
Abstract
Adoptive cell therapy using T-cell receptor (TCR)–engineered T cells is a clinically feasible and promising approach to target tumors, but is currently faced with compromised antitumor efficacies in patients. Here, we extensively validated immune-deficient mice to facilitate further development of the therapeutic potential of TCR-engineered T cells. Treatment of human melanoma-bearing SCID or NSG mice with high doses of human T cells transduced with an hgp100/HLA-A2-specific TCR did not result in antitumor responses irrespective of chemotherapeutic preconditioning. Imaging of human green fluorescent protein–labeled T cells demonstrated significant T-cell accumulation in intratumoral vasculature directly upon T-cell transfer, which was followed by loss of T cells within 72 hr. Peripheral persistence of human T cells was highly compromised and appeared related to T-cell differentiation. On the contrary, adoptive transfer (AT) of relatively low numbers of hgp100/HLA-A2 TCR-transduced mouse T cells resulted in rapid clearance of large established human melanomas. Unexpectedly and in contrast to reported studies with chimeric antibody receptor–engineered T cells, antitumor activity and homeostatic expansion of T cells were independent of TCR transgene as evidenced in two SCID strains and using two different human melanoma cell lines. Interestingly, the xeno-reactive melanoma response of mouse T cells appeared to be dictated by CD4+ tumor-infiltrating lymphocytes and did not require in vitro T-cell activation, retroviral gene transfer, or subcutaneous interleukin-2 support. Taken together, AT of human but not mouse T cells in human melanoma-bearing immune-deficient mice is in close accordance with clinical studies.
Introduction
Clinical results with TCR-engineered T cells are promising, but improvements are necessary to further develop clinical TCR gene therapy into an effective and safe treatment for cancer patients. Immune-deficient mice transplanted with a human tumor represent a widely recognized and successfully used model with human or mouse T cells gene-engineered with either chimeric antibody receptors (CARs) or TCRs. These preclinical models potentially provide a valuable basis to test novel strategies and translate receptor gene therapy to the patient. With respect to human hematological malignancies, such as lymphoma, CAR-engineered human T cells (CAR T cells) have been reported to effectively mediate antitumor responses in mice with various degrees of immune deficiency, i.e., severe combined immune-deficient (SCID), nonobese diabetic SCID (NOD.SCID), SCID Beige, and NOD.SCID/il2rg –/– (NSG) mice (Brentjens et al., 2003, 2007; Kowolik et al., 2006; Savoldo et al., 2007; Cheadle et al., 2008; Milone et al., 2009; Zhao et al., 2010). With respect to human solid tumors, high numbers of human CAR T cells injected intratumorally (i.t.) delayed tumor growth (Pinthus et al., 2003) or prolonged tumor-free survival (Gade et al., 2005) in immune-deficient mice. More interestingly, intravenously (i.v.) injected human CAR T cells inhibited growth of human solid tumors when mice were preconditioned with either cyclophosphamide or irradiation (Pinthus et al., 2004; Teng et al., 2004; Westwood et al., 2005; Zhao et al., 2009; Chekmasova et al., 2010; Craddock et al., 2010). Preconditioning appeared not to be required for tumor regression in NSG mice that were treated with an i.v. injection of T cells engineered with a CAR containing the CD137 costimulatory domain (Carpenito et al., 2009; Zhao et al., 2010; Song et al., 2011). In addition to human CAR T cells, in various studies mouse CAR T cells have been used successfully to target human solid tumors in nude or SCID mice without additional treatment (Hwu et al., 1995; Darcy et al., 2000; Haynes et al., 2001, 2002a,b). Despite numerous reports on the antitumor effects of CAR T cells, there exists only a limited number of studies on the antitumor effects of TCR-engineered T cells (TCR T cells) in immune-deficient mice. Two studies by Xue and colleagues reported that i.v. injection of human TCR T cells inhibited human lymphoma engraftment in NOD.SCID mice (Xue et al., 2005, 2010). Moreover, a single study by Bobisse and colleagues described that i.t., but not i.v., injection of human TCR T cells delayed growth of human melanoma in SCID mice (Bobisse et al., 2009). To our knowledge, there are no reports of mouse TCR T cells targeting a human tumor in immune-deficient mice. See Tables 1 and 2 for an up-to-date overview of reported studies on adoptive cell therapy with human and mouse T cells, respectively, genetically directed toward human tumors in immune-deficient mice.
ALL, acute lymphoblastic leukemia; BB, CD137 (4-1BB) costimulatory molecule; BC, breast carcinoma; BD-TCR, bidirectional TCR; BL, Burkitt lymphoma; CC, colon carcinoma; CML, chronic myeloid leukemia; EBV+LCL, Epstein Barr virus–positive large-cell lymphoma; EBV-CD30+ HD, EBV-CD30+ Hodgkin lymphoma; ErbB2, also known as HER2/Neu, human epidermal growth factor receptor 2; GC, gastric carcinoma; Ley, Lewis Y antigen; MEL, melanoma; MelanA, melanocyte antigen; MES, mesothelioma; MOv19, monoclonal antibody clone against human folate receptor α; OC, ovarian carcinoma; PC, prostate carcinoma; PSMA, prostate-specific membrane antigen; SS1, anti-mesothelin Fv; WT1, Wilms' tumor antigen.
This table provides a detailed overview of studies performed with either chimeric antibody-based receptor (CAR) or T-cell receptor (TCR)-engineered human T cells in human tumor-bearing immune-deficient mice. The studies are compared with respect to tumor cells, mouse strains, antigen-specific receptors, T-cell transfers, additional treatments, and antitumor responses.
Mouse strains: SCID, severe combined immune-deficient mice containing the Prkdc mutation; SCID Beige, mice containing the Prkdc and Beige mutation; NOD.SCID, mice containing the Prkdc mutation on a nonobese diabetic background; NOD.SCID-β–/–, NOD.SCID mice containing a b2m mutation; NSG, NOD.SCID mice containing an il2rg mutation.
Receptor format: CAR receptors are chimeric receptors that consist of various “building blocks” such as (domains of) Fc(ɛ)RIγ (in short γ) or CD3ζ (ζ), which are in some cases combined with costimulatory molecules such as CD28 (28) or 4-1BB (1BB). SS-TCR is a cysteine-modified TCR.
Route of T-cell administration: i.p., intraperitoneal; i.t., intratumoral; i.v., intravenous.
Time point(s) of T-cell transfer are after tumor transplant.
In case of an additional treatment, details are listed here per tumor model and reference: Raji cells, T-cell expansion on artificial antigen-presenting cells (AAPC) plus IL-15 (Brentjens et al., 2003); Daudi cells, 2.5 Gy total body irradiation (TBI) (Kowolik et al., 2006); autologous tumor cells and L428 cells, 230 cGy TBI and IL-2 support (Savoldo et al., 2007); Raji cells, cyclophosphamide (Cheadle et al., 2008); pre-B ALL cells, T-cell expansion on CD28 bead-based AAPC (Milone et al., 2009); CWR22 and WISH-PC14 cells, IL-2 support (Pinthus et al., 2003); COLO205 and MDA-MD-435 cells, 2.5 Gy TBI (Teng et al., 2004); WISH-PC14 cells and LuCap-35 cells, 2 Gy TBI and IL-2 support (Pinthus et al., 2004); OVCAR-3 cells, 2.5 Gy TBI (Westwood et al., 2005); BT-474 cells, cyclophosphamide and IL-2 support (Zhao et al., 2009); SK-23 cells, IL-2 support (Bobisse et al., 2009).
CD80+, but not CD80–, NALM-6 tumors were rejected.
CD19 CAR:28ζ, but not CD19 CAR:ζ, caused an antitumor response.
In this study, EBV-specific cytotoxic T cells were used as host T cells for receptor gene transfer, whereas in other studies polyclonal peripheral T cells were used.
When mice were not irradiated, receptor-engineered T cells did not cause an antitumor response.
When mice were irradiated or treated with cyclophosphamide, receptor-engineered T cells did not cause an antitumor response.
When mice received CAR T cells also engineered to express the chemokine receptor CCR2b, an antitumor response was observed.
In this study, T cells were electroporated with CAR-encoding mRNA.
MOv19 CAR:1-BBζ, but not CAR:ζ, caused an antitumor response.
CC, colon carcinoma; CEA, carcinoembryonic antigen; ErbB2, also known as HER2/Neu, human epidermal growth factor receptor 2; MOv18, mAb clone against human folate receptor α; OC, ovarian carcinoma.
This table provides a detailed overview of studies performed with chimeric antibody-based receptor (CAR)-expressing mouse T cells in human tumor-bearing immune-deficient mice. There are no in vivo studies reported so far with TCR-engineered mouse T cells directed toward human tumors in immune-deficient mice. The studies are compared with respect to tumor cells, mouse strains, antigen-specific receptors, T-cell transfers, additional treatments, and antitumor responses.
Mouse strains: Nude mice, athymic mice due to a mutation in the foxn1 gene; SCID, severe combined immune-deficient mice containing the Prkdc mutation.
Receptor format: CAR receptors are chimeric receptors that consist of various “building blocks” such as (domains of) Fc(ɛ)RIγ (in short γ) or CD3ζ (ζ), which are in some cases combined with costimulatory molecules such as CD28 (28).
Route of T-cell administration: i.p., intraperitoneal; i.t., intratumoral; i.v., intravenous.
Time point(s) of T-cell transfer after tumor transplant.
Tumor-infiltrating lymphocytes (TIL) derived from a mouse adenocarcinoma (MC38) tumor were used as host cells for receptor gene transfer, whereas in other studies polyclonal peripheral T cells were used.
T-cell transfers are given one to four times from day 0 until day 3.
Here, we evaluated immune-deficient mouse models to test the anti-human melanoma efficacy of adoptively transferred human as well as mouse T cells engineered with a human gp100/HLA-A2-specific TCR. High numbers of human TCR T cells were not efficacious against human melanoma cells in SCID or NSG mice irrespective of pretreatment with cyclophosphamide with or without busulfan. In contrast, mouse TCR T cells very efficiently cleared established human melanoma, but the antitumor responses and homeostatic expansion of mouse T cells were independent of the TCR transgene.
Materials and Methods
T cells, packaging cells, and melanoma cells
Peripheral blood mononuclear cells from healthy human donors were isolated by centrifugation through Ficoll-Isopaque (density=1.077 g/cm3; Amersham Pharmacia Biotech, Uppsala, Sweden). Transduced primary human T cells were cultured in RPMI 1640 medium supplemented with 25 mM HEPES, 200 mM L-glutamine, 10% human serum, antibiotics, and 360 IU/ml recombinant human interleukin-2 (IL-2) (Proleukin; Chiron, Amsterdam, The Netherlands) and stimulated every 2 weeks with a mixture of irradiated allogeneic feeder cells as described elsewhere (Van de Griend et al., 1984). Mouse splenocytes were cultured in complete mouse medium (CMM) consisting of RPMI supplemented with 25 mM HEPES, 200 nM L-glutamine, 10% fetal bovine serum (FBS; Greiner Bio-one Alphen a/d Rijn, The Netherlands), 1% minimum essential medium (MEM) nonessential amino acids, 1 mM sodium pyruvate, 50 μM β-mercaptoethanol, antibiotics, and 360 IU/ml IL-2. The human embryonic kidney cell line 293T and Phoenix-Ampho (Ph-A) were both used to package retroviruses carrying RNA encoding TCRαβ, and grown in Dulbecco's modified Eagle's medium with glutamine, 10% FBS, 1% MEM nonessential amino acids, and antibiotics. The same medium and supplements were used to culture the gp100/HLA-A2pos human melanoma cell lines, FM3 and BLMgp100 (BLM transfected with hgp100 cDNA).
Mice
Inbred C57BL/6 (B6), BALB/c, and BALB/cJHan™Hsd-Prkdcscid (BALB/c SCID) mice were purchased from Harlan Laboratories (Leicester, UK); B6.CB17-Prkdcscid /SzJ (B6 SCID) and NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ (NSG) mice were purchased from The Jackson Laboratory (Bar Harbor, Maine). Mice were housed according to the guidelines of the Erasmus Medical Center. Mice from 8 to 12 weeks of age were used in all our experiments following approval by the Experimental Animal Committee of the Erasmus Medical Center (DEC consult) and carried out in accordance with institutional and national guidelines.
TCR transgenes and transduction of T lymphocytes
The human gp100280–288/HLA-A2-specific (gp100/A2) TCRα and β genes were introduced separately into the retroviral vector pBullet via NcoI and XhoI (pB-TCRα and pB-TCRβ constructs) (Schaft et al., 2003). To allow detection of transduced T cells in vivo, green fluorescent protein (GFP) DNA preceded by an internal ribosomal entry site (IRES) was inserted into the XhoI-digested pB-TCRα and pB-TCRβ, resulting in pB-TCRα:GFP and pB-TCRβ:GFP constructs. Human T lymphocytes of healthy donors were activated with anti-CD3 monoclonal antibody (mAb) and transduced with retroviruses harboring TCRα:GFP and TCRβ:GFP or GFP only (Mock). The transduction procedure was performed as described by Lamers and colleagues (Lamers et al., 2006b), with the only exception that TCR-encoding retroviruses were produced by a coculture of 293T and Ph-A packaging cells. The murinized gp100/A2 TCR (hu:mo gp100/A2 TCR) was derived as described previously by Pouw and colleagues (Pouw et al., 2007) and used to transduce murine T cells. Similar to the receptors used to transduce human T cells, IRES-GFP was subcloned into the XhoI-digested hu:mo gp100/A2 TCRα and β genes. Total mouse splenocytes were isolated and activated with concanavalin A in CMM and transduced with the retroviral vector pBullet containing hu:mo gp100/A2 TCRαβ transgenes or the pSTICH:eGFP vector as control (Mock) as described previously (Pouw et al., 2007).
Flow cytometry
Gene-transduced human T cells were monitored with gp100/HLA-A2 phycoerythrin (PE)-labeled tetramers (termed gp100/A2 pMHC) followed by a cell sort for gp100/A2 pMHC binding cells using a FACS-Vantage instrument (Becton Dickinson Biosciences, San Jose, CA). Surface expression of TCR transgenes by human and mouse T cells was measured using a PE-labeled anti-TCRVβ14 mAb [Arden nomenclature (Arden et al., 1995)] (clone CAS1.1.3; Beckman Coulter, Marseille, France). For adoptive T-cell transfer studies, we have used 3–4-week-old >90% TCRVβ14-positive, and >80% gp100/A2-binding human T cells. TCR-transduced mouse T cells generally comprised about 30% TCRVβ14-positive T cells at the day of infusion. Peripheral blood, organs, and tumors were analyzed for the presence of TCR-engineered T cells by flow cytometry using the anti-TCRVβ14 mAb in combination with GFP, PerCP-labeled anti-CD3ɛ mAb (clone 145-2C11), and antigen-presenting cell (APC)-labeled anti-CD8α mAb (clone 53-6.7) (both from Becton Dickinson Biosciences). We analyzed the samples on a Cytomics FC-500 flow cytometer using CXP software (Beckman Coulter) or a FACSCalibur using CellQuest software (Becton Dickinson Biosciences).
AT models
B6 SCID, BALB/c SCID, or NSG mice were subcutaneously (s.c.) transplanted in the flank with either FM3 tumor tissue (3×3×3 mm) or 1×106 BLMgp100 cells. Mice bearing established tumors (5×5×5 mm) or mice having a tumor placed in a dorsal skinfold window chamber (see below) received two doses of TCR-transduced human (6–40×106) or mouse (1×106) T cells administered i.v. at days 0 and 7. In some experiments, mice were preconditioned with cyclophosphamide (200 mg/kg; Sigma-Aldrich, St. Louis, MO) administered intraperitoneally (i.p.) at day −1, preceded or not by busulfan (16.5 μg/kg; Duchefa Farma, Haarlem, The Netherlands) administered i.p. at day −2 (protocol kindly provided by Dr. Naomi Taylor, Montpellier, France). Human and mouse T-cell infusions were followed by 5 daily s.c. injections of recombinant human IL-2 (1×105 IU) starting at the day of T-cell transfer or as indicated otherwise. Tumor sizes were measured with a caliper along the perpendicular axes of tumors, and volumes were calculated with the formula 0.4×(A 2×B) where B represents the largest diameter and A the diameter perpendicular to B (Seynhaeve et al., 2007). In separate experiments using mouse GFP T cells, we assessed the individual and combined effects of T-cell activation, retroviral gene transfer, and s.c. IL-2 support on tumor growth. Blood from tail veins was collected weekly after the first T-cell infusion and analyzed for T cells by flow cytometry. At the indicated time points, tumors and lymphoid organs were isolated. Spleens and inguinal lymph nodes were isolated and processed to obtain single-cell suspensions by mechanic disruption over a cell strainer in PBS. Tumors were isolated, cut into small pieces, and digested in PBS containing collagenase (1 mg/ml) for 45 min at 37°C while vortexing every 5 min. Then 1 ml of 0.1 mM EDTA was added, and the tumor was disrupted over a cell strainer. Erythrocytes in blood and organs were removed by incubation in erylysis buffer (154 mM NH4Cl) for 15 min on ice. Absolute cell counts were determined using Flow-Count Fluorospheres (Beckman Coulter).
T-cell trafficking to tumor site using dorsal skinfold chamber
We prepared dorsal skinfold chambers according to Seynhaeve and colleagues (Seynhaeve et al., 2007). In short, mice were anesthetized with a mixture of saline, ketamine, and xylazine, injected s.c. Hair was removed from the skin on the back of the animal, and the skin was dissected in a circular area leaving the fascia and opposing skin intact. Next, the skinfold of the mouse was sandwiched between two frames and fixed with two light metal bolts and sutures. A small piece of tumor tissue was transplanted in the fascia, and both sides were closed with a microscopic cover glass. During surgery, the body temperature of animals was kept constant at 37°C, after which animals were housed individually at an ambient temperature of 32°C and a humidity of 70%. Twenty-one days after implantation, mice received GFP T cells. At the given time points, mice were anesthetized with 3% isoflurane and fixed to a heated stage of a Leica DM-RXA microscope. To visualize the presence of T cells, we used a Leica I3 filter set (excitation: 450–490 nm; emission: LP 515) and photographed the GFP T cells using a Sony 3CCD DXC 950 camera.
Immune histochemistry
Tumors were isolated, snap-frozen in liquid nitrogen, and stored at −80°C until use. Tissue sections were cut at 5 μm and stained for T cells with the following primary antibodies: rat anti-mouse CD3ɛ (clone GRJ01; R&D Systems, Abingdon, UK), rat anti-mouse CD4 (clone GK1.5), rat anti-mouse CD8α (clone 53-6.7) (both Biolegend, San Diego, CA), or rat IgG isotype control (clone 141945; R&D Systems). Goat anti-rat IgG Alexa Fluor 488 (Molecular Probes, Invitrogen, Breda, The Netherlands) was used as a secondary antibody. Air-dried cryosections were fixed with acetone for 10 min and blocked with PBS/1% bovine serum albumin for 30 min prior to immune staining. Next, sections were incubated with the primary antibody for 1 hr, and then washed and incubated with the secondary antibody for 1 hr, both at room temperature. Finally, we stained the nuclei with DAPI (4’,6-diamidino-2-phenylindole) and embedded the sections in mounting medium containing polyvinyl alcohol (MoWiol-488; Fluka, Zwijndrecht, The Netherlands). Sections were examined microscopically and photographed as described above. Recorded photographs were analyzed using Image Tool v3 (developed by Don Wilcox, University of Texas Health Science Center, San Antonio, TX) as described previously (Seynhaeve et al., 2007). The RGB images were converted to gray scale, and specific fluorescence intensities (range: 0–255) were measured with the threshold set at 20. Percentages of pixels with fluorescence intensity above threshold were calculated and expressed as bars.
Statistical analysis
Tumor volumes, times to tumor regression, and absolute cell counts were evaluated for statistical significance with the Mann-Whitney U test using SPSS v15 software, whereas immune histochemistry was evaluated for statistical significance with the Student's t test using GraphPad Prism v4 software. Differences with p≤0.05 were considered significant.
Results
Human TCR-engineered T cells do not control human melanoma growth in immune-deficient mice
We assessed the in vivo performance of human peripheral blood–derived T cells engineered with gp100/A2 TCR, which we had previously validated in vitro (Schaft et al., 2003). gp100/A2 TCR T cells used for AT functionally expressed TCR transgenes assessed by pMHC binding and target cell-specific cytotoxicity (Supplementary Fig. S1A and B; Supplementary Data are available at
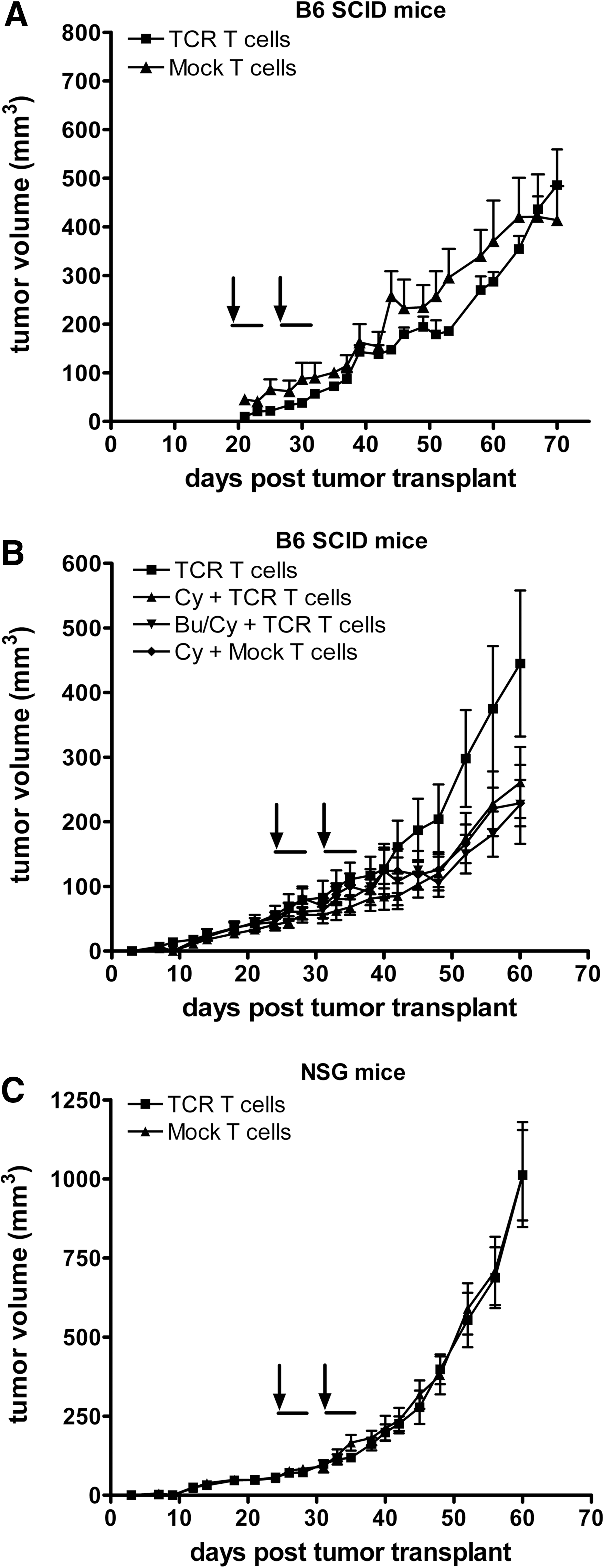
Systemic administration of gp100/A2 TCR transduced human T cells does not control growth of established human melanoma. FM3 tumor tissue (gp100+/HLA-A2+) was s.c. transplanted in the right flank of B6 SCID mice
To completely exclude “leakage” of NK cells in conventional SCID mice, and their potential adverse effect on T-cell engraftment, we have tested NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice, commonly known as NOD SCID GAMMA (NSG) (Shultz et al., 2005). The NSG mouse strain combines features of the NOD mice, the mutation SCID, and IL-2R common-γ chain deficiency. Consequently, NSG mice are severely immune-compromised, featuring the absence of mature T or B cells, lack of functional NK cells, and deficiency in cytokine signaling. T-cell engraftment was improved in NSG mice as evidenced by low but significantly higher numbers of TCR T cells in the peripheral blood at days 3 and 10 after T-cell transfer (Supplementary Fig. S3B). However, adoptively transferred T cells did not persist beyond a time period of 2 weeks past T-cell transfer and did not result in an antitumor response in melanoma-bearing NSG mice (Fig. 1C).
Human TCR-engineered T cells accumulate in tumor vasculature, but are lost shortly after AT
We used T-cell imaging to investigate whether the observed lack of peripheral persistence and antitumor response of human TCR T cells was related to their inability to migrate to the tumor site. Mice bearing an FM3 human melanoma in a dorsal skinfold window chamber received 50×106 gp100/A2 TCR:GFP T cells. Directly after injection of T cells, TCR T cells, but not mock T cells, accumulated in the tumor vasculature (Fig. 2A). Strikingly, the accumulated TCR T cells completely disappeared from the tumor site within 72 hr after adoptive T cell (Fig. 2B). Notably, human T cells predominantly showed a phenotype that corresponded to effector memory and late effector T cells at the time of AT (Supplementary Fig. S4).
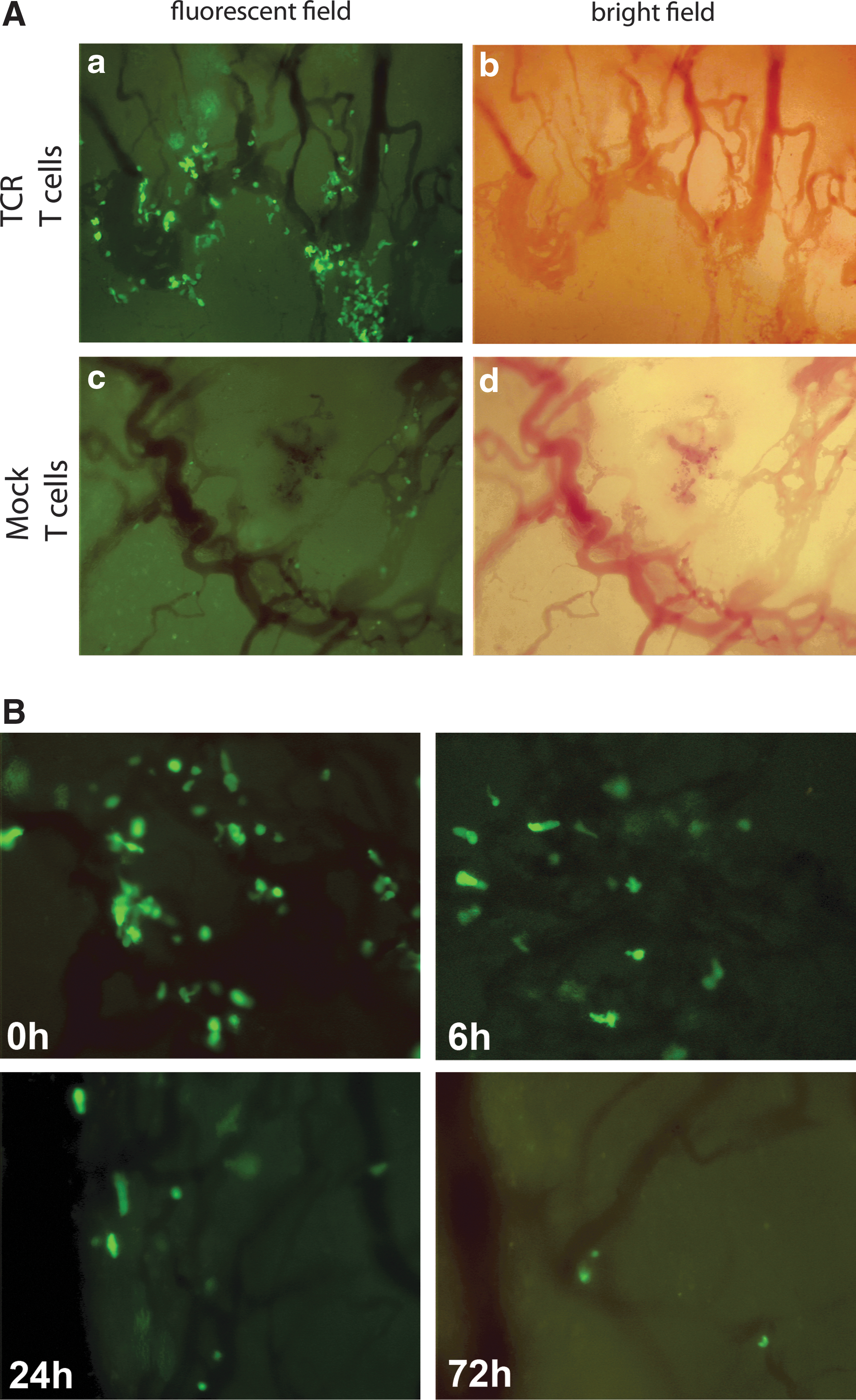
Gp100/A2 TCR-transduced human T cells initially accumulate in the tumor vasculature, but rapidly disappear. Mice bearing a human melanoma tumor in a dorsal skinfold window chamber received 50×106 TCR/GFP- or GFP-engineered human T cells i.v. at day 21 after tumor transplantation. Immediately after T-cell transfer, mice were anesthetized and fixed to a heated stage of a Leica DM-RXA microscope. T cells in the tumor vasculature were visualized with a GFP filter set (excitation wavelength: 450–490 nm; emission filter: LP 515).
Murine T cells cause complete, yet TCR transgene-independent, regression of human melanoma in immune-deficient mice
In the next series of experiments, we switched to gene-engineered murine T cells, which in the setting of CARs proved effective against solid tumors in SCID mice (Darcy et al., 2000; Haynes et al., 2001, 2002b,a). FM3 tumor-bearing B6 SCID mice were infused twice with 1×106 murine T cells expressing gp100/A2 TCR. In these mice, tumors increased in volume until day 35 (equivalent to day 11 after the first T-cell transfer) and reached a maximum average volume of 500 mm3 before rapidly regressing (Fig. 3A). Notably, Mock T cells eradicated established FM3 tumors with the same potency and kinetics as was observed for TCR T cells. Mice remained tumor-free until the end of the experiments (i.e., day 80 after tumor transplant). The antitumor responses of TCR and Mock-engineered murine T cells were confirmed with a second mouse strain (BALB/c SCID mice; Fig. 3B) and second melanoma cell line (BLMgp100 cells; Fig. 3C).
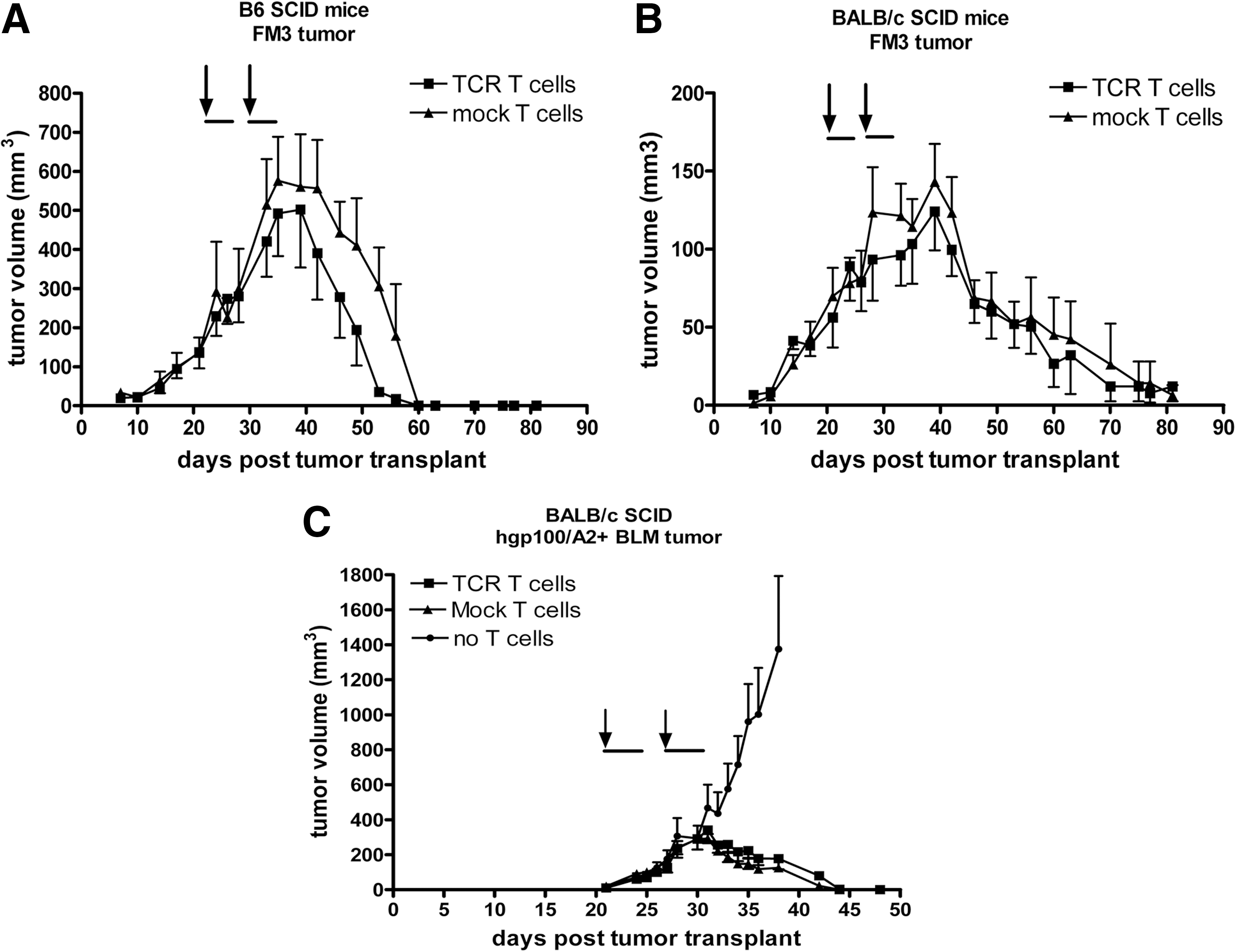
AT of murine T cells results in xeno-specific regression of human melanoma in SCID mice. FM3 tumor tissue was s.c. implanted in the right flank of B6
After AT of murine T cells, we observed that percentages of CD3+ T cells in peripheral blood increased, which was again independent of TCR transgene (Fig. 4A). T-cell expansion was not driven by the presence of tumor tissue, and percentages of peripheral T cells remained high during the complete experiment (Fig. 4A). The expression of the introduced TCR measured by anti-TCRVβ14 mAb was relatively stable during the experiment and again independent of the presence of tumor tissue (Fig. 4B). Collectively, these data demonstrate effective xeno-specific eradication of large human melanoma tumors by murine T cells and homeostatic T-cell expansion in classical SCID mice, independent of TCR transgene.
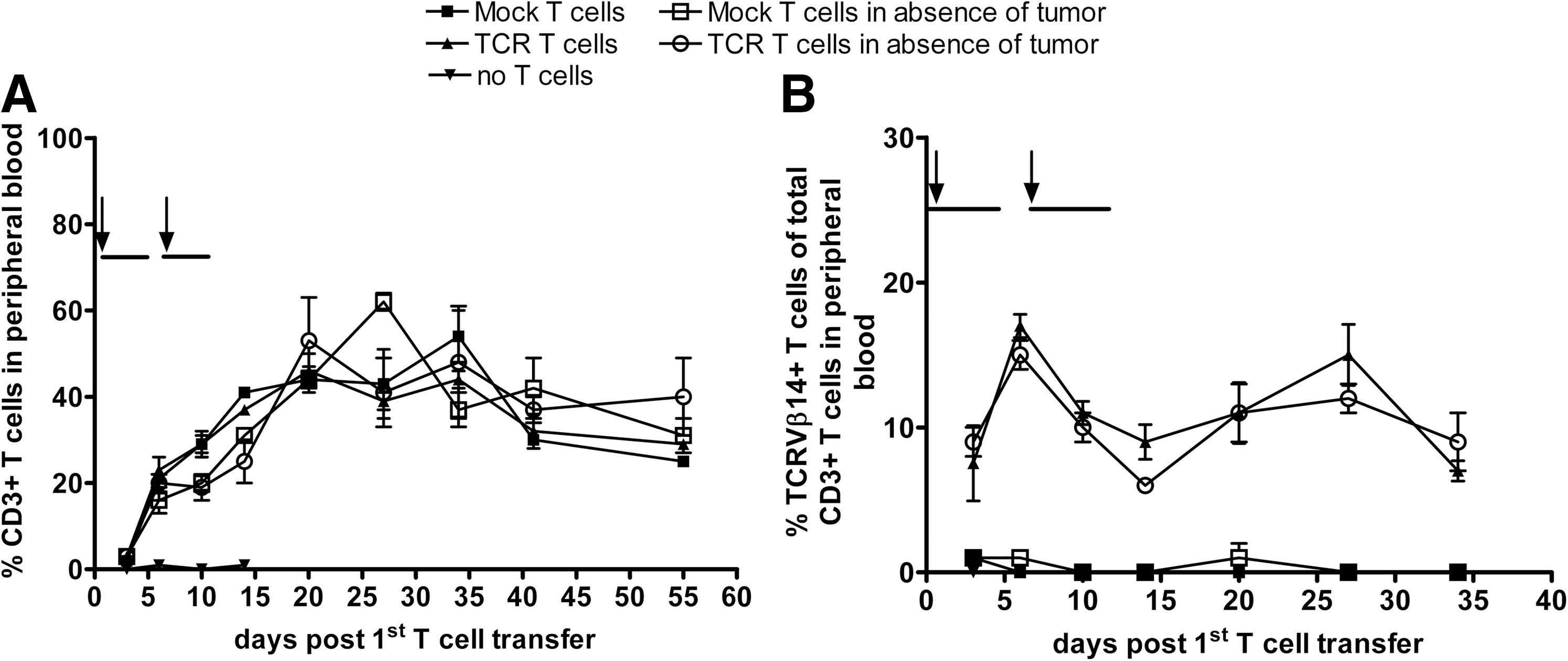
Homeostatic expansion of peripheral murine T cells upon AT into SCID mice. Human BLMgp100 melanoma cells were s.c. transplanted in the flank of BALB/c SCID mice. On days 24 and 31 after tumor transplantation, groups of six to nine mice received either 1×106 TCR/GFP- or GFP-engineered murine T cells or no T cells (arrows), followed by IL-2 injections (horizontal bars). TCR and Mock (i.e., GFP only) T cells were also administered to tumor-free mice. Peripheral blood was collected at different time points, and percentages of CD3+ T cells
Tumor-infiltrating T cells remain at the tumor margin and are predominantly of the CD4 T-cell subset
We performed two additional types of experiments to further discern the potency and rapid kinetics of the observed xeno-specific response of murine T cells. First, the migratory capacity of GFP-labeled mouse T cells and their CD8 and CD4 subsets were studied by flow cytometry and immune histochemistry. Flow cytometry allowed the detection of T cells in regressing tumors and peripheral blood, but their presence was most pronounced in spleens and tumor-draining lymph nodes (Fig. 5A). Immune histochemical staining confirmed the presence of T cells in regressing tumor tissue (Fig. 5B). Interestingly, T cells reside predominantly in the tumor margins and not in the center of the regressing tumor. Tumor-infiltrating T cells belonged to both CD8+ and CD4+ T-cell subsets, but quantification of immune histochemical stainings, summarized in Fig. 5C, revealed that the numbers of intratumoral CD4+ T cells are about fourfold higher than those of CD8+ T cells (p<0.05). We confirmed our findings on T-cell migration to the tumor with a second mouse strain (i.e., BALB/c SCID mice) and a second human melanoma cell line (i.e., BLMgp100 cells).
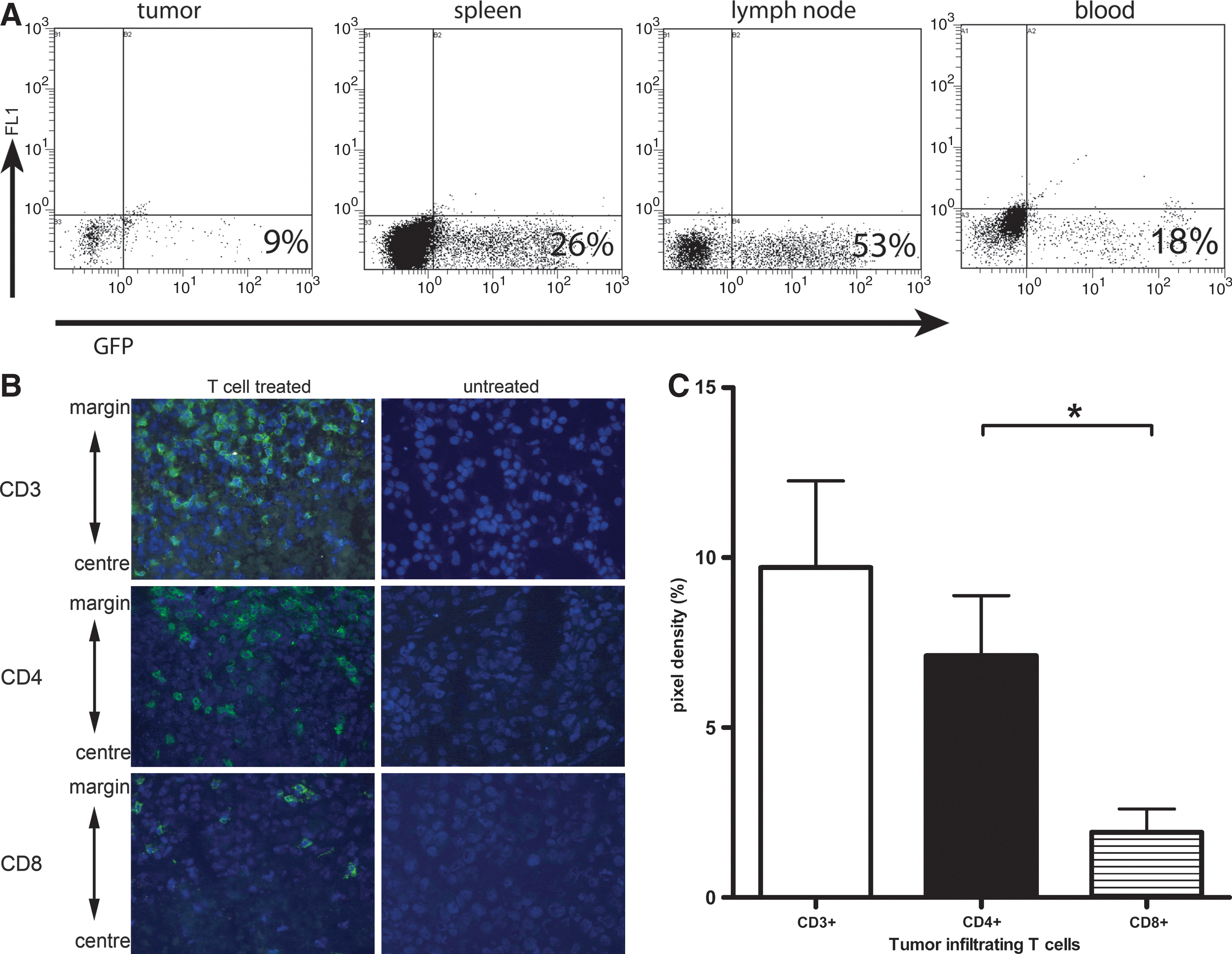
Adoptively transferred murine T cells are present in lymphoid organs, and tumor-infiltrating lymphocytes predominantly belong to the CD4+ T-cell subset. FM3 tumor-bearing BALB/c and B6 SCID mice (n=6–9) received AT of 1×106 GFP-engineered murine T cells and were treated with IL-2.
T-cell activation, retroviral transduction, and IL-2 support are not necessary per se, but enhance the potency and kinetics of antitumor T-cell responses
Next, the effects of T-cell activation, retroviral gene transfer, and s.c. IL-2 support on xeno-specific responses and peripheral persistence of murine T cells were assessed. AT of T cells that lack in vivo IL-2 support (Fig. 6A, group B), gene modification (group C), or activation (group D) resulted in clearly detectable, although less potent, antitumor activity in tumor-bearing SCID mice (Fig. 6A, compare group A versus the other groups). When T-cell activation was omitted, the time to tumor regression was significantly delayed (Fig. 6A, group D versus A, p<0.05). IL-2 support (group B) or retroviral gene transfer (group C) affected kinetics of the antitumor responses to a lesser extent, and statistical analyses showed a trend toward a delayed onset of tumor regression (Fig. 6A). In peripheral blood, T-cell numbers increased until day 37 after tumor transplant (equivalent to day 16 after first T-cell transfer) and ranged between 600 and 1,200 T cells/μl blood at the end of the experiments (Fig. 6B, day 65 after tumor transplant). Overall, T cells from the different treatment groups persisted equally well, although peripheral T-cell numbers were significantly lower when T-cell activation was omitted, or to a lesser extend when gene modification was omitted (Fig. 6B, group C versus A, p<0.05; group D versus A, p<0.01).
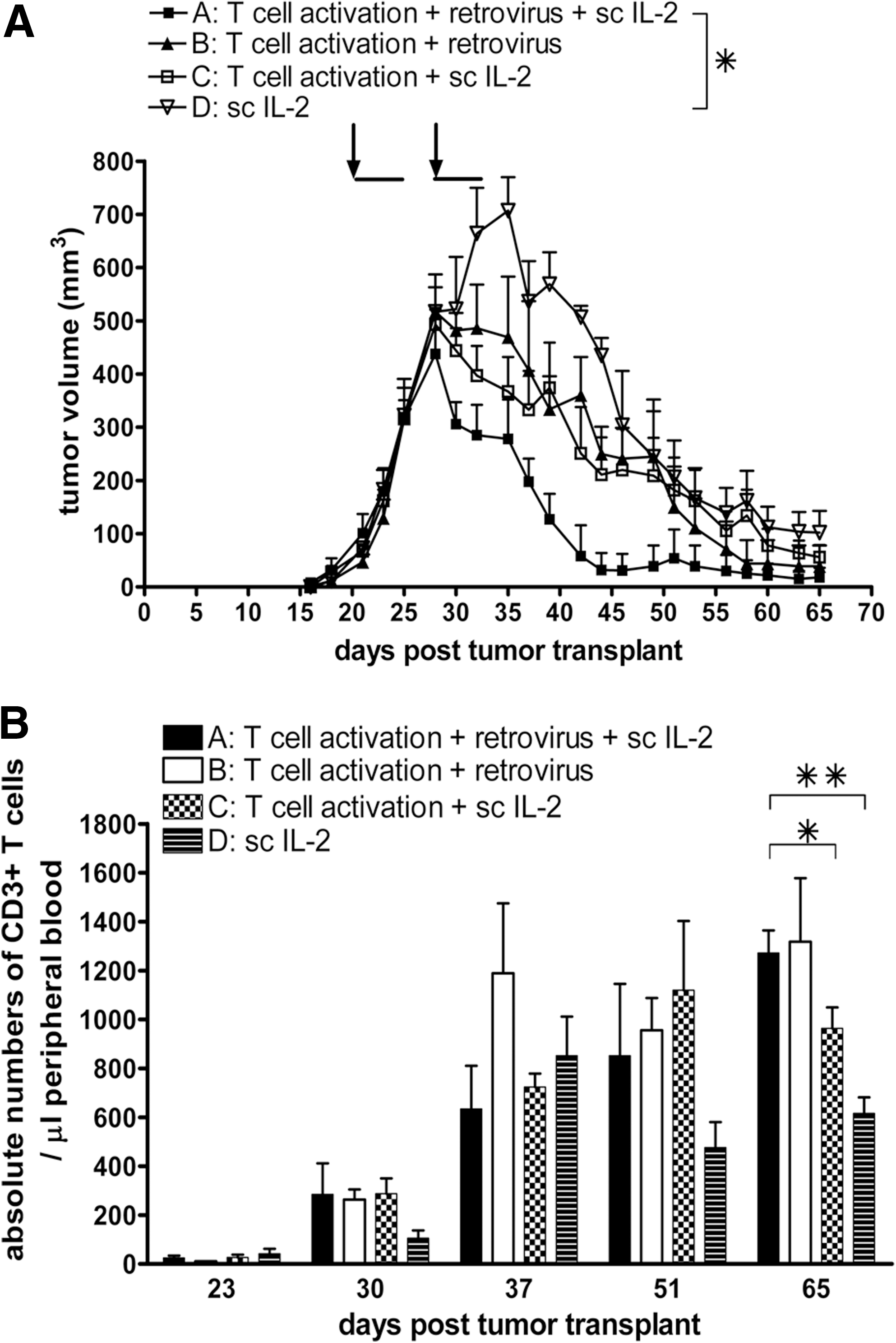
Effects of T-cell activation, retroviral transduction, and IL-2 support on the xeno-specific T-cell responses and numbers of peripheral murine T cells.
Discussion
Immune-deficient mice bearing a human tumor represent a well-recognized model to assess the therapeutic value of adoptively transferred receptor-engineered T cells. AT of both human and murine CAR-expressing T cells has been tested successfully against human lymphomas and solid tumors. However, only a limited number of studies assessed antitumor responses of TCR-expressing T cells in immune-deficient mice (Tables 1 and 2). Notably, no studies to date have reported antitumor activities of systemically administered TCR T cells, whether human or murine T cells, toward human solid tumors in immune-deficient mice. Here, we have tested the antitumor effect and persistence of adoptively transferred human and murine TCR-transduced T cells in immune-deficient mice transplanted with hgp100/HLA-A2-positive human melanoma.
We observed that transfer of human T cells in SCID mice with doses up to 40×106 TCR T cells was not able to control growth of established tumors (Fig. 1A). This lack of antitumor response was due to neither an inability of gp100/A2 TCR T cells to respond toward FM3 tumors nor the immune status of the recipient mice. TCR T cells were routinely tested in vitro in advance of our AT experiments; they demonstrated high (>80% pMHC binding) and stable surface expression (>3 months; data not shown) of TCR transgene and yielded significant antigen-specific responses (FM3-specific cytotoxicity). Also, FM3 tumors that were isolated after T-cell treatment expressed levels of HLA-A2 and gp100 protein that were comparable to those measured in FM3 cells cultured in vitro. To exclude “leakiness” of immune reactive cells, a potential problem of SCID mice, we tested both pretreatment with chemotherapy and the use of more immune-deficient mice. Although pretreatment with cyclophosphamide or irradiation may enhance antitumor activity of human T cells in SCID Beige or NOD.SCID mice (Pinthus et al., 2004; Teng et al., 2004; Westwood et al., 2005), we did not observe an enhanced antimelanoma effect in SCID mice pretreated with cyclophosphamide (Fig. 1B). Also, a combination of cyclophosphamide and busulfan, which results in transient yet complete depletion of T cells, B cells, NK cells, and dendritic cells in immune-competent mice (personal communication, Dr. Naomi Taylor, Montpellier, France; Straetemans et al., manuscript submitted), did not enhance the antimelanoma activity of TCR-engineered human T cells in SCID mice (Fig. 1B). In more immune-deficient NSG mice (Shultz et al., 2005), we again observed no antimelanoma response following treatment with TCR T cells (Fig. 1C). Notably, immediately after T-cell transfer, TCR, but not Mock, T cells accumulated in FM3 tumor vasculature, but these T cells were rapidly (within 72 hr) lost (Fig. 2). T-cell persistence in peripheral blood was negligible and not enhanced by preconditioning and, at best, only marginally enhanced in NSG versus SCID mice (Supplementary Fig. S3). Our data confirm a study by Bobisse and colleagues, in which TCR-engineered T cells showed antimelanoma activity and peripheral persistence following i.t. but not i.v. administration in immune-deficient mice (Bobisse et al., 2009).
Another parameter that is potentially related to T-cell persistence is T-cell exhaustion. Flow cytometric analysis of markers of T-cell memory, activation, and costimulation showed that the majority of T cells at the day of AT have a late T-cell differentiation phenotype (Supplementary Fig. S4). In fact, using two sets of definitions of T-cell subsets, one including the adhesion molecule CD62L (according to Kaneko et al., 2009; Wang et al., 2011) and another including both costimulatory markers CD27 and CD28 (according to Appay et al., 2008; Kalos et al., 2011), we observed that the sum of effector memory and end-stage T cells minimally represented about 90% of T cells. This finding may argue that under-representation of less differentiated cells (i.e., naive and central memory T cells) relates to an inability of human T cells to persist and act against the tumor. In fact, clinical studies demonstrate that T-cell differentiation is associated with T-cell persistence (Huang et al., 2005), which in turn is associated with antitumor T-cell efficacy (Robbins et al., 2004). In our current study, anti-CD3 mAb stimulation and expansion with IL-2 may have created a bias toward TCR T cells with a late differentiation phenotype. Notably, in a clinical phase I trial to treat metastasized renal cell carcinoma with anti-CD3 mAb-activated and IL-2-cultured CAR T cells (Lamers et al., 2006a), we observed that transduced T cells of patients, who all showed progressive disease, consistently have a late differentiation CD8 T-cell phenotype at the time of AT (Lamers, unpublished observations). Collectively, clinical trials with either CAR or TCR gene-transduced T cells using similar T-cell activation and culture regimens together with in vivo IL-2 support demonstrate that peripheral T-cell persistence, although variable, is generally limited (Kershaw et al., 2006; Lamers et al., 2006a; Morgan et al., 2006; Johnson et al., 2009; Parkhurst et al., 2011). The present model, in which NSG mice with human melanoma were treated with TCR-engineered T cells, closely recapitulates the cell-processing and patient-treatment methods used in clinical receptor gene therapy studies and may represent a valid model to test strategies to improve T-cell persistence and antitumor efficacy.
Two recent clinical trials provide evidence that alternative methods for T-cell stimulation and cytokine support may benefit clinical receptor gene therapy (Butler et al., 2011; Kalos et al., 2011; Porter et al., 2011). In one study, Butler and colleagues adoptively transferred MART1/A2-specific T cells that were stimulated with CD80- and CD83-expressing artificial APCs and cultured in the presence of both IL-2 and IL-15 into patients with metastatic melanoma without preconditioning or in vivo IL-2 support (Butler et al., 2011). This study resulted in three mixed responses and long-lasting disease stabilizations and one complete response out of seven patients. In another study, Kalos and colleagues adoptively transferred T cells that were stimulated with anti-CD3 and CD28 mAb-coated beads and gene-engineered with a CD19-specific CAR, which contained the costimulatory domain CD137 and the accessory domain CD3ζ, into patients with chronic lymphocytic leukemia who were preconditioned, but did not receive IL-2 support (Kalos et al., 2011). This study resulted in complete remissions in two out of three patients. Strikingly, in both studies post infusion, T cells persisted long-term in the circulation, were enriched for T cells with a central memory T-cell phenotype, and retained functional responses toward target antigens. Although patient numbers were low, these studies clearly demonstrate the potential value of T-cell costimulation and exposure to IL-15 with respect to the persistence of adoptively transferred T cells. In extension to these clinical studies, there is also ample preclinical evidence that supports the use of naive or central memory T-cell subsets to enhance T-cell persistence and antitumor efficacy of transferred T cells (Berger et al., 2008; Hinrichs et al., 2009; Wang et al., 2011). Along these lines, we are currently testing T-cell stimulations with anti-CD3 and CD28 mAbs, T-cell culture with IL-15 and IL-21, and gene engineering with modified TCR formats, such as TCRs coupled to CD3ɛ, CD3ζ, and costimulatory domains, for enhanced in vivo persistence and performance (Schaft et al., 2006; Pouw et al., 2010a,b).
In the next series of AT experiments, we used mouse T cells, in analogy to earlier studies with CAR-engineered mouse T cells (Table 2). Our studies show that syngeneic mouse T cells can very potently and rapidly eradicate large established human melanomas in SCID mice (Fig. 3). In addition, mouse T cells expanded and persisted in vivo for >6 weeks after the first T-cell transfer (Fig. 4). Unexpectedly and in contrast to studies with CAR T cells, the antitumor activity and expansion of T cells were independent of the receptor transgene and represented xeno-specific T-cell responses. In fact, T-cell expansions were of a homeostatic nature and independent of the cognate tumor antigen. These findings were confirmed with two SCID strains, B6 SCID and BALB/c SCID, and two human melanoma cell lines, FM3 and BLMgp100. Notably, MHC class I expression on FM3 and BLMgp100 tumor cells is high (>90% of cells in vitro and directly after in vivo isolation; data not shown) and potentially drives the antimelanoma activity of mouse T cells. Smyth and colleagues reported that cytotoxicity of mouse T cells derived from human tumor-immunized mice can partially be blocked with an anti-human MHC class I antibody in vitro (Smyth et al., 1997). Along these lines, MHC class Ilow tumor cells may be considered to lower the extent of xeno-specific responses, but recognition by TCR-engineered T cells is also likely to be compromised. Alternatively, mouse T cells with a restricted or monoclonal TCR repertoire can be used as recipient cells for TCR gene transfer. This may prevent xeno-specific activity of transferred T cells, but the translational value of such a model should be considered with caution.
To further investigate and better understand the potency and kinetics of the xeno-specific effect of murine T cells, we studied T-cell localization and demonstrated that mouse T cells migrate to lymph node and spleen and persist for >6 weeks in peripheral blood (Fig. 5A). In regressing human tumors, murine T cells infiltrated the tumor site and formed a ring at the tumor margin, but were not present in the center of the tumor (Fig. 5B). This finding was in line with observations reported by Hanson and colleagues (Hanson et al., 2000), who showed that in regressing tumors T cells encircled the tumor site, but in progressively growing tumors T cells were scattered in patches of necrotic tumor tissue. CD4+ but not CD8+ T cells constitute the predominant T-cell subset that infiltrated the regressing human tumors (Fig. 5C). Remarkably, this was found in both BALB/c and B6 SCID mice despite the fact that B6 T cells predominantly consist of CD8+ T cells (peripheral blood T cells of BALB/c and B6 mice comprise on average 30% and 80% CD8+ T cells, respectively). Although we cannot rule out an early involvement of CD8+ T cells, our immune histochemistry data suggest that rejection of human tumors in SCID mice is mediated by CD4+ T cells, confirming reports on the CD4 dependence of T cell-mediated xenograft rejections (Pierson et al., 1989; Friedman et al., 1999). In another series of experiments, we observed that T-cell stimulation and, to a lesser extent, retroviral transduction and s.c. IL-2 support contributed to the potency and kinetics of xeno-specific responses of mouse T cells (Fig. 6A). T cells that were not stimulated prior to T-cell transfer persisted well in vivo, but generally the lack of T-cell stimulation adversely affected T-cell numbers in peripheral blood (Fig. 6B). Although T-cell stimulation is not necessary for the ultimate rejection of tumors and peripheral T-cell persistence per se, it may be of importance in case a suboptimal TCR is used.
In conclusion, we have demonstrated that, despite reports with CAR transgenes, immune-deficient mice have limited value to assess the therapeutic efficacy of mouse T cells engineered with TCR transgenes. With respect to TCR-engineered human T cells, stimulated with anti-CD3 mAb and cultured and supported in vivo with IL-2, T-cell persistence is highly compromised and antitumor responses are lacking in immune-deficient mice. These observations are in close accordance with clinical studies, are related to T-cell differentiation, and confirm the validity of NSG mouse models to test strategies to enhance T-cell persistence and antitumor efficacy of receptor-engineered human T cells.
Footnotes
Acknowledgments
The authors would like to thank Sabine van Steenbergen, Dr. Cor Lamers, and Dr. Zsolt Sebestyen for flow cytometric stainings, analyses, and interpretations. Dr. Naomi Taylor (Institut de Génétique Moléculaire de Montpellier, Montpellier, France) is thanked for providing and discussing the busulfan and cyclophosphamide preconditioning protocols. This work was financed in part by Erasmus Medical Center Translational Research (TR-2004) and by the European Union Framework Program 6 “Adoptive Engineered T Cell Targeting to Activate Cancer Killing” (EUFP6 ATTACK) project (number FP6-018914).
Author Disclosure Statement
There are no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.