
Abstract
Multipotent mesenchymal stromal or stem cells (MSCs) are likely to be agents of connective tissue homeostasis and repair. Because the hallmark of osteoarthritis (OA) is degeneration and failure to repair connective tissues it is compelling to think that these cells have a role to play in OA. Indeed, MSCs have been implicated in the pathogenesis of OA and, in turn, progression of the disease has been shown to be therapeutically modulated by MSCs. This review discusses current knowledge on the potential of both marrow- and local joint-derived MSCs in OA, the mode of action of the cells, and possible effects of the osteoarthritic niche on the function of MSCs. The use of stem cells for repair of isolated cartilage lesions and strategies for modulation of OA using local cell delivery are discussed as well as therapeutic options for the future to recruit and appropriately activate endogenous progenitors and/or locally systemically administered MSCs in the early stages of the disease. The use of gene therapy protocols, particularly as they pertain to modulation of inflammation associated with the osteoarthritic niche, offer an additional option in the treatment of this chronic disease. In summary, elucidation of the etiology of OA and development of technologies to detect early disease, allied to an increased understanding of the role MSCs in aging and OA, should lead to more targeted and efficacious treatments for this debilitating chronic disease in the future.
Osteoarthritis
Stem Cells
Stem cells represent an important element in regenerative strategies for tissue repair by virtue of their availability in large numbers and relative ease of preparation and delivery.
Embryonic stem cells (ESCs), although pluripotent with the potential to address regeneration of tissues in all germ lines, have ethical issues associated with their use. These issues may be overcome by the use of induced pluripotent stem cells (iPSCs) generated by reprogramming of human somatic cells (Jaenisch and Young, 2008; Nishikawa et al., 2008; Tweedell, 2008). iPSCs have a similar, although not identical, phenotype to ESCs. Reports highlight major differences between both cell lines: ESCs generated from human embryos carrying the fragile X mutation demonstrated silencing of the FMR1 gene after differentiation, whereas this gene was not activated in iPSCs derived from fibroblasts of affected individuals carrying the mutation despite successful reprogramming to pluripotency (Urbach et al., 2010). Human iPSCs, although capable of differentiating to hematopoietic derivatives, did so with significantly decreased efficiency compared with ESCs. Furthermore, the iPSC-derived hemangioblasts demonstrated increased apoptosis, limited growth and expansion capability, and rapid onset of senescence (Feng et al., 2010). Clinical use of both cell types may require the development of predifferentiation protocols and depletion of the residual parent population before introduction into patients. Alternatively, there is increasing evidence that most adult tissues have a resident stem or progenitor cell population with a critical role in homeostasis and tissue repair. Adult stem cells have a more restricted differentiation potential than pluripotent stem cells, but are currently used in clinical practice (e.g., hematopoietic stem cells for marrow transplantation) and clinical testing of other adult stem cells, including multipotent mesenchymal stromal or stem cell (MSC) preparations, has progressed rapidly (Ankrum and Karp, 2010).
MSCs were first described by Friedenstein, who identified a subpopulation of cells within the stromal compartment of bone marrow with osteogenic potential (Friedenstein et al., 1966, 1970, 1974). These adherent, fibroblastic-like cells were shown to be capable of forming colonies from a single cell (colony-forming unit fibroblastic [CFU-F]) and had the capacity to form multiple skeletal tissues in vivo (Owen and Friedenstein, 1988; Friedenstein, 1995). Subsequently, individual, clonal populations of stem cells in human bone marrow were identified that retained the potential to differentiate into chondrocytes, osteocytes, and adipocytes (Pittenger et al., 1999). Since 1999, MSCs have been isolated and characterized from many other human sources including adipose tissue (Gimble et al., 2007; Meliga et al., 2007; Bunnell et al., 2008), umbilical cord blood, and Wharton's jelly (Weiss and Troyer, 2006; Weiss et al., 2006; Flynn et al., 2007; Troyer and Weiss, 2008). They have the capacity to differentiate into cells of connective tissue lineages, including bone, fat, cartilage, and muscle. Bone marrow-derived MSCs have an additional potential in providing the stromal support system for hematopoietic stem cells (Barry and Murphy, 2004; Delorme and Charbord, 2007; Bianco et al., 2008). It has also been suggested that stromal cells derived from bone marrow may be capable of vascular smooth muscle differentiation in long-term cultures (Dennis and Charbord, 2002) and cells with MSC characteristics isolated from arteries and microvessels have been described as multipotential “pericyte-like” stem cells (Mody et al., 2003; Tintut et al., 2003; Abedin et al. 2004; Tavian et al., 2005; da Silva Meirelles et al., 2006). These cells have the capacity to differentiate to multiple lineages (Shi and Gronthos, 2003; Tintut et al., 2003) and various reports have suggested a perivascular niche as the source of MSCs derived from bone marrow, skeletal muscle, brain, and fat (Shi and Gronthos, 2003; Tavian and Peault, 2005; Sacchetti et al., 2007; Zannettino et al., 2008). Prospective isolation techniques identified progenitor cells from blood vessels in various tissues including skeletal muscle and adipose tissue that exhibited multipotentiality at a clonal level and expressed MSC markers in both cultured and noncultured cells. Perivascular cells were proposed as the precursors for MSCs and other adult stem cells (Crisan et al., 2008), leading to the hypothesis that all MSCs are pericytes (Caplan, 2008).
MSCs from joint tissues
Isolation of MSCs from joint tissue was first reported in 2001 when multipotent mesenchymal cells with the capacity for chondrogenesis, osteogenesis, adipogenesis, and sporadic myogenesis were grown from a digest of adult human synovial membrane (De Bari et al., 2001b). As for marrow-derived MSCs, clonal heterogeneity has been demonstrated for MSCs isolated from synovium with progenitors of varying proliferative capacity and distinct mesenchymal differentiation potency described (Karystinou et al., 2009). Interestingly, we have shown that marrow-derived MSCs, whether injected into a healthy or injured joint, engrafted primarily to synovial tissue or perichondrium with no cells detected at the surface of normal or damaged articular cartilage (Murphy et al., 2003). De Bari and coworkers also demonstrated in vivo myogenesis with synovium-derived MSCs contributing to myofibers and functional satellite cells in regenerating nude mouse muscle (De Bari et al., 2003). However, a more recent report suggested that synovium-derived MSCs had a limited capacity to undergo myogenic differentiation in vitro or to contribute to muscle regeneration in vivo (Meng et al., 2010). MSCs have also been isolated from normal and early osteoarthritic synovial fluid; these authors acknowledged that the source of the cells was likely degrading synovium. Interestingly, in comparison with matched bone marrow-derived cells, synovial fluid cells showed greater clonogenicity and chondrogenic differentiation capacity (Jones et al., 2008). In general, synovium-derived MSCs seem to have a more chondrogenic phenotype than those derived from bone marrow or infrapatellar fat pad (Sakaguchi et al., 2005; Lee et al., 2010).
Progenitor cells at the surface of healthy articular cartilage were originally described in bovine tissue (Dowthwaite et al., 2004). When prepared as clonal populations these cells had greater growth potential and higher telomerase activity than dedifferentiated chondrocytes isolated from the same source (Khan et al., 2009). Mesenchymal-like cells with progenitor characteristics were subsequently identified in normal and osteoarthritic human articular cartilage (Alsalameh et al., 2004; Fickert et al., 2004) and were shown to undergo chondrogenic and osteogenic, but not adipogenic, differentiation (Grogan et al., 2009). Pacifici and co-workers have linked Gdf5 expression with progenitor potential by crossing a ROSA-LacZ-reporter with Gdf5-Cre mice to track events at prospective joint sites. LacZ-positive cells initially constituted the interzone and persisted in joint-forming areas throughout development. These progenitor cells formed articular cartilage and synovial lining as well as other joint tissues. However, they contributed minimally if at all to underlying growth plate cartilage and bone growth, indicating that a population of Gdf5-expressing mesenchymal progenitors uniquely contributed to articular cartilage (Koyama et al., 2008).
Mode of action
MSCs remain at the forefront of current translational efforts in cellular therapy for a broad spectrum of diseases. However, some controversy exists as to the primary effect of the cells in the injured environment: do MSCs undergo tissue-specific differentiation or act in a paracrine manner to produce soluble reparative factors, or is the therapeutic effect a consequence of both mechanisms? Data suggest that the therapeutic potential of these cells, at least in some applications, is related to paracrine effects such as the release of factors that (1) modulate the immune response, (2) mobilize or promote host cell survival, (3) recruit and induce mitosis of endogenous tissue progenitor cells at the site of injury while stimulating an angiogenic response, or (4) prevent an inappropriate fibrotic response (for reviews see Caplan, 2009; Oh et al., 2010). However, studies in the area of cardiac repair question the concept that MSCs act solely through paracrine mechanisms. A study in pigs using allogeneic MSCs for the treatment of chronic ischemic cardiomyopathy suggested that long-term engraftment and trilineage differentiation to cardiomyocytes, vascular smooth muscle cells, and endothelial cells contributed at least in part to an improvement in cardiac function (Quevedo et al., 2009). However, the trilineage differentiation shown was minimal.
Immunomodulatory effects
Factors produced by MSCs in response to the inflammatory environment include interleukin (IL)-10, IL-1 receptor antagonist (IL-1Ra), and transforming growth factor (TGF)-β (Pittenger, 2009). Indeed, the establishment of an immunosuppressive local milieu has been proposed as a major factor in the immune privilege of MSCs (English et al., 2008, 2009). There is evidence that hematopoietic cells may provide the “licensing” signal for MSCs (English et al., 2007; Polchert et al., 2008) to deliver immunosuppressive signals including IL-10 and prostaglandin E2 (PGE2). Upregulation of IL-10 in the brain has been associated with the therapeutic potential of MSCs in reducing neuronal injury after an ischemic injury in rats (Liu et al., 2009) and increased IL-10 production by host macrophages in response to MSCs has been described to occur through a PGE2-dependent mechanism in treating sepsis in mice (Németh et al., 2009). These results and others have led to studies assessing gene therapeutic strategies. In particular, systemic administration of genetically modified MSCs overexpressing IL-10 in a model of experimental arthritis resulted in the inhibition of symptoms through suppression of the autoimmune response as well as the production of cytokines such as IL-4 (Choi et al., 2008).
Paracrine effects
The beneficial paracrine effects of MSCs have been demonstrated in the articular joint via administration of cells after surgically induced injury. Local delivery of autologous caprine MSCs in a solution of hyaluronan to a meniscectomized stifle joint resulted in significantly increased regeneration of meniscal tissue and chondroprotection when compared with meniscectomized joints treated with hyaluronan alone (Murphy et al., 2003). However, it was readily apparent that the green fluorescent protein (GFP)-labeled MSCs used in the study colonized just a small proportion of the regenerated meniscus. It was therefore concluded that the implanted MSCs induced a host repair response through the release of paracrine factors to replace the resected medial meniscus. Similarly, a model involving subcutaneous implantation of hydroxyapatite scaffolds loaded with MSCs into syngeneic, allogeneic, or immunocompromised mice was used to assess the origin of bone routinely found in the scaffold after an extended period in vivo. Interestingly, no bone was formed in the allogeneic setting as a result of rapid destruction of the cells. However, in syngeneic and immunocompromised recipients the implanted cells were found to be pivotal to bone formation, but tissue formation was dependent on recruited recipient osteoprogenitors (Tasso et al., 2009). The accumulation of human MSCs has also been associated with an increased number of oligodendrocytes in lesion areas of mice with experimental allergic encephalomyelitis (Bai et al., 2009).
The antifibrotic action of MSCs was first described in a bleomycin-induced lung injury model in which murine MSCs, administered after exposure to the bleomycin, homed to the injured lung and reduced not only the inflammatory response but also collagen deposition (Ortiz et al., 2003). IL-1Ra, secreted by the MSCs, was found to be integral to this process and acted by blocking tumor necrosis factor (TNF)-α and IL-1 in the lung (Ortiz et al., 2007). An immunosuppressive mechanism of action for MSCs was also demonstrated in carbon tetrachloride-induced liver injury resulting in suppressed fibrosis, an effect specific for MSCs as administration of hematopoietic stem cells resulted in acute inflammation and subsequent fibrosis (Pulavendran et al., 2010).
Stimulation of angiogenesis
Increased angiogenesis has been shown to occur in late OA. MSCs can promote early angiogenic events, a mechanism known to contribute to tissue repair, by increasing endothelial cell (EC) proliferation and migration in vitro as well as significantly increasing the stability of vessels formed by ECs through a cell contact-mediated mechanism (Duffy et al., 2009). Similarly, MSC differentiation to endothelial and smooth muscle cells was found to occur in support of new blood vessel formation in the kidney (J. Chen et al., 2008). Other studies support a paracrine mechanism for MSC reconstitution of the microcirculation or collateral perfusion (Kinnaird et al., 2004; Ladage et al., 2007) through molecules such as secreted frizzled-related protein-1 or Cyr61 (Dufourcq et al., 2008; Estrada et al., 2009).
In contrast to the paradigm of MSCs influencing healing or repair through paracrine mechanisms, early tissue engineering principles of cell therapy were based on the ability of the delivered cells to engraft, differentiate, and contribute to the formation of repair tissue. Differentiation remains an important avenue of research for tissue engineering applications and MSCs remain at the forefront of current translational efforts in cellular therapy for a broad spectrum of diseases. Because our understanding of the mode of action of MSCs in specific applications is incomplete, it is not possible to rule out cell differentiation, whether overt as may be the case for connective tissue applications or minimal in other scenarios, as a factor in therapeutic efficacy.
MSCs in osteoarthritis
Inherent differences between MSCs derived from healthy or osteoarthritic bone marrow have been well documented. A number of years ago MSCs isolated from the marrow of patients undergoing joint replacement surgery were compared with cells from healthy, age-matched control subjects with no evidence of OA. The proliferative rates of MSCs derived from osteoarthritic marrow were significantly reduced compared with healthy controls. Furthermore, the differentiation profile of the osteoarthritic MSCs was altered, with reduced chondrogenic and adipogenic activity but increased osteogenesis (Murphy et al., 2002). These changes were systemic in that osteoarthritic MSCs isolated from iliac crest or tibial and femoral components were comparable. Although these cells were cultured in selected fetal calf serum without supplemental growth factors, studies using growth factor-complemented medium for the establishment and growth of MSC populations did not show decreased proliferation of or chondrogenesis by cells isolated from the patients with OA (Scharstuhl et al., 2007), although it must be noted that quantitation of proteoglycan deposition per cell was not performed in this study. Indeed, the continuing controversy about the effects of age and disease on proliferation and functional properties of MSC populations may be attributed to culture conditions and the inability to recapitulate either the aging or indeed the osteoarthritic niche in vitro. For example, human periosteal MSCs have spontaneous chondrogenic activity in cultures from donors younger than 30 years that is lost with passaging and absent from older donors (De Bari et al., 2001a), a deficiency overcome with fibroblast growth factor (FGF)-2 supplementation (Im et al., 2006).
Synovial fluid, the milieu to which cells within the joint are exposed, changes dramatically with OA onset (for review see Goldring and Goldring, 2007). Chondrocytes, synovial cells, and perhaps even joint progenitor cells contribute and respond to cytokines, chemokines, and antioxidant levels in the diseased synovial fluid, resulting in oxidative stress. For example, the inflammatory mediators IL-1β and TNF-α are increased in OA synovial fluid and have dramatic effects on chondrogenesis of stem cells. Figure 1 shows the effect of 5% osteoarthritic synovial fluid on chondrogenesis of goat bone marrow MSCs. Chondrogenesis induced by TGF-β3 was significantly inhibited by the addition of OA synovial fluid and preexposure of the early micromass culture to the fluid before addition of TGF-β3 had an even more dramatic effect. Inhibition of Sox9, the putative master chondrogenic transcription factor, by inflammatory cytokines present in synovial fluid was first described in 2000 (Murakami et al., 2000). More recently, the inhibitory effect of the cytokines on MSC chondrogenesis was shown to be mediated through NF-κB-dependent pathways (Wehling et al., 2009). Interestingly, the NF-κB pathway was also implicated in inflammation-associated responses of fibroblast-like synoviocytes (FLSs) in rheumatoid arthritic pannus. A major proportion of FLSs was shown to be marrow-derived, with the capacity to differentiate to mesenchymal lineages. These researchers showed increased proliferation and repression of the osteogenic and adipogenic differentiation of FLSs with inhibition of NF-κB, and proposed that arthritic FLSs were MSCs with arrested differentiation in response to the arthritic inflammatory environment (Li and Makarov, 2006).
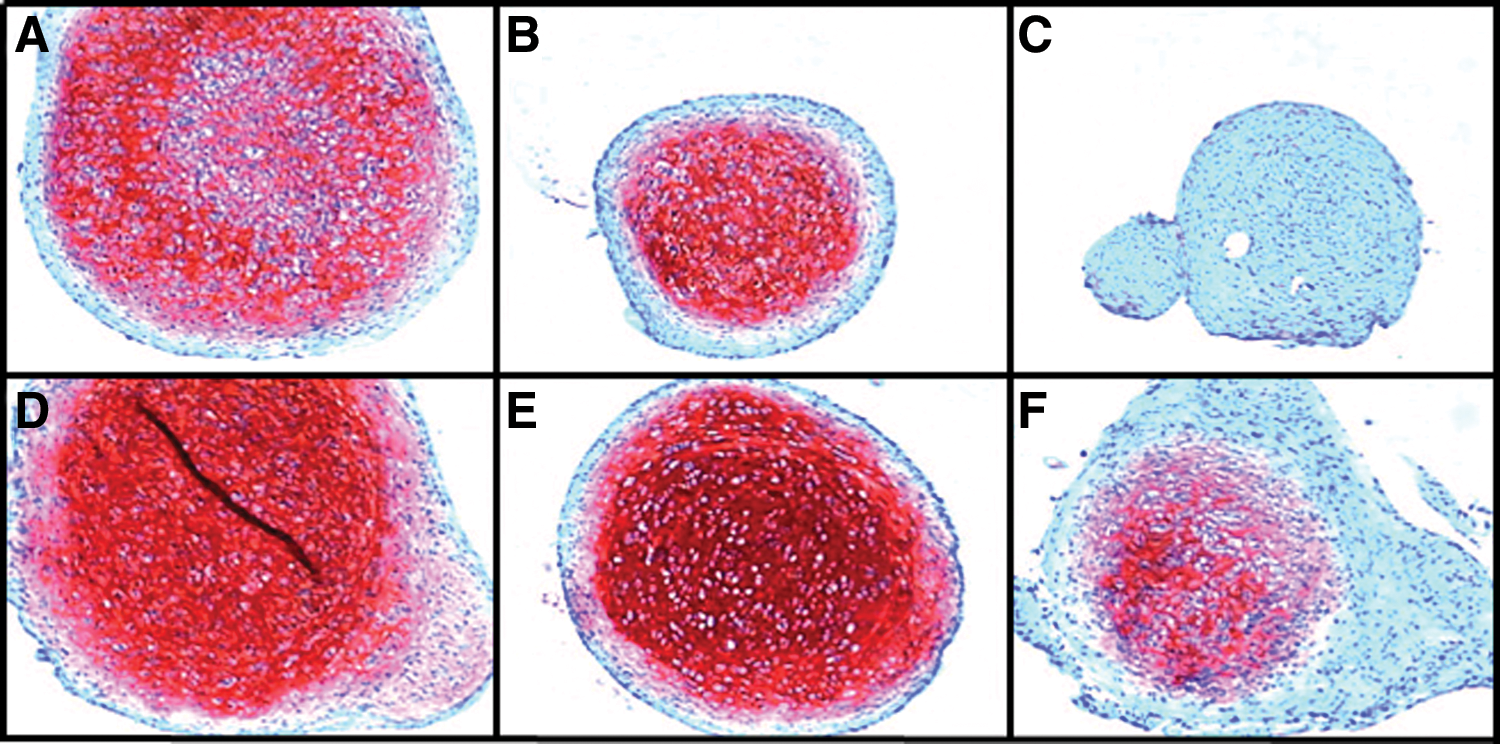
Potential effects of the osteoarthritic joint niche on chondrogenesis of MSCs. MSCs, isolated from goat bone marrow, were placed in pellet culture and induced to differentiate in the presence of TGF-β3 for 7 days (top) and 14 days (bottom) as described previously (Murphy et al., 2003). Synovial fluid was harvested from osteoarthritic goat knees and maintained at 5% (v/v) throughout the culture with 10-ng/ml TGF-β3
Aberrant progenitor function has also been suggested as pertinent to the osteoarthritic changes that occur in articular cartilage. Notch-1-positive cartilage progenitor cells have been isolated from immature cartilage and a number of groups have described an increased incidence of these cells in osteoarthritic cartilage (Alsalameh et al., 2004; Fickert et al., 2004; Hiraoka et al., 2006). However, one study found that more than 45% of cells in normal and osteoarthritic cartilage were positive for the putative progenitor markers Notch-1, Stro-1, and vascular cell adhesion molecule (VCAM)-1. In contrast, the progenitor cartilage side population isolated by fluorescence-activated cell sorting (FACS) represented just 0.14% of the cellular complement in both tissues and this population had chondrogenic and osteogenic capacity. These data led to the suggestion that the markers may not be useful for identification of cartilage progenitors but rather contribute to the abnormal cell activation and differentiation process characteristic of OA (Grogan et al., 2009).
Intriguingly, migratory cells not normally found in healthy articular cartilage add to the complexity of progenitor populations in degenerating cartilage. A clonal, multipotent population in repair tissue associated with late-stage OA (Koelling et al., 2009) was hypothesized to migrate from blood vessels occupying fissures or breaks in the tidemark of vascularized cartilage tissue. Although maintaining an osteochondroprogenitor phenotype, they do not differentiate into fully committed chondrocytes in situ, but instead are possibly restricted to a fibrocartilaginous phenotype providing temporary repair due to the absence of appropriate differentiation cues (Khan et al., 2009).
MSCs may also play a role in osteophyte formation, perhaps another attempt at repair to counteract the altered architecture of the injured or OA joint. Osteophytes are thought to be derived from precursors in the periosteum or perichondrium in response to growth factors such as TGF-β and develop through endochondral ossification (van der Kraan and van den Berg, 2007). Mice with a targeted disruption of mitogen-inducible gene 6 develop early-onset OA characterized by significant joint enlargement and deformity associated with osteophyte formation. Furthermore, proliferation of mesenchymal progenitors followed by chondrogenesis was shown to be associated with the osteophyte formation (Zhang et al., 2005).
Stem Cell Therapy for Osteoarthritis
Maintenance or restoration of a fully functional joint with biomechanically stable articular cartilage remains the holy grail of therapeutic or regenerative strategies in OA. In reality, complete cartilage degeneration requires total joint replacement through artificial implants and clinical intervention for cartilage repair is beneficial only when focal lesions are small enough to be reconstituted with cells or reparative tissue from various sources (Redman et al., 2005). Operative interventions for the treatment of cartilage lesions include the transplantation of osteochondral grafts (mosaicplasty), microfracture, and autologous chondrocyte implantation (ACI) with or without a scaffold matrix (MACI; matrix-induced autologous chondrocyte implantation) to deliver reparative cells (Bentley et al., 2003; Hangody and Fules, 2003; Knutsen et al., 2004; Bartlett et al., 2005). A major limitation of all these strategies is the inability to treat large defects (Steinert et al., 2007), thus excluding patients with OA.
Much of the early work on the preclinical evaluation of MSCs in musculoskeletal applications also focused on repair of articular cartilage and bone. Cells were delivered to defects in cartilage or bone in procedures requiring the application of a cell-loaded three-dimensional solid scaffold implanted through an open surgical procedure. As early as 1994, researchers described repair of defects in articular cartilage in rabbits (Wakitani et al., 1994) and humans (Wakitani et al., 2002) and segmental-defect bone repair (Bruder et al., 1998). The first human trial assessing the ability of MSCs to promote repair a large musculoskeletal defect was reported in 2001 with successful healing of a large bone defect (Quarto et al., 2001). This group performed a 6- to 7-year follow-up on four patients and reported integration of implants and no evidence of late fractures in the implant zone, indicating long-term durability of this tissue engineering approach with autologous bone marrow MSCs loaded on a macroporous bioceramic scaffold (Marcacci et al., 2007).
Efforts have focused on the use of MSCs as a therapy for traumatic OA. Murphy and colleagues have used a somewhat simpler scaffold-free approach for the treatment of OA associated with meniscal injury. Delivery of autologous MSCs, retrovirally transduced to express green fluorescent protein, to caprine joints subjected to total meniscectomy and resection of the anterior cruciate ligament resulted in regeneration of meniscal tissue and significant chondroprotection (Murphy et al., 2003). Evidence of cell engraftment to the synovium and the perichondrium as well as the surface lining of other joint tissues including the lateral meniscus and fat pad was noted. Although a significant number of labeled cells was detected at the surface of regenerated meniscus, and to a much lesser extent embedded in the repair tissue, the primary reparative response was host-derived and presumably resulted from interaction between the implanted stem cells and synoviocytes or endogenous progenitors at the site of injury. MSCs derived from synovium have been similarly used to treat meniscal defects. These cells adhered to meniscal lesions and differentiated into meniscal or collagen type II-expressing fibrocartilage cells. In one study the cells promoted meniscal regeneration (Horie et al., 2009) but no therapeutic benefit was found in an earlier study (Mizuno et al., 2008). The same strategy of direct intraarticular injection of MSCs resulted in improved repair of chondral defects in a pig model (Lee et al., 2007).
Delivery of allogeneic MSCs to the destabilized joint after total or partial meniscectomy also resulted in protection against the development of osteoarthritic symptoms (our unpublished results). However, host protection by transplanted MSCs in the OA models was not complete and all treated joints displayed some degree of damage to the articular cartilage.
Stem cell targeting
Adequate tissue repair strategies may require specific cellular targeting to the site of injury as retention and engraftment of transplanted cells are inadequate. Although cartilage has an easily accessible repertoire of chondrogenic progenitor cells that are capable of mitotic division and chondrogenic differentiation (Dowthwaite et al., 2004; Koelling et al., 2009), it is incapable of self-regeneration in adults (Wei and Messner, 1999). Conceivably, these progenitor cells that localize to the surface zone in normal cartilage are compromised or lost early in the osteoarthritic process. We therefore proposed that targeted delivery of progenitor cells in early OA may increase MSC engraftment and impact progression of the osteoarthritic process. Although no studies have been performed on cell targeting to osteoarthritic cartilage to date, prechondrocytes have been targeted to an osteochondral defect in a rabbit model using “cell-painting” techniques (Dennis et al., 2004) whereby the cells were coated with lipidated protein G to enable attachment of selected antibodies that could in turn bind specific cartilage matrix molecules. In another study, CD44 present on MSCs was enzymatically converted to confer potent E-selectin/L-selectin-like binding affinity to enhance targeting of MSCs to bone marrow (Sackstein et al., 2008). Although MSCs introduced into the normal or osteoarthritic joint do not engraft to the cartilage surface (Murphy et al., 2003) these cells will attach and populate deep fissures of fibrillated cartilage after a 20-min incubation with gentle agitation, using an ex vivo cartilage explant model. The attached MSCs will ultimately form a regenerated surface after exposure to chondrogenic differentiation medium (Fig. 2).
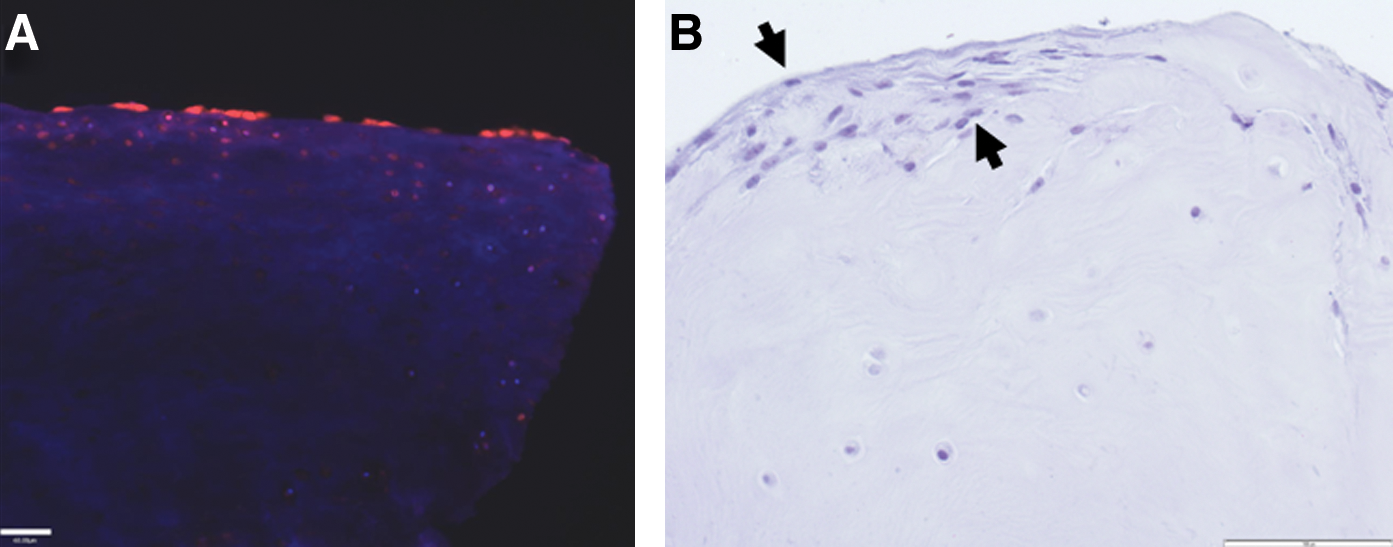
Targeting of MSCs to human osteoarthritic cartilage.
Gene Therapy for Osteoarthritis
As OA is not a systemic disorder, but is instead limited to enclosed joints, it is uniquely suited to therapeutic interventions delivered via gene therapy. Gene delivery therefore offers a method for localized, continued overexpression of a therapeutic agent with the aim of suppressing OA-associated chondrocyte apoptosis, supporting cell viability and stimulating the deposition of a healthy cartilage-like extracellular matrix (ECM) containing collagen type II and sulfated glycosaminoglycans (GAGs).
Genetic modification of therapeutic cells, or of host tissue directly, to continually overexpress anabolic factors may support the adaptation of therapeutic progenitor cells to a chondrocyte-like phenotype, as well as enhanced deposition of a cartilaginous ECM. For example, virally induced expression of Sox9 alone (Tew et al., 2005; Cucchiarini et al., 2007), or in combination with Sox5 and Sox6 (Ikeda et al., 2004), resulted in the induction of a chondrogenic phenotype, even in OA chondrocytes or in nonchondrogenic cell lines. Further, coexpression of Sox5, Sox6, and Sox9 will suppress the hypertrophic or osteogenic differentiation of transformed cells, offering an enhancement over direct cell therapy for modulation of OA. Similarly, the introduction of β-1,3-glucuronosyltransferase 1, a GAG-synthesizing enzyme, into chondrocytes has been demonstrated to stimulate an abundance of GAG production even in the presence of IL-1β-induced proteoglycan depletion (Venkatesan et al., 2004).
Stimulation of cellular proliferation by viral overexpression of mitogenic factors, or suppression of cell death via overexpression of antiapoptotic factors, may both inhibit OA disease progression as well as stabilize the articular tissue. Viral overexpression of FGF-2 in chondrocytes has been demonstrated to stimulate proliferation in vitro as well as improved tissue repair when transfected cells are delivered in vivo (Cucchiarini and Madry, 2005; Yokoo et al., 2005; Kaul et al., 2006). Further, the combination of FGF-2 and Sox9 overexpression in human OA chondrocytes stimulated mitogenesis, stimulated GAG and collagen type II deposition, and inhibited cellular hypertrophy (Cucchiarini et al., 2009). Alternatively, the overexpression of antiapoptotic Bcl-2 (Surendran et al., 2006), or the serine proteinase inhibitor kallistatin, offers a means by which native chondrocytes can be protected from OA-induced cell death (Wang et al., 2005; Hsieh et al., 2009).
As the TGF-β superfamily is paramount for the development of cartilage and supports cartilage-like ECM deposition by chondrocytes, it is only logical to genetically overexpress TGF-β superfamily members in an effort to induce the secretion of a hyaline-associated matrix. Several alternative methodologies to overexpress TGF-β1 have been investigated including light-activated gene transduction (Ito et al., 2004) or Lipofectamine (Guo et al., 2007). These studies resulted in successful protein overexpression, the enhancement of cartilaginous ECM protein production, and enhanced chondral repair in conjunction with the suppression of the cartilage-degrading matrix metalloproteinases MMP-1 and −3 (Guo et al., 2007). Adeno-associated virus (AAV)-based overexpression of TGF-β1 in MSCs (Pagnotto et al., 2007) or healthy or OA chondrocytes (Ulrich-Vinther et al., 2005) also resulted in the synthesis of type II collagen and aggrecan, the suppression of MMP-3 (Ulrich-Vinther et al., 2005), and the repair of osteochondral defects (Pagnotto et al., 2007). Similar results were obtained by retroviral (Lee et al., 2005) or adenoviral vector expression, whereby proteoglycan synthesis was enhanced with TGF-β1 alone (Blaney Davidson et al., 2007) or in combination with insulin-like growth factor (IGF) overexpression (Smith et al., 2000). Similarly, production of bone morphogenetic protein (BMP)-7, a member of the TGF-β superfamily, by adenovirus in bovine chondrocytes (Hidaka et al., 2001) or by retrovirus in MSCs (Mason et al., 1998) has been demonstrated to result in enhanced GAG and type II collagen secretion as well as the suppression of chondrocyte hypertrophy (Smith et al., 2000; Hidaka et al., 2001), resulting in bone and articular cartilage regeneration in osteochondral defects (Mason et al., 1998).
Modification of the inflammatory cascade in OA has been extensively investigated through genetic modification. Suppression of NF-κB via adenoviral overexpression of small interfering RNA (siRNA) in the synovium and cartilage has been demonstrated to result in the inhibition of synovial inflammation and cartilage degradation by suppressing Cox-2, nitric oxide synthase (NOS)-2, and MMP-9 in IL-1β- and TNF-α-induced arthritis (Lianxu et al., 2006; L.X. Chen et al., 2008). A similar inhibition of inflammation was observed after IL-4 overexpression in a canine model, resulting in suppression of inflammatory mediators prostaglandins and MMPs (Rachakonda et al., 2008).
Adenoviral overexpression of IGF-1 has been demonstrated to be safe in an equine model when directly administered into the joint (Goodrich et al., 2006), making it an attractive vector for OA human gene therapy. Transfection of articular chondrocytes with AdIGF-1 results in cartilage ECM gene and protein expression, as well as the beneficial inhibition of chondrocyte dedifferentiation (Nixon et al., 2000; Brower-Toland et al., 2001). In addition, AdIGF-1-transfected chondrocytes, when administered intraarticularly, contribute to improved cartilage morphology and the deposition of hyaline-like tissue in cartilaginous lesions (Goodrich et al., 2007). Simple plasmid-based overexpression of IGF-1 in rabbit chondrocytes, when encapsulated in alginate and delivered in vivo, results in improved cartilage repair and accelerated subchondral bone formation in osteochondral defects (Madry et al., 2005).
By far, the most extensively investigated mechanism for regulating inflammation is the overexpression of IL-1Ra to inhibit IL-1-mediated OA inflammation. Overexpression of IL-1Ra by retrovirus in equine, canine, or lapine synovial tissues regularly results in reduced lesion severity or cartilage degradation (Pelletier et al., 1997; Frisbie and McIlwraith, 2000; Zhang et al., 2004) in the setting of OA, an effect enhanced by the coadministration of IL-10 (Zhang et al., 2004). Similarly, AAV-based overexpression of IL-1Ra has been demonstrated to reduce OA-associated lameness and to improve cartilage morphology in an equine model (Frisbie et al., 2002). Adenoviral overexpression of IL-1Ra similarly suppresses inflammation (Kay et al., 2009) and protects OA chondrocytes from IL-1-induced GAG degradation (Baragi et al., 1995). Alternatively, delivery of the IL-1Ra gene via chitosan nanoparticles (Zhang et al., 2006) or liposomes (Fernandes et al., 1999) demonstrated a similar suppression of cartilage lesion development without the use of a viral vector. The delivery of IL-1Ra via a gene-based mechanism was shown to be beneficial over treatment with recombinant protein (Gouze et al., 2003), possibly due to sustained protein expression.
The coadministration of IGF-1- and IL-1Ra-containing vectors offers a unique proanabolic response while suppressing catabolic mediators where the suppression of IL-1α, IL-1β, and associated MMPs by IL-1Ra, is accompanied by IGF-1-stimulated type II collagen and GAG deposition (Haupt et al., 2005). In the setting of a microfractured full-thickness chondral defect, the coadministration of AdIL-1Ra and AdIGF-1 demonstrated increased type II collagen and proteoglycan deposition (Morisset et al., 2007).
Future Considerations
MSCs offer the potential to open new frontiers in the practice of medicine. However, it is critical to increase our understanding of the mechanisms by which these cells impact the progression of OA, or contribute to the pathogenesis of disease, to enable the development of innovative therapeutic options. Other avenues of research that need to be addressed include (1) establishing the optimal conditions for MSC engraftment in the OA joint, (2) identifying the optimal therapeutic cell by comparing chondrocytes, local synovium-derived or circulating marrow-derived MSC populations, and (3) interrogating the underlying mechanisms of action that contribute to attenuation of osteoarthritic symptoms after joint injury. Current cellular therapies in the joint focus on repair of isolated cartilage lesions and do not as yet address the widespread damage associated with OA. With continued improvements in early diagnosis of the disease, it is conceivable that cellular therapies may be applied to resurfacing of minimally damaged cartilage. Progenitor cells have been isolated from the cartilage surface zone, but this layer is lost with the destruction of the cartilage surface in early OA. Strategies that target regeneration of this layer may ultimately reestablish a functional surface zone and delay progression of the disease. It is also conceivable that targeting strategies using nanomaterials could attract either endogenous stem cells or culture-expanded autologous or allogeneic MSCs applied either locally or systemically. Gene therapies also offer some promise particularly in the modulation of inflammatory mediators associated with the osteoarthritic niche. In summary, the convergence of research into the root causes of OA and the role of stem cells in this etiology should lead to more targeted and efficacious treatment for this debilitating disease in the future.
Footnotes
Acknowledgments
This work was funded by Science Foundation Ireland, Centre for Science Engineering and Technology award (CSET) in Regenerative Medicine. The authors gratefully acknowledge the contribution of Mr. William Curtin, Department of Orthopaedic Surgery, Merlin Park Hospital Galway, to this research.
Author Disclosure Statement
The authors have no conflict of interest to disclose.