
Abstract
Using conditionally replicating adenoviral vectors (CRAds) is a promising strategy in the treatment of solid tumors. The prospective of this study was to design a novel CRAd for the treatment of gastrointestinal cancer and show its efficacy in vitro, as well as in vivo. To determine if aberrant wnt signaling in tumor cells can be used to selectively drive viral replication, we analyzed six colorectal and hepatocellular cell lines, as well as 13 colorectal tumors and 17 gastric tumors, for β-catenin mutation status or aberrant wnt signaling, both of which were found frequently. Based on these findings, a novel CRAd (Ad5F11.wnt-E1A-hIL24) containing an E1A expression cassette driven by an artificial wnt promoter and delivering an apoptosis-inducing gene, interleukin-24 (IL24), was engineered. To enhance infection efficiency, the virus was pseudotyped by replacing adenovirus serotype 5 (Ad5) with Ad11 fiber. Ad5F11.wnt-E1A-hIL24 virus exhibited high selectivity toward cells with aberrant wnt signaling both in vitro and in mouse xenograft tumors. Transduction efficiency was significantly improved compared with that of nonpseudotyped control viruses. The proliferation of tumor cell lines, as well as tumor growth, in mouse xenografts could be profoundly inhibited by viral infection with Ad5F11.wnt-E1A-hIL24. The therapeutic effect was associated with increased apoptosis through caspase-3 activation. In addition, Ad5F11b vector exhibited a more favorable biodistribution, blood clearance, and transgene expression compared with conventional Ad5 vector after systemic or intratumoral injection in human gastrointestinal cancer xenografts. We think that our approach is a promising strategy in the treatment of gastrointestinal cancer, warranting further clinical investigation.
Introduction
Wingless/wnt pathway reactivation has been found in a number of malignancies (Morin, 1999), including colon cancer. β-Catenin (CTNNB1) is an important component of the wingless/wnt pathway and can act as a transcription factor for cellular oncogenes. In the absence of a wingless/wnt signal, free cytoplasmic β-catenin is degraded by phosphorylation-mediated ubiquitination and consequent proteolytic degradation (Chen et al., 1995; Clevers, 2006), thus maintaining a low steady-state level. β-Catenin can bind to the T-cell factor/lymphoid enhancer factor (TCF/LEF) binding site in the promoter region of oncogenes such as c-myc and cyclin D1, inducing their transcription.
Accumulation of β-catenin has been found to be associated with mutations in the β-catenin gene in a number of malignancies (Morin, 1999), such as colon cancer (Ilyas et al., 1997), hepatocellular carcinoma (de La Coste et al., 1998; Wong et al., 2001), and melanoma (Rubinfeld et al., 1997). Thus, an artificial promoter containing a TCF/LEF binding site sequence can be used to selectively drive high-level transgene expression in tumor cells with deregulated β-catenin/wnt signaling (Brunori et al., 2001; Lipinski et al., 2001, 2004; Fuerer and Iggo, 2002; Dihlmann and von Knebel Doeberitz, 2005; Rahmani et al., 2005; Metzl et al., 2006).
Oncolytic adenoviruses have been widely tested in clinical trials for the treatment of solid tumors, but their therapeutic efficacy is still limited. More recently, oncolytic adenoviruses have been designed to carry a therapeutic gene—the so-called “armed” oncolytic adenoviruses—and have shown promising results in preclinical studies (Crompton and Kirn, 2007; Liu et al., 2007).
In the present study, we have engineered a novel armed oncolytic adenovirus for the treatment of gastrointestinal cancer. Tumor-selective viral replication is controlled by TCF/LEF elements that complement aberrant wnt signaling, which we have found to be present in colorectal, as well as gastric, tumors. It is armed with the mda-7/IL24 [interleukin 24 (IL24)] therapeutic gene, which has been shown to drive apoptosis, and pseudotyped with adenovirus serotype 11 (Ad11) to enhance infection efficiency.
Materials and Methods
Tumor samples
Human colorectal and gastric tumor tissues and their adjacent normal tissues were obtained from 13 colorectal cancer patients and 17 gastric cancer patients who underwent surgery.
Cell lines and culture conditions
The human hepatocellular carcinoma cell line HepG2 (wild-type p53), BEL7402 (mutant p53), SMMC7721 (mutant p53), and human normal liver cell line L02 (wild-type p53) were obtained from the cell bank of the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. The human embryonic kidney cell line HEK293 was purchased from Microbix Biosystems Inc. (Toronto, ON, Canada). Human colorectal cancer cell lines HCT116/4016 (wild-type p53) and HCT116/379.2 (p53 knockout) were prepared by Dr. B. Vogelstein's laboratory (Johns Hopkins University, Baltimore, MD). Cells were maintained in a humidified 37°C atmosphere containing 5% CO2 and cultured in Dulbecco's modified Eagle medium (GIBCO, Grand Island, NY) supplemented with 10% fetal bovine serum (GIBCO), 2 mmol/L L-glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin.
Analysis for β-catenin mutation and CD46 or CAR expression
Total RNAs were isolated from frozen human colorectal or gastric tumor samples and adjacent normal tissues, as well as a variety of tumor cell lines. The cDNA was synthesized using SuperScript (Invitrogen, Carlsbad, CA) and analyzed for β-catenin mutations by direct sequencing, and for CD46 (cluster of differentiation 46) or CAR (coxsackievirus and adenovirus receptor) expression by quantitative real-time PCR (qRT-PCR).
Luciferase activity assay
Seventy-two hours after transfection into tumor cells via Lipofectamine (Invitrogen), luciferase activity was determined using a luciferase reporter assay kit (Promega, Madison, WI).
Luciferase activity of living animals and excised tissues was measured using a NightOwl LB 981 Molecular Imaging System (Ji et al., 2009). Luciferase activity of excised tissues was normalized to protein content in corresponding samples.
Adenoviral vectors
The wnt reporter plasmid M50 Super 8X TOPflash containing eight tandem duplications of TCF/LEF binding sites (AGATCAAAGGgggta) in the promoter region of the luciferase coding sequence was kindly provided by Dr. Randall Moon (University of Washington, Seattle, WA).
Ad5F11.wnt-E1A-hIL24 and Ad5F11.wnt-E1A were engineered by amplifying the modified wnt promoter by PCR using the M50 TOPflash plasmid as a template, and inserting it into the E1 region of an adenoviral shuttle plasmid p74. A constitutive MDA-7/IL24 expression cassette was then inserted into the E3 region of plasmid p74 (P74/Wnt-E1A/CMV-IL24). For pseudotyping with Ad11 fiber, the shuttle plasmids P74/Wnt-E1A/CMV-IL24 or P74/Wnt-E1A and the Ad11 fiber containing PPE3 plasmid (Ad5 backbone) were cotransformed into competent DH5α cells, and then recombinants were verified by restriction-enzyme digestion and further transfected into HEK293 cells.
Ad5F11b-Luc containing cytomegalovirus (CMV)-driven luciferase reporter gene shares the same capsid with Ad5F11b.wnt-E1A-hIL24 and Ad5F11b.wnt-E1A, but is replication-defective due to deleted E1A. The construction of Ad5.TERT-E1A/E1B has been published elsewhere (Huang et al., 2004). All viruses were propagated and purified on a CsCl gradient using standard protocol. Viral infectious titer [50% tissue culture infective dose (TCID50)] was determined using the TCID50 assay on HEK293 cells.
Viral progeny assay
Viral replication capacity was determined according to protocols as published previously (Luo et al., 2008).
Cytotoxicity assay
The fraction of viable cells was determined at the indicated time points after viral infection using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay or by visualizing the density of living cells using crystal violet staining.
Apoptosis assay
After viral treatment, cells were collected, stained with annexin V–fluorescein isothiocyanate and propidium iodide, and counted by flow cytometry. On tumor sections, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed according to the manufacturer's instructions (Takara Biotechnology, Dalian, China).
Western blotting
Western blot analysis was performed as published previously (Liu et al., 2010). The antibodies used in this study include the following: rabbit polyclonal anti-E1A antibody (1:3,000), anti-MDA-7/IL24 (1:500), and anti-β-catenin (1:500) (Santa Cruz Biotechnology, Santa Cruz, CA); anti-caspase-3 (1:200), anti-caspase-7 (1:200), anti-poly(ADP-ribose) polymerase (PARP) (1:500), anti–cleaved caspase-3 (Asp175) (1:200), anti–cleaved caspase-7(Asp198) (1:200), anti–cleaved PARP (Asp214) (1:500), anti-Bax (1:500), and anti-Bcl-2 (1:500) (Cell Signaling Technology Inc., Danvers, MA); and anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:5,000) (Kang Chen, Shanghai, China).
In vivo analysis
The mice used in this study were maintained in accordance with the institutional guidelines approved by the Committee for Animal Experimentation of Shanghai Jiaotong University.
Human colon cancer xenograft tumors were established by subcutaneous injection of HCT116/379.2 cells (5 × 106) into the flanks of Balb/c nude mice. When tumors grew to a size of 80–100 mm3, the tumor-bearing mice were randomly divided into four groups. Each group was treated by intratumoral injection of phosphate-buffered saline (PBS) or 5 × 108 TCID50 of adenoviral vector every other day for a total of four times. Tumors were measured twice per week, and tumor volume was calculated according to the following equation: V = width2 × length/2. Mice with tumors larger than 15 mm were considered deceased.
Morphologic analysis
Xenograft tumors were excised, fixed in phosphate-buffered formalin, and paraffin-embedded. The tumor sections were stained with anti-MDA-7/IL24 antibody (1:20; Cell Signaling Technology), or TUNEL.
Statistical analysis
All statistical analyses were conducted using SPSS 11.5 software (SPSS, Chicago, IL). One-way ANOVA followed by post-hoc Dunnett's t test was performed for the analysis of tumor volume. Differences in p values of <0.05 were considered significant.
Results
Evaluation of β-catenin mutations in tumor cell lines and specimens
To determine if aberrant wnt signaling in tumor cells can be used to selectively drive viral replication in these cells, we have analyzed six colorectal and hepatocellular cell lines, as well as 13 colorectal tumors and 17 gastric primary tumors and their adjacent normal tissue, for β-catenin mutations. Alterations in the coding sequences and the predicted amino acid substitutions are shown in Fig. 1A. Four of six tested tumor cell lines showed mutations in the β-catenin coding sequence, including a 348-bp deletion (codon 25–140; Fig. 1B) in HepG2, which resulted in a frame shift and an 80-kDa truncated protein (Fig. 1C); a 3-bp deletion (codon 33) in HCT116/4016 and HCT116/379.2, which resulted in a loss of a serine residue; and three point mutations (D32E, S33K, and I35V) in BEL7402 (Fig. 1A). No mutations were identified in the control cell line L02 and in SMMC7721.
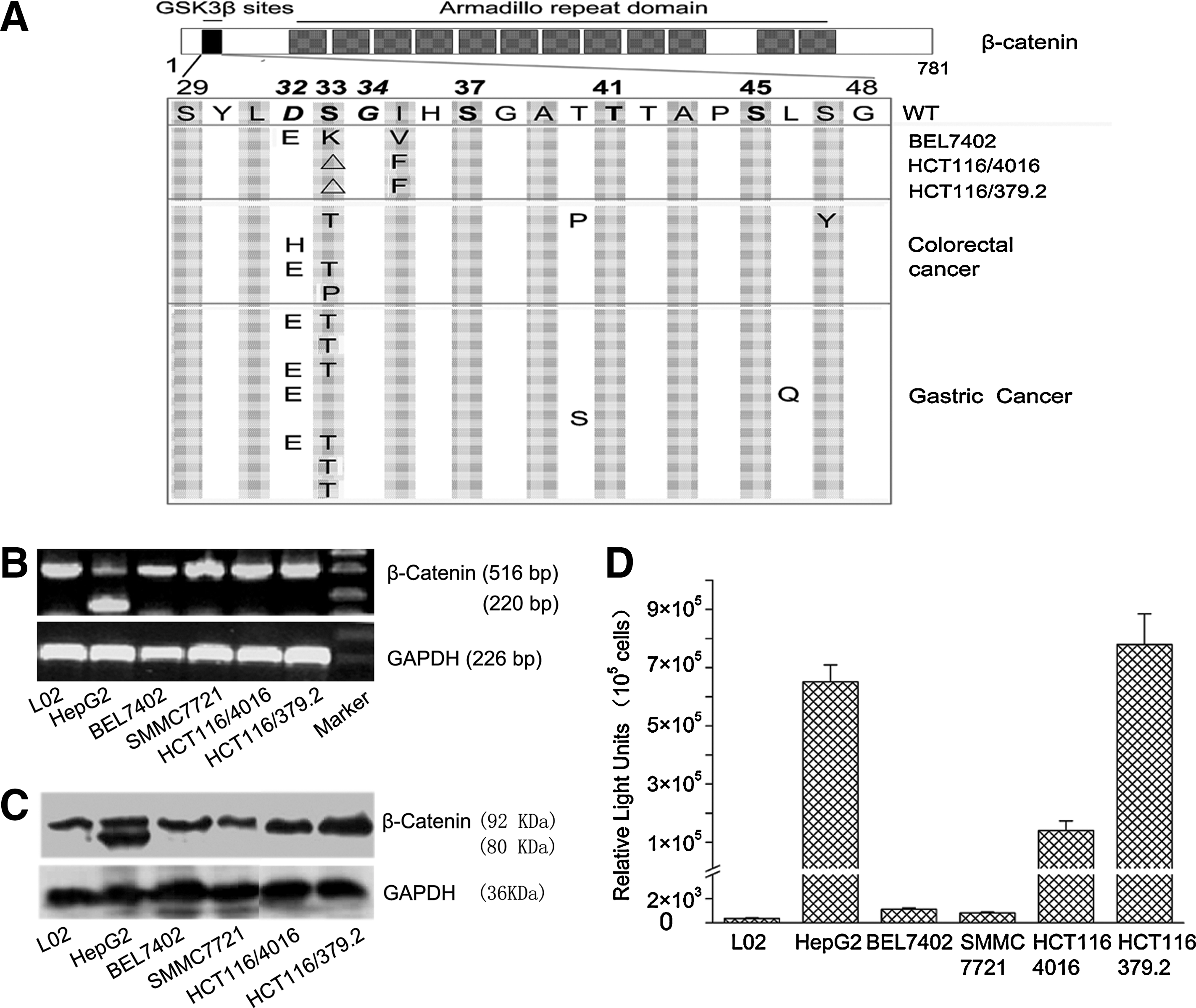
β-catenin mutation and activity in tumor tissues and cell lines. (
In the analysis of primary human tumors, five of 13 (38.5%) colorectal and eight of 17 (47.1%) gastric tumors showed mutations in tumor tissues, but not in their adjacent normal tissues. All mutations were heterozygous and predominantly located in exon 3 of the β-catenin gene, which harbors all potential glycogen synthase kinase-3β phosphorylation sites (S33, S37, T41, and S45) as well as the D32 and G34 ubiquitination sites that are required for proteasome-dependent degradation (Yost et al., 1996; Aberle et al., 1997). Mutations at these sites have been shown to stabilize β-catenin (Giles et al., 2003).
Overall β-catenin protein levels and expression of truncated variants were assessed in the tumor cell lines using Western blot analysis. The predicted 80-kDa β-catenin protein was found to be expressed in HepG2 cells, and both the HCT116/4016 and HCT116/379.2 cell lines expressed high overall levels of β-catenin (Fig. 1C).
Wnt activity in tumor cell lines
In order to assess β-catenin activity in cultured tumor cell lines, a luciferase-based reporter plasmid (M50 Super 8X TOPflash) containing eight tandem duplications of TCF/LEF binding sites (AGATCAAAGGgggta) in the promoter region of the luciferase expression cassette was transfected into each tumor cell line, and luciferase activity was measured after 72 hr. Cell lines carrying β-catenin mutations (HepG2, HCT116/4016, and HCT116/379.2) exhibited a 100–700-fold higher luciferase activity compared with cell lines with wild-type β-catenin (Fig. 1D).
Characterization of Ad5F11b.wnt-E1A-hIL24 viral plasmids
A wnt-controlled chimeric oncolytic adenoviral vector armed with the mda-7/IL24 gene (Ad5F11b.wnt-E1A-hIL24) was engineered by inserting eight tandem repeats of the TCF/LEF binding sites into the E1 promoter region driving E1A gene expression, which is required for viral replication. A constitutive MDA-7/IL24 expression cassette was inserted into the E3 region. To enhance viral transduction efficiency, the virus was pseudotyped by replacing Ad5 fiber with Ad11 fiber. In addition, a virus containing no MDA-7/IL24 expression cassette (Ad5F11b.wnt-E1A) was generated as a control. A schematic representation of Ad5F11b-Luc, Ad5F11b.wnt-E1A, and Ad5F11b.wnt-E1A-hIL24 is shown in Fig. 2A.
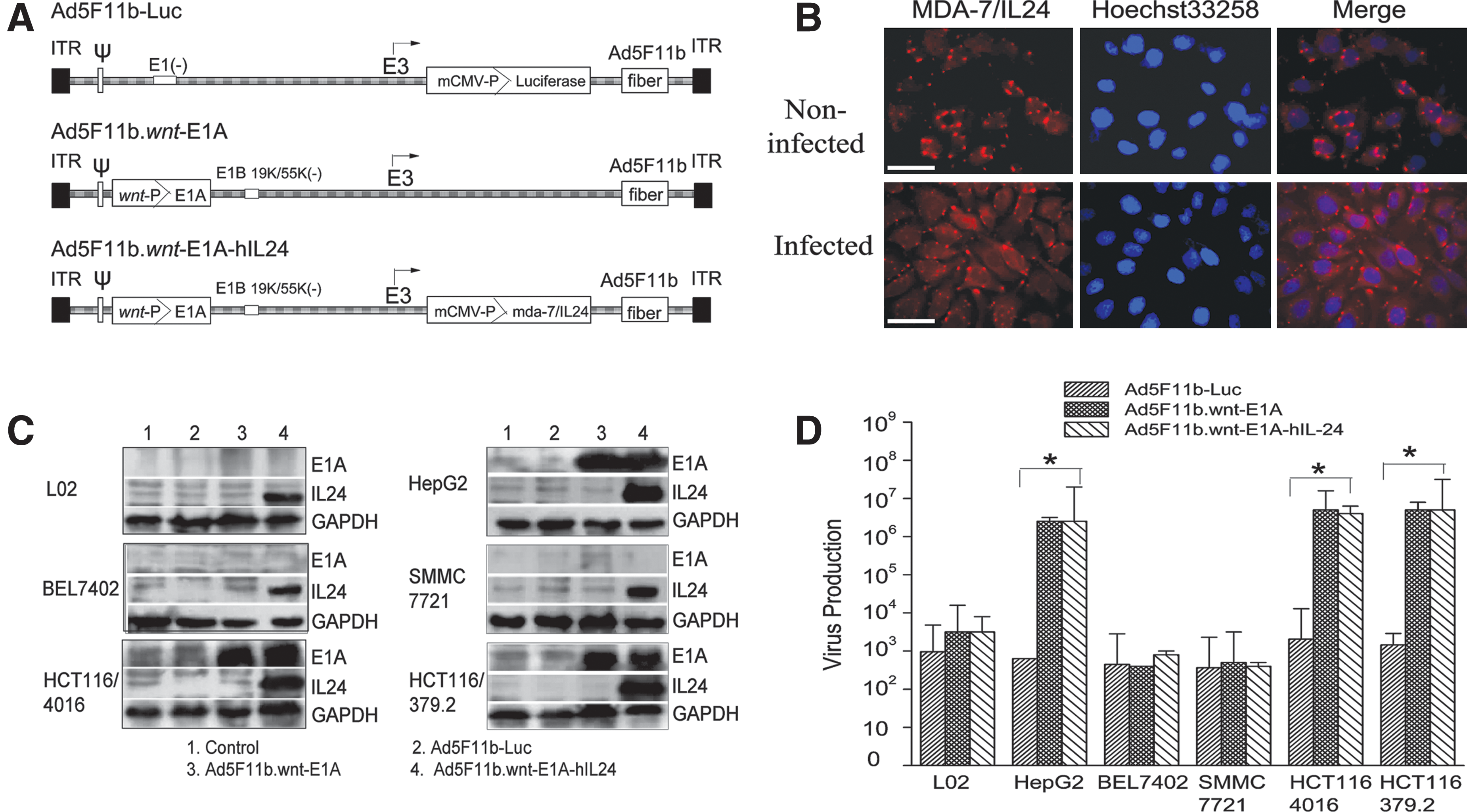
Structure, expression, and replication of adenoviral vector. (
Expression and subcellular localization of hIL24 in Ad5F11b.wnt-E1A-hIL24–infected SMMC7721 cells were analyzed by immunofluorescence staining. Endogenous hIL24 became visible as perinuclear dots, whereas exogenous hIL24 was equally distributed in the cytoplasm (Fig. 2B).
Total expression levels of hIL24 and E1A after infection with Ad5F11b.wnt-E1A or Ad5F11b.wnt-E1A-hIL24 were analyzed by Western blotting. E1A expression was detected in cell lines (HepG2, HCT116/4016, and HCT116/379.2) that had been shown to contain mutations in their β-catenin gene, but not in cell lines with a wild-type β-catenin gene (Fig. 2C). IL24 protein expression was detected in HepG2, HCT116/4016, and HCT116/379.2 cells, which was consistent with E1A expression (Fig. 2C). These results indicated that Ad5F11b.wnt-E1A-hIL24 selectively replicated in tumor cells with an activated wnt pathway.
To further confirm wnt-selective viral replication, viral progeny was quantified using TCID50 assay. The wnt activated cell lines HepG2, HCT116/4016, and HCT116/379.2 infected with either Ad5F11b.wnt-E1A or Ad5F11b.wnt-E1A-hIL24 showed two to three orders of increase in viral particles compared with L02, BEL7402, and SMMC7721 cells infected with the same amount of adenovirus (Fig. 2D). The control vector Ad5F11b-Luc did not replicate in any of the tested cells regardless of their wnt activity status.
Differential gene transduction efficiency of Ad5 and Ad5/F11
In order to be able to quantify and compare viral infection potency of Ad5 and the pseudotyped Ad5/F11, respective replication-incompetent viruses containing a CMV-driven luciferase expression cassette were constructed. They share the same capsid with the therapeutic Ad5F11b.wnt-E1A-hIL24 and Ad5F11b.wnt-E1A, but are rendered replication-defective by removal of the E1A gene that is required for viral replication, thus allowing viral transduction efficiency to be exclusively assessed.
Ad5- and Ad5/F11-mediated gene transduction in tumor cells was investigated both in vitro and in vivo. In vitro, HCT116/379.2 cells infected with Ad5F11b-Luc showed a 2.5-fold induction of luciferase activity compared with cells infected with Ad5-Luc (Fig. 3A, left panel). Similar results were obtained from normal cells L02 infected with Ad5-Luc and Ad5F11b-Luc. Expression levels of the primary receptors for Ad5 (CAR) and Ad11 (CD46) were examined by qRT-PCR. CD46 was ubiquitously expressed in almost all tested tumor cell lines, as well as colorectal or gastric tumor tissues. Interestingly, the CD46 expression level in tumor tissue was significantly higher than that in its adjacent normal tissues. In contrast, CAR expression levels were similar in tumor tissues and their adjacent normal tissues (Fig. 3A, middle and right panels).
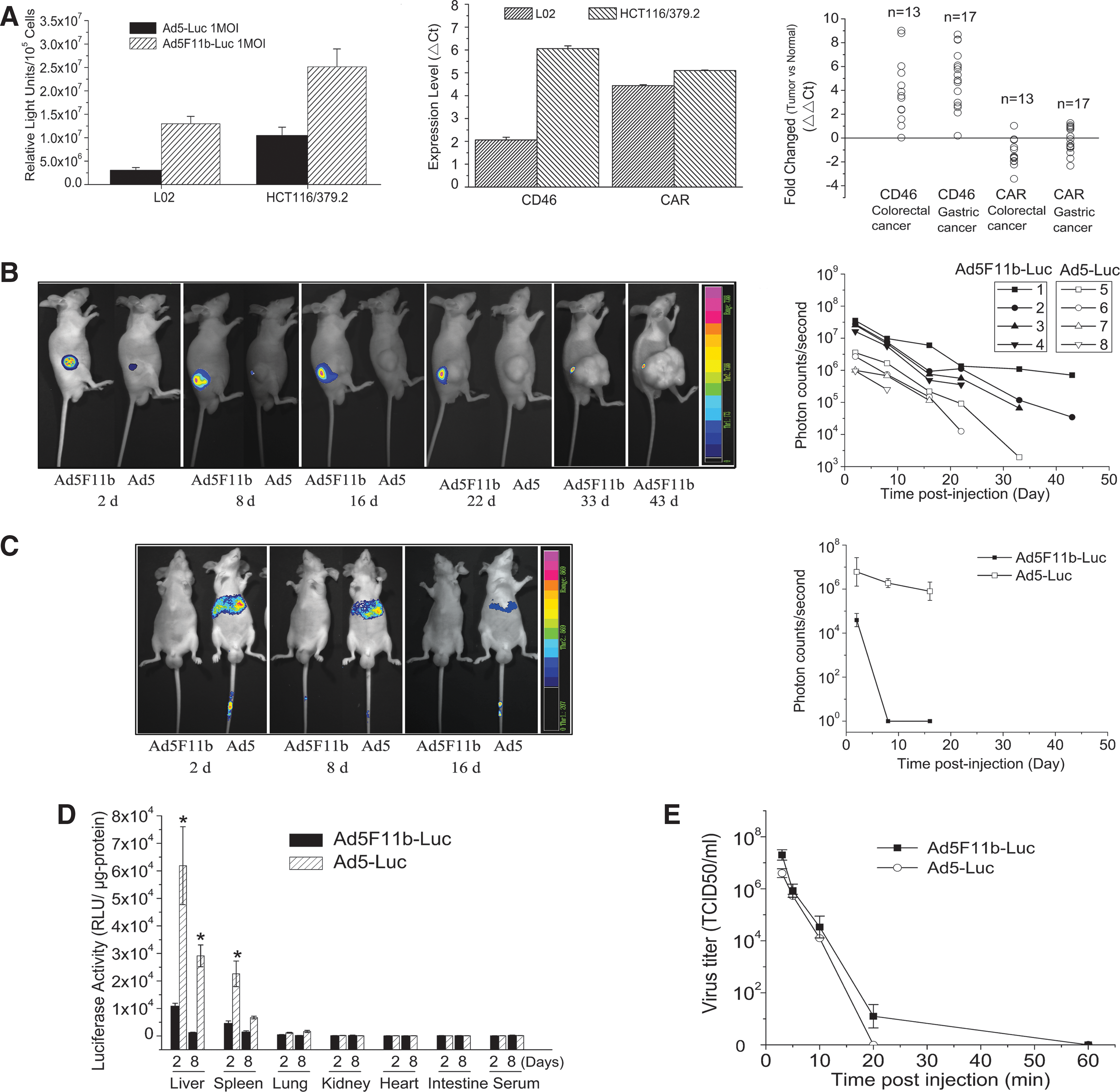
Differential expression patterns of Ad5 and Ad5/F11. (
The differential gene transduction of Ad5- and Ad5/F11-infected cells was further determined in vivo using live cell imaging. Nude mice bearing human colon cancer xenografts were intratumorally injected with 1 × 108 TCID50 of Ad5-Luc or Ad5F11b-Luc, and luciferase activity in tumors was quantified by bioluminescent imaging. As can be seen in Fig. 3B, photon counts in tumors injected with Ad5F11b-Luc were significantly higher than in tumors injected with Ad5-Luc (p < 0.001) at all time points. At day 2 post injection, average photon counts of tumors injected with Ad5F11b-Luc reached about 12-fold higher levels than those injected with Ad5-Luc. In addition, the luciferase signal could not be detected in any of the Ad5-Luc–injected tumors on day 33 post injection (0/4), but was still detectable on day 43 in 50% of Ad5F11b-Luc–injected tumors (2/4) (Fig. 3B).
Differential biodistribution of Ad5 and Ad5/F11 adenoviral vectors
The biodistribution of Ad5 and Ad5/F11 after intravenous injection of adenoviral vectors was examined by live cell imaging of normal nude mice that had been injected with either Ad5 or Ad5/F11. The majority of Ad5-Luc accumulated in the liver and resulted in high levels of transgene expression in the liver. Ad5-Luc–mediated transgene expression in the liver was detectable 16 days after intravenous injection of Ad5-Luc (1 × 108 TCID50). In contrast, luciferase signals of the livers of Ad5F11b-Luc–injected mice were very weak and rapidly dropped to baseline (Fig. 3C).
Furthermore, liver, spleen, lung, kidney, heart, intestine tissues, and serum were collected on days 2 and 8 post intravenous injection of either Ad5 or Ad5/F11, and luciferase activity was measured. Livers generally showed the highest luciferase levels, followed by spleen. Other tissues, such as lung, kidney, heart, intestine, and serum, exhibited negligible luciferase levels in both Ad5-Luc– and Ad5F11b-Luc–injected mice. Luciferase signals of the livers and spleens of Ad5-Luc–injected mice were 100-fold and 10-fold higher, respectively, compared with those injected with Ad5F11b-Luc (Fig. 3D).
Clearance of Ad5F11b and Ad5 from circulation was assessed by collecting blood samples retro-orbitally at 3 min, 5 min, 10 min, 20 min, or 60 min post tail vein injection of 1 × 108 TCID50 of Ad5-Luc or Ad5F11b-Luc in normal Balb/c nude mice. Serum was subsequently isolated and applied to HEK293 to measure residual viral particles in serum. Both Ad5 and Ad5/F11 were rapidly eliminated from circulation, i.e., no residual viral particles in serum were detected 20 min after intravenous injection (Fig. 3E) of either Ad5 or Ad5/F11.
Ad5F11b.wnt-E1A-hIL24 suppressed tumor cell growth by inducing apoptosis
The tumor cells HepG2, BEL7402, SMMC7721, HCT116/4016, and HCT116/379.2 and normal cells L02 were plated onto 96-well plates and infected with Ad5F11b.wnt-E1A, Ad5F11b.wnt-E1A-hIL24, or the replication-defective Ad5F11b-Luc as a control. Cytotoxic effects were detected using the MTT assay. Ad5F11b-Luc showed little or no cytotoxicity in all tested cell lines (Fig. 4A). In contrast, replication-competent adenoviruses Ad5F11b.wnt-E1A or Ad5F11b.wnt-E1A-hIL24 exhibited significant toxicity in infected cells. Ad5F11b.wnt-E1A-hIL24–induced toxicity was more apparent than that induced by Ad5F11b.wnt-E1A and correlated with MDA-7/IL24 expression levels as detected in the supernatant by ELISA (Fig. 4B). In addition, cytotoxicity was much more pronounced in HepG2, HCT116/4016, and HCT116/379.2 cells, which had been shown to have elevated wnt signaling. The cytotoxic effect was a function of viral load and incubation time of Ad5F11b.wnt-E1A-hIL24.
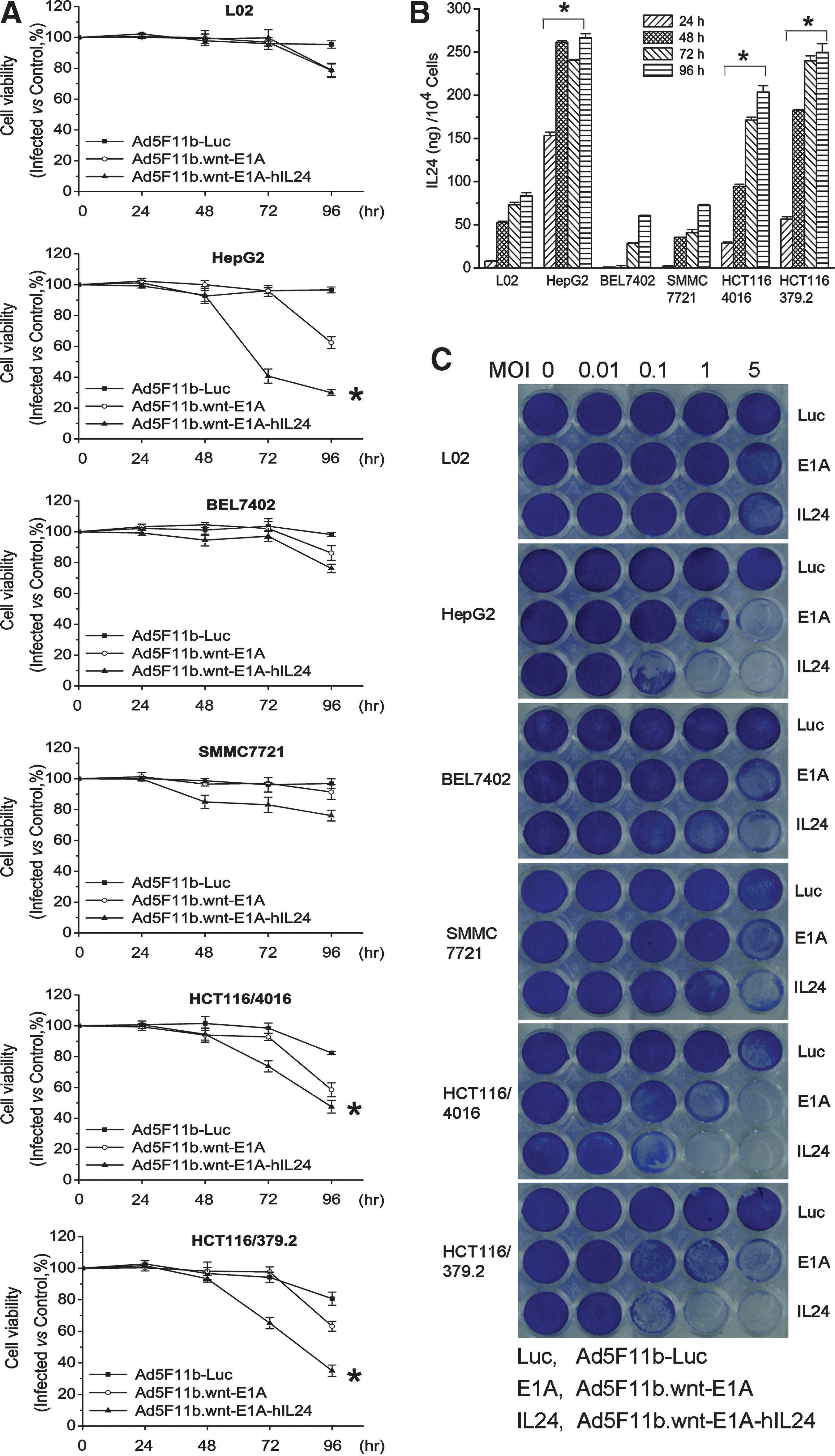
Cytotoxicity of oncolytic adenoviral vectors in vitro. (
The cell-killing effect of Ad5F11b-Luc, Ad5F11b.wnt-E1A, and Ad5F11b.wnt-E1A-hIL24 was further evaluated by infecting L02, HepG2, BEL7402, SMMC7721, HCT116/4016, and HCT116/379.2 cells at various multiplicities of infection (MOIs). After 96 hr, cells were stained with crystal violet, allowing the assessment of cell density. The results confirmed the data obtained by the MTT assay mentioned above and are shown in Fig. 4C.
To verify that Ad5F11b.wnt-E1A-hIL24 kills by inducing apoptosis in infected cells, L02, HepG2, BEL7402, SMMC7721, HCT116/4016, and HCT116/379.2 cells were infected with Ad5F11b-Luc, Ad5F11b.wnt-E1A, or Ad5F11b.wnt-E1A-hIL24 and analyzed by annexin V staining. In cells infected with Ad5F11b.wnt-E1A-hIL24 or Ad5F11b.wnt-E1A, a larger fraction of apoptotic cells was detected than in Ad5F11b-Luc–infected cells (Fig. 5A). Again, β-catenin/TCF mutation-carrying cell lines exhibited stronger annexin V staining after infection with Ad5F11b.wnt-E1A-hIL24 than cell lines with wild-type β-catenin, supporting the results from the previous experiment.
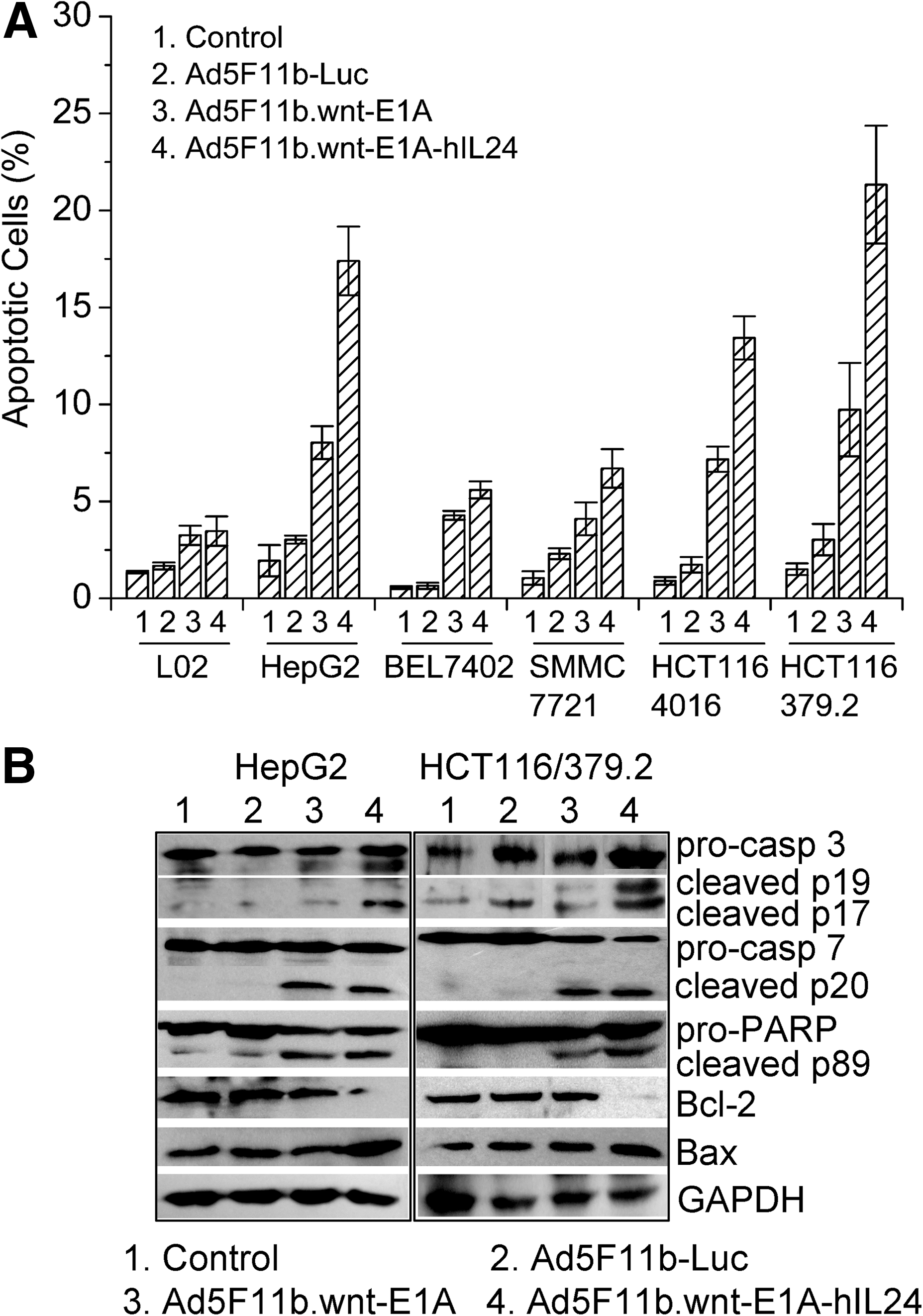
Apoptosis in Ad5F11b.wnt-E1A-hIL24–treated tumor cells. (
Apoptosis induction by Ad5F11b.wnt-E1A-hIL24 was further characterized by quantifying procaspase-3 and procaspase-7 cleavage, as well as cleavage of the caspase substrate PARP, by Western blot analysis. As shown in Fig. 5B, in HepG2 and HCT116/379.2 cells treated with Ad5F11b.wnt-E1A-hIL24, a larger fraction of cleaved caspase-3, caspase-7, and PARP could be detected compared with cells treated with Ad5F11b.wnt-E1A, Ad5F11b-Luc, or PBS.
In addition, increased expression of the pro-apoptotic molecular Bax and reduced expression of the anti-apoptotic Bcl-2 protein were detected in Ad5F11b.wnt-E1A-hIL24–infected cells (HepG2 and HCT116/379.2), but not in Ad5F11b.wnt-E1A– or Ad5F11b-Luc–infected or uninfected cells (Fig. 5B).
Ad5F11b.wnt-E1A-hIL24 suppressed human xenograft tumor growth in animal models
To determine whether Ad5F11b.wnt-E1A-hIL24 could effectively suppress tumor growth in vivo, we established human HCT116/379.2 subcutaneous tumors in nude mice.
When tumors reached a size of 80–100 mm3, they were injected with PBS, Ad5F11b-Luc, Ad5F11b.wnt-E1A, or Ad5F11b.wnt-E1A-hIL24, and tumor size was measured over a time period of 24 days. At the end of the observation period, tumors were excised and weighed. The average tumor volume and weight in the Ad5F11b.wnt-E1A-hIL24–treated group were significantly smaller (p < 0.01) compared with those of the PBS-, Ad5F11b-Luc–, and Ad5F11b.wnt-E1A–injected tumors (Fig. 6A, left and middle panels). In addition, Ad5F11b.wnt-E1A-hIL24 treatment resulted in a longer survival time of mice (p < 0.01) compared with other treated counterparts or controls (Fig. 6A, right panel).
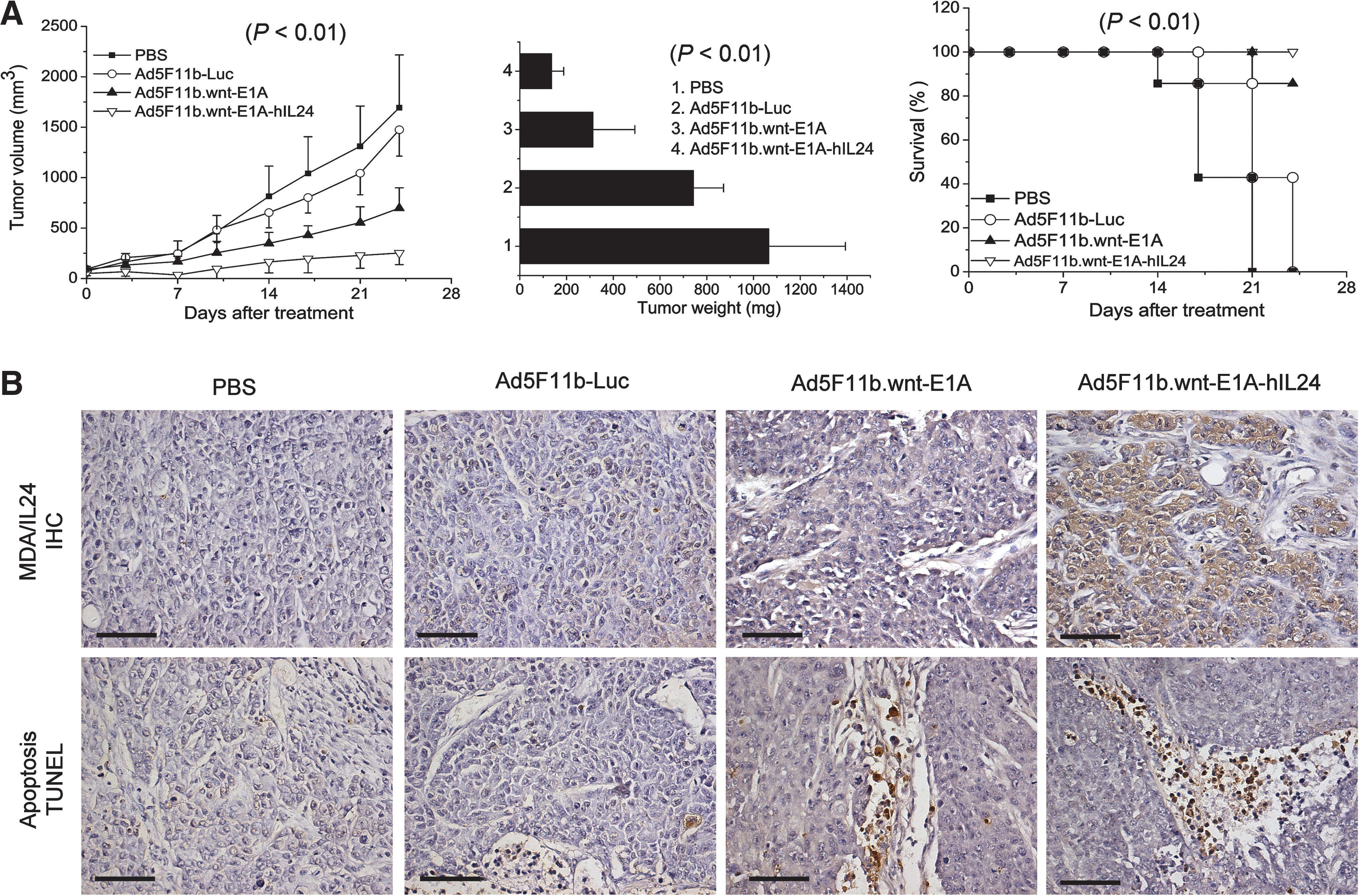
Tumor growth inhibition mediated by Ad5F11b.wnt-E1A-hIL24 in vivo. HCT116/379.2 human colon cancer xenografts in nude mice were treated with PBS or 5 × 108 TCID50 Ad5F11b-Luc, Ad5F11b.wnt-E1A, and Ad5F11b.wnt-E1A-hIL24. (
To verify that the therapeutic effect of Ad5F11b.wnt-E1A-hIL24 was, in fact, induced by the toxic effect of IL24, immunohistochemical staining and ELISA for IL24 expression in tumor tissues were performed. Tumors treated with Ad5F11b.wnt-E1A-hIL24 exhibited a strong IL24 staining (Fig. 6B, upper panel). In contrast, cells infected with IL24-deficient viruses did not exhibit any staining of IL24. IL24 protein concentration in Ad5F11b.wnt-E1A-hIL24–treated tumors was about 690 ± 184 pg/μg of total tumor protein. In concordance with our in vitro data, tumors that had been treated with Ad5F11b.wnt-E1A-hIL24 exhibited more extensive apoptosis as assessed by TUNEL assay (Fig. 6B, lower panel). In addition, IL24 expression and apoptosis were evaluated in liver sections from mice treated with PBS, Ad5F11b-Luc, Ad5F11b.wnt-E1A, and Ad5F11b.wnt-E1A-hIL24, and in all of these no staining could be detected (data not shown).
Discussion
The therapeutic effectiveness of commonly used oncolytic adenoviruses is mainly limited by their tumor specificity, transduction efficiency, half-life, and cytotoxic effectiveness.
In our strategy to design a novel oncolytic adenovirus for the treatment of gastrointestinal cancer, we have tried to optimize these properties by: (1) increasing specificity by linking viral replication to aberrant wnt signaling by replacing E1A promoter with β-catenin–responsive elements and deleting E1B coding sequence; (2) increasing viral transduction efficiency by pseudotyping with Ad11 fiber; and (3) increasing the cell-killing potency by introducing a therapeutic gene, IL24, which has been shown to effectively induce apoptosis in cancer cells.
It has been reported that one third to one half of gastrointestinal cancers harbor mutations in the β-catenin gene, resulting in increased nuclear β-catenin levels and transcriptionally activated expression of genes containing TCF binding sites (Morin, 1999; Giles et al., 2003). In addition, the wnt signaling pathway has been shown to be highly activated in cancer stem cells (Vermeulen et al., 2010). This has provided the rationale for us and others to use promoters containing TCF-responsive elements to control genes essential for adenoviral replication, as well as therapeutic genes, such as the apoptosis-inducing gene fadd. In early studies, this strategy has been shown to increase selective transgene expression in colon cancer cell lines by 50–100-fold (Chen and McCormick, 2001; Dihlmann and von Knebel Doeberitz, 2005).
To further optimize selectivity, the E1B region of Ad5F11b.wnt-E1A-hIL24 that encodes for two proteins, E1B-19kDa and E1B-55kDa, was deleted. The adenoviral E1B-19kDa protein, a Bcl-2 homologue, functions as a potent apoptosis inhibitor to protect infected cells from apoptosis by preventing Bax–Bak heterodimerization and mitochondrial pore formation. The E1B-55kDa gene product interacts directly with p53 and mediates its inactivation, translocation to the cytoplasm, ubiquitination, and degradation. Deletion of E1B-55kDa has been reported to increase viral selectivity toward p53-defective tumor cells due to delayed viral RNA export mediated by the loss of E1B-55kDa (O'Shea et al., 2004). The first recognized E1B-55kDa–deleted adenovirus, ONYX-015, can be regarded as the prototype of oncolytic adenoviral therapy (Doloff et al., 2008). In addition, E1B-19kDa and E1B-55kDa double-deleted oncolytic adenoviruses have been shown to exhibit an enhanced antitumor effect as compared with either E1B-19kDa or E1B-55kDa single-deleted oncolytic adenoviruses (Kim et al., 2002, 2009).
By replacing Ad5 fiber with Ad11 fiber, the viral tropism is shifted from cells expressing CAR receptors to cells expressing CD46 receptors. As CAR expression has been reported to be down-regulated whereas CD46 is up-regulated in most tumor cells (Coyne and Bergelson, 2005; Ravindranath and Shuler, 2007; Pache et al., 2008), we anticipated a higher transduction efficiency that we could confirm by live cell imaging using a luciferase-based replication-incompetent Ad11 pseudotyped adenovirus.
In addition to making use of a favorable CD46 expression profile, Ad5 represents a very common pathogen, and anti-Ad5 antibodies can be detected in more than 90% of the adult population. Thus, the serotype switch might reduce clearance by preexisting antibodies or immune cells directed against Ad5, thus increasing their half-life in vivo. However, as we have used immunocompromised mice in this study, we were not able to correlate prolonged transgene expression in Ad5/F11b-injected tumors with Ad11 pseudotyping.
Replication-incompetent adenoviruses armed with MDA-7/IL24 are currently being tested in clinical trials with promising results and little toxicity (Fisher et al., 2003; Cunningham et al., 2005; Lebedeva et al., 2007; Eager et al., 2008). MDA-7/IL24 is a multifunctional cancer-specific apoptosis-inducing cytokine (Gupta et al., 2006). It exerts strong antitumor activity through a variety of apoptotic signaling pathways. As expected, our oncolytic adenovirus armed with MDA-7/IL24 killed tumor cells more effectively than the same virus lacking the mda-7/IL24 expression cassette. Our results indicated that the enhanced cell-killing effect of Ad5F11b.wnt-E1A-hIL24 was indeed mediated by apoptosis induction, as demonstrated by cleavage of caspase-7, caspase-3, and PARP, as well as by activation of pro-apoptotic molecules such as Bax and inhibition of anti-apoptotic Bcl-2 family members such as Bcl-2.
In summary, we could show that our modified oncolytic adenoviruses led to significantly improved tumor cell transduction, long-lasting therapeutic gene expression, and potent growth inhibition in human gastrointestinal xenograft tumors. We think that selectively targeting colorectal tumor cells with aberrant wnt signaling using an armed oncolytic adenovirus is a promising new approach in the treatment of colorectal cancer, warranting further clinical investigation. In addition, our strategy could be modified to work in a variety of solid tumors.
Footnotes
Acknowledgments
This work was supported by Grant of State Key Laboratory of Oncogenes and Related Genes (80-07-03), National Basic Research Project of China (2010CB529902), National High-Tech R&D Program (2007AA021202), and National Natural Science Foundation for Outstanding Youth (30325043, 30428015).
Author Disclosure Statement
No competing financial interests exist for all authors.