded to, in vivo HSC niches may be of two kinds: osteoblastic and vascular. Transplantation of human MSCs into immunodeficient mice has shown that the vascular component is the pericyte located on the abluminal side of the endothelial cell (Sacchetti et al., 2007). Four weeks after transplantation of human CD146+ bone marrow CFU-Fs into immunodeficient mice, the few human cells that retained the expression of CD146 were located on the abluminal side of mouse-derived endothelial cells forming incipient sinuses. By week 8, foci of hematopoietic cells were clearly associated with the CD146+ parasinusal cells. These cells are anatomically and morphologically similar to alkaline phosphatase+ adventitial reticular cells (ARCs) described in the 1960s (Westen and Bainton, 1979; Lichtman, 1981) and to parasinusal “myoid” cells expressing α-SM actin (Schmitt-Graff et al., 1989) found in fetal and adult human bone marrow (Galmiche et al., 1993; Charbord et al., 1996). As observed with ARCs and pericytes, parasinusal myoid cells extend cytoplasmic processes deep into the marrow space, making contact with numerous hematopoietic cells. Remarkably, CD146+ cells are α-SM actin+ and express additional smooth muscle markers such as calponin, desmin, and leiomodin. However, direct and regular overlap in situ between alkaline phosphatase+ cells and α-SM actin+ cells has not been demonstrated. Pericytes from sites other than bone marrow may also give rise to MSC-like cells (Crisan et al., 2008).
HSC niches can be easily conceived within the framework of MSC differentiation (Fig. 1C) because MSCs give rise to both osteoblasts and pericytes that belong to the family of vascular smooth muscle cells (reviews in Sims, 1986; Hirschi and D'Amore, 1996; da Silva Meirelles et al., 2008). Other evidence of the role of MSCs in HSC niche formation has been provided by Muguruma and colleagues, who reported observations made after intrabone injection of labeled culture-amplified human bone marrow MSCs into immunodeficient mice (Muguruma et al., 2006). Ten weeks after transplantation 60% of the labeled cells were α-SM actin+ and located in the vicinity of sinuses, and 30% were alkaline phosphatase+ and located primarily in the endosteal region. Transplantation of human cord blood cells after that of MSCs revealed the frequent interaction of MSC progenies and CD34+/CD38– hematopoietic precursors.
The immunomodulatory property is likewise a property unrelated to stemness. Various reports have shown that dermal fibroblasts are able to suppress the mixed lymphocyte reaction similarly to bone marrow MSCs, leading to the hypothesis that immunosuppression is a property of the mesenchyme whatever its location (Haniffa et al., 2007, 2009; Jones et al., 2007).
Taken together, the data suggest that MSCs represent a specific type of adult tissue stem cell that, in addition to stem cell attributes, has the capacity to support hematopoiesis and to be immunosuppressive (Fig. 2). The stromal property might be specific for bone marrow MSCs, whereas the immunomodulatory capacity might be shared by mesenchymal cells distributed throughout the body.
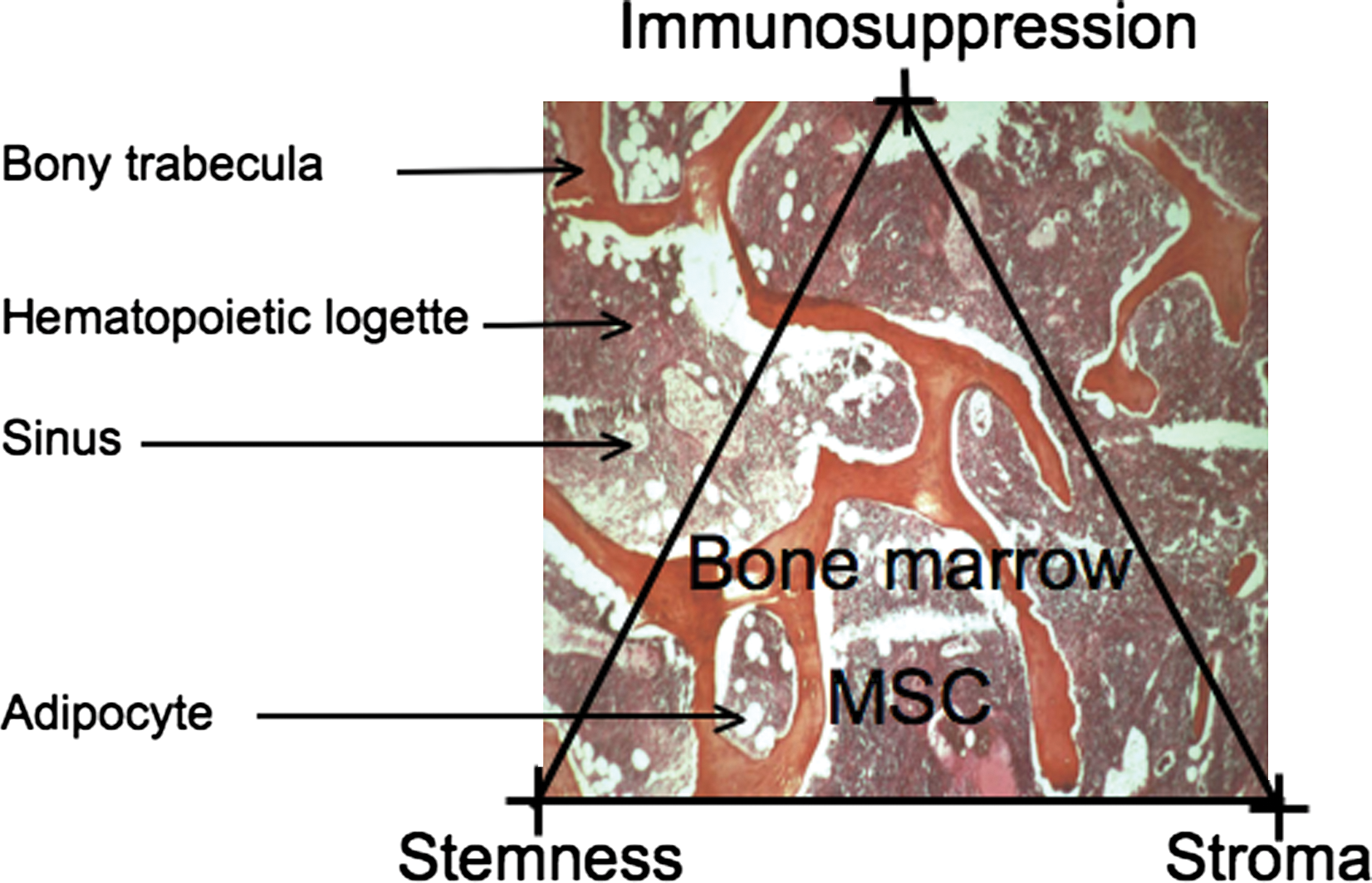
The MSC in its context. There is no precise information about the location of the MSCs in the marrow logettes; what is known is their progeny: osteoblasts, abluminal cells in the marrow sinuses and adipocytes. The three major properties of the MSC are emphasized: stemness, stromal capacity (stroma), and immunosuppressive potential (immunosuppression). Color images available online at
A Step Further: Are MSCs Pluripotent Cells?
In addition to the mesenchymal lineages described previously, MSCs have been reported to give rise to endothelial cells (Oswald et al., 2004), skeletal and cardiac muscle cells (Umezawa et al., 1992; Wakitani et al., 1995; Makino et al., 1999; Schulze et al., 2005; Rose et al., 2008), neural cells (Cho et al., 2005; Wislet-Gendebien et al., 2005; Tropel et al., 2006; Trzaska et al., 2007; Tondreau et al., 2008), hepatocytes (reviews in Prindull and Zipori, 2004; Snykers et al., 2009), and epithelial cells (Spees et al., 2003; Phinney and Prockop, 2007).
These reports raise a number of critical issues: fusion in vivo, criteria for differentiation, capacity for reprogramming, and selection by nonstandardized culture conditions of rare cell populations.
Some of the differentiations observed in vivo may be due to fusion with cells in contact with MSCs, which is the case, in one report, for hepatocytes, cardiomyocytes, and Purkinje neurons (Alvarez-Dolado et al., 2003). Similar cases of fusion have been reported with other types of adult tissue stem cells, primarily HSCs.
Criteria for differentiation need to be rigorously defined. It appears difficult to conclude as to a differentiation process from the expression of a number of markers without that of the key transcription factors. For example, we have found that MSCs expressed cytoskeletal proteins usually expressed in neural stem cells (nestin), hepatocytes (cytokeratin-8 and -18), biliary cells (cytokeratin-19), and sarcomeric muscle (troponins, α-C-actin), without expression of proneural or neuronal, prohepatocytic, or myogenic key transcription factors (Delorme et al., 2009). Although cytoskeletal markers remain adequate indicators of a differentiation pathway, there are numerous exceptions to the rule, such as the expression of cytokeratin-18 in vascular smooth muscle cells in the synthetic phase. Similar misleading expression of factors has also been reported by other investigators (Montzka et al., 2009).
Some of the observed differentiations may result from reprogramming. Dezawa and colleagues have shown that rodent and human bone marrow MSCs can be reprogrammed into cells with skeletal muscle potential after specific treatment comprising first cytokines and then gene transfer of the notch intracellular domain (Dezawa et al., 2005). The cells thus treated expressed master skeletal transcription factors and were able to regenerate muscle after transplantation in animal models of muscle injury. Remarkably, the cells could not be reprogrammed if the treatment sequence was reversed (gene transfer and then cytokines administration). Similar reprogramming may take place when treating cells with DNA-demethylating agents such as 5-azacytidine, a procedure often used to induce sarcomeric muscle differentiation (Wakitani et al., 1995; Makino et al., 1999), and delivery of key transcription factors has also been used to reprogram MSCs into neural cells, pancreatic beta cells, and endothelial cells (review in Barzilay et al., 2009).
When using standardized culture conditions set up within the European FP6 research program “Genostem,” we have generated human bone marrow MSCs whose differentiation lineages were restricted to the four lineages usually characterizing MSCs (Charbord et al., 2010). This does not preclude the obtaining, by other culture methods, of a rarer population of cells with greater differentiation potential. Many factors may contribute: extensive screening of the serum batch, strict control of the degree of confluency, oxygen tension, stress such long-term trypsin incubation, and so on.
Some of the amplification protocols may select for MSCs of neuroectodermal origin that can differentiate into neural cells. There are many arguments indicating that some MSCs may derive from the neuroectodermal layer. Cell-tracking studies have shown that the connective tissue above the aortic arch derives from the neuroectoderm. Neural crest stem cells generated from embryonic stem cells can give rise under appropriate conditions to vascular smooth muscle cells, adipocytes, osteoblasts, and chondrocytes (Lee et al., 2007). The neuroepithelium supplies the earliest wave of MSC differentiation in the mouse embryo (Takashima et al., 2007). Last, few mouse bone marrow MSCs have been proven to be neuroectodermal in origin (Nagoshi et al., 2008; Morikawa et al., 2009). Whether anti-nerve growth factor receptor (NGFR)/CD271 or anti-GD2 antibodies select for these neuroectodermal precursors in human bone marrow remains an open question (Quirici et al., 2002; Martinez et al., 2007).
Another population that may be selected under special conditions is that of pluripotent cells, which are akin to embryonic stem cells regarding their differentiation potential and even gene expression (Jiang et al., 2002; Beltrami et al., 2007; Greco et al., 2007; Kuroda et al., 2010). This is the case for MAPCs giving rise to mesodermal (including mesenchymal), neuroectodermal, and endodermal lineages. Some strains express telomerase and Octamer-4/POU domain class 5 transcription factor-1 (OCT4/POU5F1), a transcription factor essential for the maintenance of pluripotency in embryonic stem cells. Other investigators have reported the expression in bone marrow MSCs of the pluripotency core transcription factors OCT4, NANOG, and SOX2, and have found similar regulatory circuitries for OCT4 in MSCs and embryonic stem cells (Greco et al., 2007).
Taken together, the data indicate that the culture procedure may select for cells with significant differentiation potential. Some of the differentiations are compatible with the mesodermal origin of the MSCs whereas others fit with the well-evidenced neuroectodermal origin of an MSC subpopulation. Differentiation into cells of endodermal origin might indicate that MSCs have undergone a mesenchymal-to-epithelial transition (MET), an instance common during development but less frequent in the adult (Prindull and Zipori, 2004). Indeed, such METs have been described in MSC-like cells generated from the fetal liver (Chagraoui et al., 2003; Dan et al., 2006; Inada et al., 2008). Last, the generation of pluripotent cells might result from the selection of rare vestigial embryonic stem cells having homed to the bone marrow. Alternatively, this population might result from reprogramming, similar to what occurs for induced pluripotent cells from dermal fibroblasts (Takahashi and Yamanaka, 2006).
Senescence and Transformation of MSCs
MSCs present a high capacity of proliferation in vitro. However, many data suggest that MSCs cannot proliferate beyond the Hayflick limit of approximately 50 population doublings (PDs). This limit usually takes into account the total number of cell divisions that had occurred in vivo and after culture initiation. This might explain why cells in culture become senescent and why senescence may occur earlier in cultures from older individuals.
Senescent MSCs are characterized by a flat hypertrophic phenotype with accumulation of β-galactosidase+ cells. Several studies have pointed out a number of mechanisms responsible for senescence: karyotypic abnormalities (Izadpanah et al., 2008), reduction in telomere length (Banfi et al., 2002; Baxter et al., 2004), induction of expression of the cyclin inhibitor p16 INK4A (Shibata et al., 2007; Lee et al., 2009), activation of progerin, a mutant form of lamin A and subsequent activation of the notch pathway (Scaffidi and Misteli, 2008), induction of microRNAs that may impinge on cell cycle factors (Wagner et al., 2008), hypermethylation of promoters of the polycomb complex target genes (Teschendorff et al., 2010), and reduction of antioxidant production and decrease in actin microfilament formation dynamics (Kasper et al., 2009; Lee et al., 2009).
Telomerase is usually not detected in MSCs (Banfi et al., 2002; Guillot et al., 2007; Serakinci et al., 2008). Expression of telomerase has been shown to slow down or abrogate the process of senescence. Remarkably, although extending the life span beyond 50 PDs, it also maintains or increases the osteogenic and stromal potential of the cells (Shi et al., 2002; Simonsen et al., 2002; Kawano et al., 2003). Telomerase therefore proved able to immortalize MSCs without the chromosomic instability and crises characteristic of transfection with oncogenes such as those encoding H-Ras, T antigen of SV40, or E6/E7 of papilloma virus (Kawano et al., 2003).
However, telomerase gene transfer may also lead to malignant transformation. This has been shown by Moustapha Kassem's team, which observed in extensively passaged telomerase+ lines the deletion of the p16 INK4A/ARF locus, an activating mutation of K-RAS, and expression of cancer germline antigens (Serakinci et al., 2004; Gjerstorff et al., 2009). These data indicate that cells into which telomerase has been transferred represent an adequate model with which to study transformation and some of the differentiation pathways, but do not constitute an appropriate cell source for cell therapy.
Ex vivo amplification of MSCs may lead to clonal selection and subsequent malignant transformation. The frequency of this process appears to be high in mouse MSCs, where extensive passaging leads to cytogenetic aberrations and sarcoma development (Miura et al., 2006; Aguilar et al., 2007; Tolar et al., 2007). Spontaneous transformation has also been reported in human MSCs even after the cells had reached the postsenescence phase (Rubio et al., 2005; Rosland et al., 2009). However, one report suggests that the transformation reported by Rubio and colleagues (2005) was an artifact resulting from contamination with another tumor epithelial cell line (Garcia et al., 2010). Moreover, karyotypic abnormalities are not consistently observed in transformed cells; on the contrary, aneuploidy does not implicate that a malignant clone is generated (Tarte et al., 2010).
This latter conclusion does not rule out concerns about potential transformation. Strong arguments indicate that the fusion gene EWS–FLI1, resulting from translocation of the EWS DNA-binding domain with the ETS gene family member FLI1, may induce the transformation of MSCs into Ewing's sarcoma cells (Tirode et al., 2007; Burns et al., 2008; Kauer et al., 2009). Tirode and colleagues have shown that the profile of Ewing's sarcoma lines converged to that of normal MSCs after EWS–FLI1 abrogation. Moreover, silenced lines could recover part of their differentiation potential, primarily toward adipocytes and osteoblasts. A population of CD133+ tumor cells appears to constitute the Ewing's sarcoma stem cells because they are able to generate the tumor in serial transplantations in immunodeficient mice (Suva et al., 2009). Remarkably, the CD133+ cell population retains its mesenchymal differentiation potential and expresses higher levels of OCT4/POU5F1 and NANOG than the CD133– subset. Other data have indicated that Ewing's sarcoma cells express neural markers, which is compatible with the transformation of neuroectodermally derived MSCs or might result from EWS–FLI1-driven neuroectodermal differentiation in transformed MSCs (Riggi et al., 2009). Last, transformed MSCs, either spontaneously or after transfer of the telomerase gene, express the CD99 antigen characteristic of Ewing's sarcoma cells (Burns et al., 2008; Rosland et al., 2009).
Ewing's sarcoma is not the only cancer type that may be generated by MSCs (Tang et al., 2008). In particular, the fusion gene SYT–SSX1 induces in transduced MSCs a transcriptomic profile similar to that of synovial sarcoma (Cironi et al., 2009). It has also been reported that some gastric cancers may originate from MSCs (Houghton et al., 2004); in these cases MSCs might have fused with gastric mucosa cells infected with Helicobacter pylori.
As for senescence, the mechanisms of transformation appear to be diverse: induction of telomerase, deletion of CDKN2A/p16 INK4A , an activating mutation of the K-RAS gene (Serakinci et al., 2004; Miura et al., 2006; Burns et al., 2008; Rosland et al., 2009), and loss of p53 in the absence or not of the cyclin inhibitor CDKN1A/p21 CIP1/WAF1 (Armesilla-Diaz et al., 2009; Rodriguez et al., 2009). Remarkably, similar mechanisms are implicated in the induction of pluripotent cells, underscoring the relationship between reprogramming, aging, and cancer (Krizhanovsky and Lowe, 2009).
Taken together, the data suggest that MSCs are able to transform into malignant cells. Sarcomas of diverse types may represent for mesenchyme what leukemias represent for hematopoiesis. Target cells in each system may range from most immature cells (MSCs or HSCs) to mesenchymal or hematopoietic cells at various stages of differentiation.
Future Considerations
A systems biology approach needs to be taken to understand the dynamics of the stem cell attributes of MSCs. In particular, the molecular basis for plasticity remains poorly understood. Plasticity may be related to lineage priming, because differentiation in the primed lineages would not entail the setup of a whole molecular program but the upregulation of only a few of the program components. Moreover, lineage priming indicates that promoters of the key transcription factors are in an open configuration, and therefore accessible to certain chromatin remodelers/transcriptional coactivators, such as SETDB1 (SET domain protein bifurcated-1) (Takada et al., 2007) or TAZ (transcriptional coactivator with PDZ-binding motif ) (Hong et al., 2005), that modify the balance in favor of one or the other pathway. Further studies on such molecules, studies of the epigenetic status of the promoters of key transcription factors before and during differentiation (such as reported in Noer et al., 2009, for adipose-derived stem cells), and the development of adequate biomathematical models are required at this step.
It is essential to understand in each clinical setting the precise mechanism of repair in order to optimize the procedure of administration (route, schedule, dose, pretreatment with cytokines or chemokines, etc.). It is also critical to standardize amplification procedures such that cells with similar properties are delivered to each patient, and to make possible the comparison of clinical results. The multiple characteristics of the MSCs account for the versatility of the mechanisms of injured tissue repair (Phinney and Prockop, 2007). Unlike HSCs, it is only in a few situations that regeneration by repopulation and differentiation is taking place; for example, MSCs implanted in bone or cartilage lesions may repair by direct differentiation into osteoblasts or chondrocytes. In other disease models, including vascular diseases, cardiac infarcts, or immune diseases, benefit from MSC injection might result from the secretion of cytokines that would induce the proliferation and/or differentiation of nearby differentiated cells or resident stem cells. Alternatively, because of the known immunomodulatory properties of MSCs, MSC-dependent repair might be secondary to impairment of migration and proliferation or increased apoptosis of inflammatory cells. Last, it remains possible that reprogramming occurs in certain situations, after fusion with local cells, or because of a specific cytokinetic context of the diseased/injured tissue.
Last, one must not overlook the potential risks of MSC administration. The phenotypic instability related to plasticity raises concerns about the outcome of transplanted cells that may undergo unwanted and deleterious differentiation after implantation/engraftment at specific sites (Breitbach et al., 2007). The transformation potential is also a major concern. The mechanisms of transformation need to be understood (Richter et al., 2009; Riggi et al., 2010) so as to define strict release criteria for culture-amplified cells with minimal risk of transformation.
In conclusion, bone marrow MSCs constitute a specific adult tissue stem cell population. Although MSC administration may be extremely useful in a number of clinical applications, their transplantation is not without risks that must not be overlooked when developing cell therapy protocols.
Footnotes
Acknowledgments
This work was supported by the European Community (Key Action 1.2.4-3 Integrated Project “Genostem,” contract 503161) and by a grant from the Institut National du Cancer (INCA Project “Bortes”).
Author Disclosure Statement
The author declares no conflict of interest.