
Abstract
Currently, lentiviral vectors for research and gene therapy are produced from 293-T cells that are transiently transfected with plasmids encoding the vector and helper functions. However, transiently transfected vectors as well as the presence of SV40 virus large T-antigen (T-Ag) cause serious technical and safety considerations. We aimed to exploit single copy integration sites in the HEK293 genome supporting lentiviral vector production. We found that lentiviral vectors result in minimal infectious particle production from single copy integrants in HEK293. Moreover, once this cell line harbors single copy integrations of lentiviral vectors, its ability to transiently produce lentiviral vectors becomes strongly impaired. T-Ag has a dramatic effect on virus production. Low levels of constitutive T-Ag expression can overcome the production restriction imposed by integrated lentiviral vectors copies. Interestingly, T-Ag does not exert its role at the level of transcriptional activity of the vector; rather, it seems to impose an indirect effect on the cell thereby enabling lentiviral vector production. Altogether, our study highlights the restrictions for integrated lentiviral vectors that are relevant for the establishment of stable and safe producer cell lines.
Introduction
For transduction of the vector, DNA is either introduced by classical DNA transfection (Ikeda et al., 2003) or by viral transduction (Kafri et al., 1999; Klages et al., 2000; Ikeda et al., 2003; Ni et al., 2005; Cockrell et al., 2006). The latter strategy has been the method of choice for the generation of the majority of cell lines described to date. Viral transduction allows the integration of higher numbers of vector copies by employing high virus titers and/or successive rounds of infection. However, this approach is not applicable for self-inactivating (SIN) vectors, because the inactivation of the 5′ long terminal repeat (LTR) upon infection would compromise the generation of packagable viral transcripts. Therefore, stable production of SIN-derived vectors can only be performed by transfection of vector DNA. Introduction of a vector by transfection has been observed to be accompanied by silencing of full-length vector mRNA expression upon extended passaging. One reason for this effect might be the tandem pattern of genomic insertion (Garrick et al., 1998). This so-called repeated-induced gene-silencing phenomenon is related to an increase of chromatin condensation and methylation and can frequently result in variegated pattern of expression in a population of transfectants. A novel strategy for lentiviral DNA transfection involves the introduction of hundreds of concatamerized vector copies into the cellular genome. This results in satisfactory titers for a certain period of cultivation time (Throm et al., 2009). In general, a linear correlation between copy number of the vector and virus production does not apply. Higher copy numbers might increase the probability for beneficial integration sites, albeit at the expense of genetic stability. It is well known that a critical parameter is the nature of the vector integration site, which defines the expression and thus the titer. This has been studied for single copy vector integration events (Coroadinha et al., 2006; Schucht et al., 2006).
Conceptually, safe (i.e., defined and genetically stable) production systems for γ-retroviral and lentiviral vectors require a low copy number or, most beneficially, a single vector copy integrated into the producer cell genome. Only in such a scenario can the sequence of the produced viral mRNA can be predicted. In particular, the generation of deleterious read-through transcripts can be largely excluded. Such RNAs can arise due to the inherent leakiness in the polyadenylation signal of retroviruses including lentiviruses (Furger et al., 2001; Zaiss et al., 2002; Schambach et al., 2007) and can potentially result in oncogene transduction (Swain and Coffin, 1992; Uren et al., 2005).
γ-Retroviral modular producer cell lines have been established that allow production of high titer vectors from single, defined chromosomal sites (Gama-Norton et al., 2010; Coroadinha et al., 2006; Schucht et al., 2006; Loew et al., 2010). This permits production of γ-retroviral vectors in a predictable and controlled manner (Coroadinha et al., 2006; Schucht et al., 2006; Loew et al., 2010; Gama-Norton et al., 2010). However, the development of cellular systems that exploit single chromosomal loci for the production of lentiviral vectors in a predictable and controlled manner has not been achieved so far. In this study, we evaluated the feasibility of lentiviral vectors that are commonly used in transient production protocols to generate infectious viral particles upon low copy integration in the HEK293 genome. This work represents the first systematic study on lentiviral genome expression in HEK293 upon chromosomal integration. The results show that HEK293 cells as such do not support efficient lentiviral vector production when the vector is integrated at a low copy number in the cellular genome. Intriguingly, this impairment is partially circumvented by prolonged expression of T-Ag. We envision that functional replacement of T-Ag could help convert a nonproductive to a productive cell line for lentiviral vectors with high safety standards.
Material and Methods
Lentiviral vectors
All vectors represented in Fig. 1A were derived from the HIV-1 genome. Vector I is derived from pRLL.cPPT.PGK.eGFP.PRE (kindly provided by L. Naldini, Milano, Italy). Vector II is a derivative of vector I and harbors a green fluorescence protein (GFP) gene driven by the 5′ LTR and an RFP cassette downstream from the PGK promoter. The GFP gene was cloned into the EcoRV site upstream of the cPPT sequence. Vector III is a Tat-dependent non-SIN lentiviral vector derived from pHR′-CMVLacZ plasmid (kindly provided by D. Trono, Lausanne, Switzerland). Vector IV is a targeting lentiviral vector used for recombinase-mediated cassette exchange (RMCE) and is based on pRLL.cPPT.PGK.eGFP.PRE. Briefly, the lentiviral cassette was cloned in pEMTAR vector (Verhoeyen et al., 2001) in order to create a targetable lentiviral vector. It contains Flp recombination targets F-WT and F-5 flanking the viral genome. EMCV-IRES elements and an ATG start codon are positioned upstream to the F-5 site to complement the ATG-deficient neomycin-phosphotransferase expression upon targeting (for details consult Verhoeyen et al., 2001; Schucht et al., 2006).
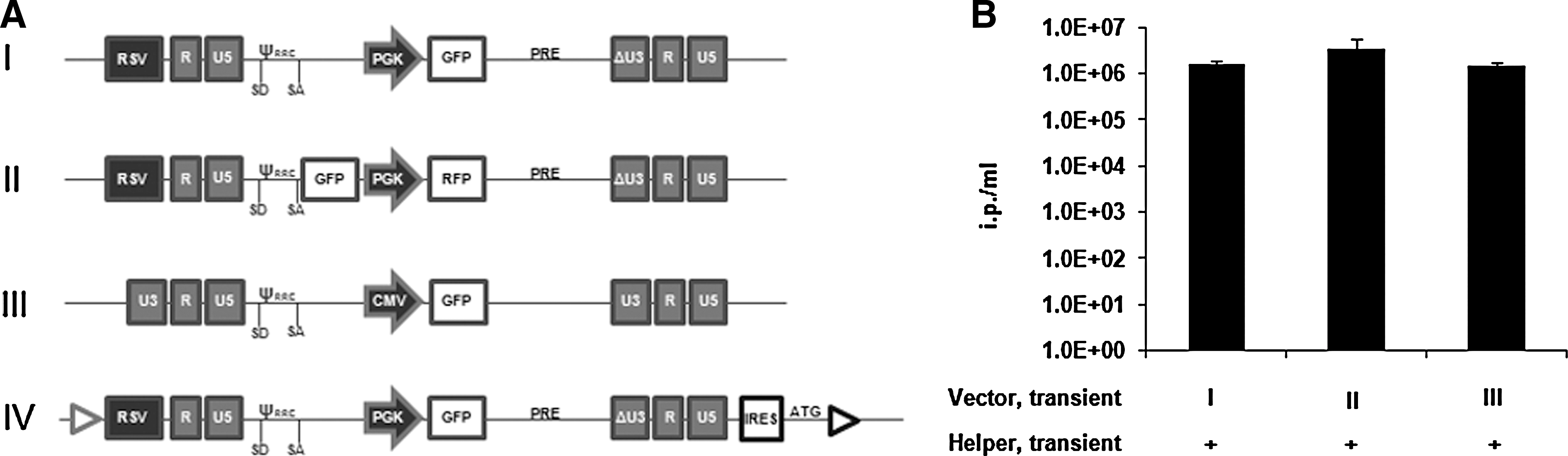
Sequences or maps as well as additional features and cloning details of the vectors are available upon request.
Mammalian cell culture
HEK293-derived 1B2 and 293-3 cell lines were previously described elsewhere (Coroadinha et al., 2006; Schucht et al., 2006). Briefly they contain a single targetable site flanked by two heterospecific FRT sites (F-WT and F-5) and an ATG-deficient neo gene at the 3′ end. The chromosomal location of each tagging locus is described elsewhere (Gama-Norton et al. 2010).
HEK293 cells (BioReliance), 293-T (ATCC CRL-11268), and all the derived lentiviral tagged clones were cultivated at 37°C in a humidified atmosphere with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Sigma) with 10% fetal calf serum (Cytogen), 2 mM
NIH3T3 cells (ATCC CRL-1658) were used to titer the lentiviral vector particles with cultivation conditions similar to the ones for the other cultures.
Lentiviral tagging
DNA transfection
Plasmid transfer was performed by using a conventional electroporation protocol or a nucleofection-based method as described in Nehlsen et al. (2009). HEK293 cells were electroporated using the GenePulser® electroporator (BioRad). For this purpose 1×106 cells were transfected with 2.6 μg of the tagging lentiviral vector. For the nucleofection-based method, 2 μg of lentiviral plasmid DNA was used, using Nucleofector® Kit V (Amaxa®, Lonza). Individual cell clones expressing eGFP were isolated and expanded.
Viral transduction
Lentiviral tagging vectors were generated upon transient transfection of 293-T following a standard protocol. Briefly, 293-T cells were transfected with vector III, together with packaging plasmids pLP1, pLP2, pLP/VSV-G (all from Invitrogen), and pCMV-TAT by using the calcium phosphate precipitation method. Upon filtration (45 μm) and supplementation with polybrene (8 μg/ml, Sigma), the supernatant was used to infect HEK293 and 293-T cells at a multiplicity of infection (MOI) of 0.01. Forty-eight hours after infection, single transduced clones expressing GFP were isolated by fluorescent activated cell sorting (FACS).
Viral vector production
HEK293- and 293-T–derived cell clones with integrated lentiviral genome were transiently transfected with plasmids encoding the packaging functions, pLP1, pLP2, and pLP/VSV-G (Invitrogen) by using the standard calcium phosphate precipitation method. In order to monitor the overall capacity of these clones to produce lentiviral vectors from episomal copies, the packaging helper functions were co-transfected in an independent experiment with pRLL.cPPT.PGK.eGFP.PRE (vector I) or other vectors as indicated in the figures.
Flow cytometry
Flow cytometric analysis was performed on a FACSCalibur® (Becton Dickinson). For titration of GFP-expressing virus, the cells were washed, trypsinized, and stained with propidium iodide (50 μg/ml) to exclude dead cells.
Single cell sorting was performed on a FACSVantage SE® (Becton Dickinson).
Recombinase-mediated cassette exchange
For site-specific cassette exchange, HEK293-derived tagged clones 1B2 and 293-3 were co-transfected with Flp recombinase–expressing vector (pFlpe) and the targeting vector by using GenePORTER® 2 transfection reagent (Genlantis) and following a protocol described elsewhere (Gama-Norton et al., 2010). Correctly targeted clones were isolated upon selection in G418- and ganciclovir-containing medium for 14 days. G418- and ganciclovir-resistant single clones were evaluated for correct targeting by PCR or Southern blot analysis.
Integration of T-Ag–encoding plasmid
pRITA (May et al., 2004) encoding the SV40 T-Ag was stably integrated into the genome of HEK293-derived cells upon co-transfection with a plasmid encoding hygromycin as a selectable marker (ratio 10:1) by using the calcium phosphate precipitation method. Selection started 48 hr after transfection and was carried out for about 14 days, after which individual colonies were picked, expanded, and confirmed for SV40 T-Ag integration by PCR.
Real-time PCR
For the relative quantification of SV40 T-Ag and lentiviral packaging sequence (ψ) containing transcripts, human β-actin mRNA was used for normalization and to allow a comparison between experiments.
Total mRNA was isolated using the RNeasy Kit® (Qiagen) with DNase digestion to remove genomic DNA from the sample, following the instructions from the supplier. Synthesis of first-strand cDNA templates from total RNA was generated using the Ready-To-Go® You-Prime First-Strand Beads (GE Healthcare) with oligo dT primers, following the manufacturer's instructions.
Relative real-time (RT)-PCR was performed using QuantiTect SYBR Green® PCR Kit (Qiagen). Briefly, for each sample, 10 μl of SYBR Green master mix was added to 1 μl of a 10 mM forward and reverse primer. Diluted cDNA sample and RNase-free water were combined to a final volume of 20 μl. For each sample, duplicates or triplicates were performed and a no-template reaction was included as the negative control. Standard conditions were used for the PCR amplification (15 min at 95°C, then 60 cycles of 95°C for 15 sec, 58°C for 20 sec, and 72°C for 30 sec, followed by a cooling step). RT-PCR was performed using the 96-well LightCycler® 480 Real-Time PCR System (Roche). For detection of the T-Ag gene, the primers used were as follows: T-Ag1 5′-TAGTGGCTGGGCTGTTCTTT-3′ and T-Ag2 5′-GGTGGGTTAAAGGAGCATGA-3′; for detection of ψ sequence in lentiviral genome, the primers used were as follows: LentiPsiFwd1 5′-ATCTCTAGCAGTGGCG-3′ and LentiPsiRev1 5′-CTCCCCCGCTTAATAC-3′; for detection of the housekeeping gene human β-actin, the primers used were as follows: hActβ Fwd 5′-TCTTCCCCTCCATCGTG-3′ and hActβ rev 5′-TTCAGGGTGAGGATGCC-3′.
All bars in graphs of Figs. 1–5D represent average values obtained from at least two independent experiments. Error bars represent SD associated with at least three measurements.



Relation of infectious titer and full-length viral mRNA level of HEK293-derived clones integrated with a single lentiviral genome (TAR293LV) not expressing or expressing T-Ag (“−T-Ag” and “+T-Ag”, respectively). As control, the results obtained for 293-T are included. aValues indicate the level of Psi mRNA/β-Actin mRNA. Values of individual samples were normalized to the value obtained from 293-T which was set to 5×104.
Results
HEK293-derived cell clones harboring chromosomal integrants of lentiviral genomes have impaired ability to produce infectious viral particles
We applied a strategy to identify single chromosomal sites supporting high titer virus production of lentiviral vectors because it was successfully done for γ-retroviral vectors (Coroadinha et al., 2006; Schucht et al., 2006; Loew et al., 2010). In the first step, HEK293 cell clones that express lentiviral vectors encoding GFP were created. We anticipated that upon integration the expression of GFP would allow identifying chromosomal loci that support high levels of 5′ LTR promoter activity and concomitantly sustain high levels of lentiviral vector expression. These cell clones were transiently transfected with plasmids encoding HIV-1–derived gag-pol, rev, and VSV-G envelope (in the figures designated as “Helper, transient”). Infectious viral particles produced under these conditions were determined (designated in the figures as “Vector, integrated”).
The lentiviral vectors I, II, and III described in Fig. 1A were used for this tagging. In standard production protocols based on transient transfection these vectors gave rise to titers that were above 1×106 ip/ml (Fig. 1B). For tagging, three different strategies were applied. In the first approach, a conventional SIN-lentiviral vector encoding GFP (Fig. 1A, vector I) was transfected into the cells, and stable clones expressing high levels of GFP were identified. The expression of the reporter gene was supposed to identify chromosomal loci that support a high level of transcription, reflected by the activity of the internal promoter. In this strategy, high GFP expression does not necessarily correspond to high performance of the 5′ LTR promoter, a prerequisite for generation of high levels of full-length lentiviral RNA. Hence, in the second approach a dual reporter gene screening vector was employed. In this vector GFP is positioned downstream of the LTR, followed by the internal PGK promoter that drives RFP as a second reporter (Fig. 1A, vector II). Both SIN-vectors were randomly integrated into HEK293 cells, following either a conventional electroporation protocol or a nucleofection-based method. Both methods have been previously shown to provide a high percentage of single or low copy integration of different DNA cassettes (Nehlsen et al., 2009). After isolation of cell clones on the basis of GFP expression strength, their capacity to produce infectious particles was evaluated (Fig. 2). More than 40 independent cell clones were analyzed (for GFP expression profiles of representative clones see Supplementary Fig. S1A, B; Supplementary Data are available online at
Since plasmid transfection might impose a bias towards specific integration sites that are unfavorable with respect to viral vector production, we employed lentiviral transduction of screening vectors to tag “natural” integration sites. For this purpose, we utilized the Tat-dependent non-SIN lentiviral vector III depicted in Fig. 1A. Upon infection with a MOI of 0.01, GFP-expressing clones were isolated. Cell clones with different levels of stable reporter gene expression (representatives are given in Fig. S1C) were analyzed for lentiviral vector production. Among 20 of these cell clones, none was able to produce relevant amounts of infectious lentiviral particles (Fig. 2, light gray triangles). From these results we conclude that lentiviral vectors after random chromosomal integration at a low copy number do not support high titer vector production. This might be explained by the inability to produce enough viral RNA from integrated vector copies. Alternatively, the potential of the cells to produce virus is compromised once the vector is integrated into the genome. We proved the latter assumption by transient co-transfection of a lentiviral vector in 49 of the cell clones (in the figures designated as “Vector, transient”). We found that these clones showed only low levels (<1×104 ip/ml) of lentiviral vector production (Fig. 2, dark gray circles). This was irrespective of the vector, the level of GFP expression of the integrated cassette, or the transfer method applied. In these clones the titer was about 100-fold less than the titer achieved with the standard vector I represented in Fig. 1A upon transient transfection in wild-type HEK293 cells (Fig. 2B). We thus concluded that lentiviral vector production from episomal copies is impaired when lentiviral genomes are stably integrated into the chromosomal DNA of the HEK293 cells.
Exploitation of well-defined HEK293 genomic loci towards lentiviral vector expression
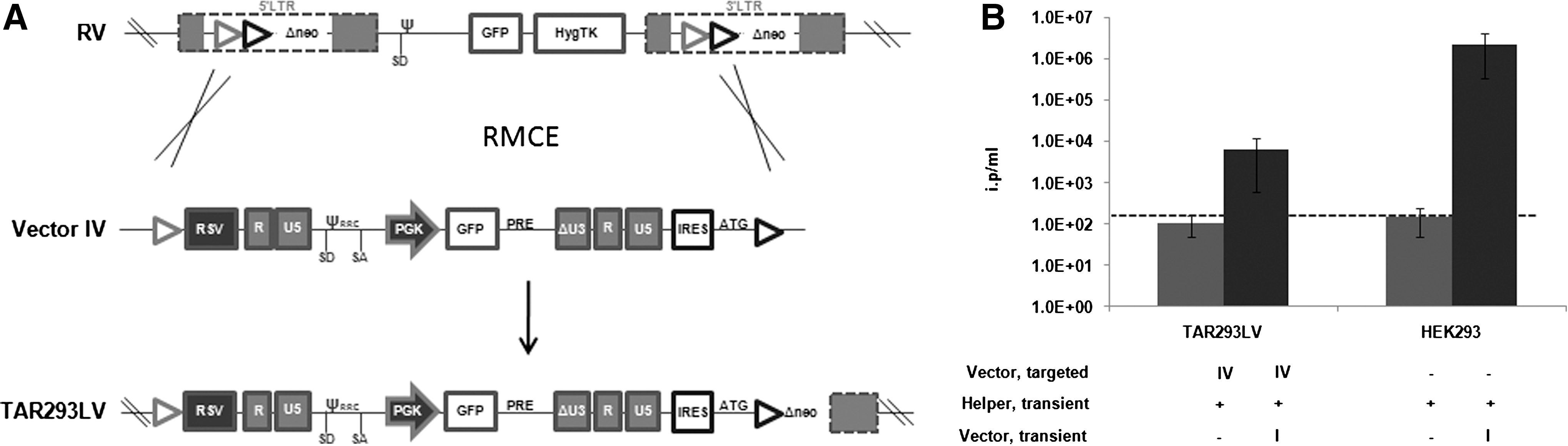
We previously established stable production systems for γ-retroviral vectors that are based on single copy integration of the vector in HEK293 cells (Coroadinha et al., 2006; Schucht et al., 2006). Through the use of flp-RMCE (Verhoeyen et al., 2001) many different γ-retroviral vectors were integrated into the established chromosomal sites of the helper cell lines (Coroadinha et al., 2006; Schucht et al., 2006; Loew et al., 2010; Gama-Norton et al., 2010). In all cases very high titers were obtained. We evaluated the capacity of these two well-described HEK293 loci to support lentiviral expression. Taking advantage of RMCE, we integrated the lentiviral vector IV represented in Fig. 1A into the tagged loci of HEK293-derived 1B2 and 293-3 cells (Coroadinha et al., 2006; Schucht et al., 2006), as described in Fig. 3A. Upon selection and molecular characterization of correctly targeted clones (data not shown), infectious vector particle production was measured as described above. Lentiviral vector production was below the detection level, both from the tagged locus of 1B2 cells (TAR293LV cell line, Fig. 3B) and the tagged locus of 293-3 cells (not shown), despite very high levels of GFP expression (Supplementary Fig. S1D). Moreover, the capacity of these clones to transiently produce lentiviral vectors was impaired if compared with HEK293 cells (Fig. 3B) and 1B2 cells (data not shown). This observation is in line with the results described above.
Furthermore, we evaluated if such inefficient vector production from targeted lentiviral sequences could be overcome by employing more favorable promoter combinations in this specific chromosomal site. To this end, we designed a panel of several targetable lentiviral vectors with different promoter constellations (Supplementary Fig. S2). For this purpose we selected promoters that were previously confirmed for high expression in this specific chromosomal locus (Gama-Norton et al., 2010). However, as depicted in Supplementary Fig. S2B, the modulation of the nature of the promoters within the 1B2 integration site did not result in a significant increase in vector production capacity.
We confirmed that this effect is also obvious if a lentiviral vector different from the integrated vector is transiently expressed. For this purpose, we transfected TAR293LV cells with an RFP-encoding lentiviral vector based on a different viral backbone, as depicted in Supplementary Fig. S3A. This vector also gave low titers in TAR293LV cells (Supplementary Fig. S3B).
To further exclude that the just described phenomena result from specific features of a particular vector family, an unrelated Tat-dependent SIN-vector was included in the study (Supplementary Fig. S4A). This vector was previously shown to give high titers upon transient transfection in 293-T cells (Ikeda et al., 2003). Upon targeted integration of this vector, the production of lentiviral vectors from its proviral DNA was also impaired (Supplementary Fig. S4B). Finally, we tested non–HIV-1–derived vectors, namely a Tat-independent SIN simian immunodeficiency virus (SIV)-derived vector (Supplementary Fig. S5A). The SIV-derived vector was also accompanied by a reduction in titer upon stable integration to the same extent as the HIV-1–derived vectors. However, in this case the effect on transient vector production was less pronounced than the one observed for the HIV-1–derived vectors (Supplementary Fig. S5B). At the same time, upon integration of a control retroviral vector, transient transfection of lentiviral helper and vector resulted in a lentiviral production comparable (95%) to HEK293 cells. Together, these results corroborate the observations presented in Figs. 2 and 3.
The cellular ability to produce lentiviral vectors upon chromosomal integration is improved in 293-T cells
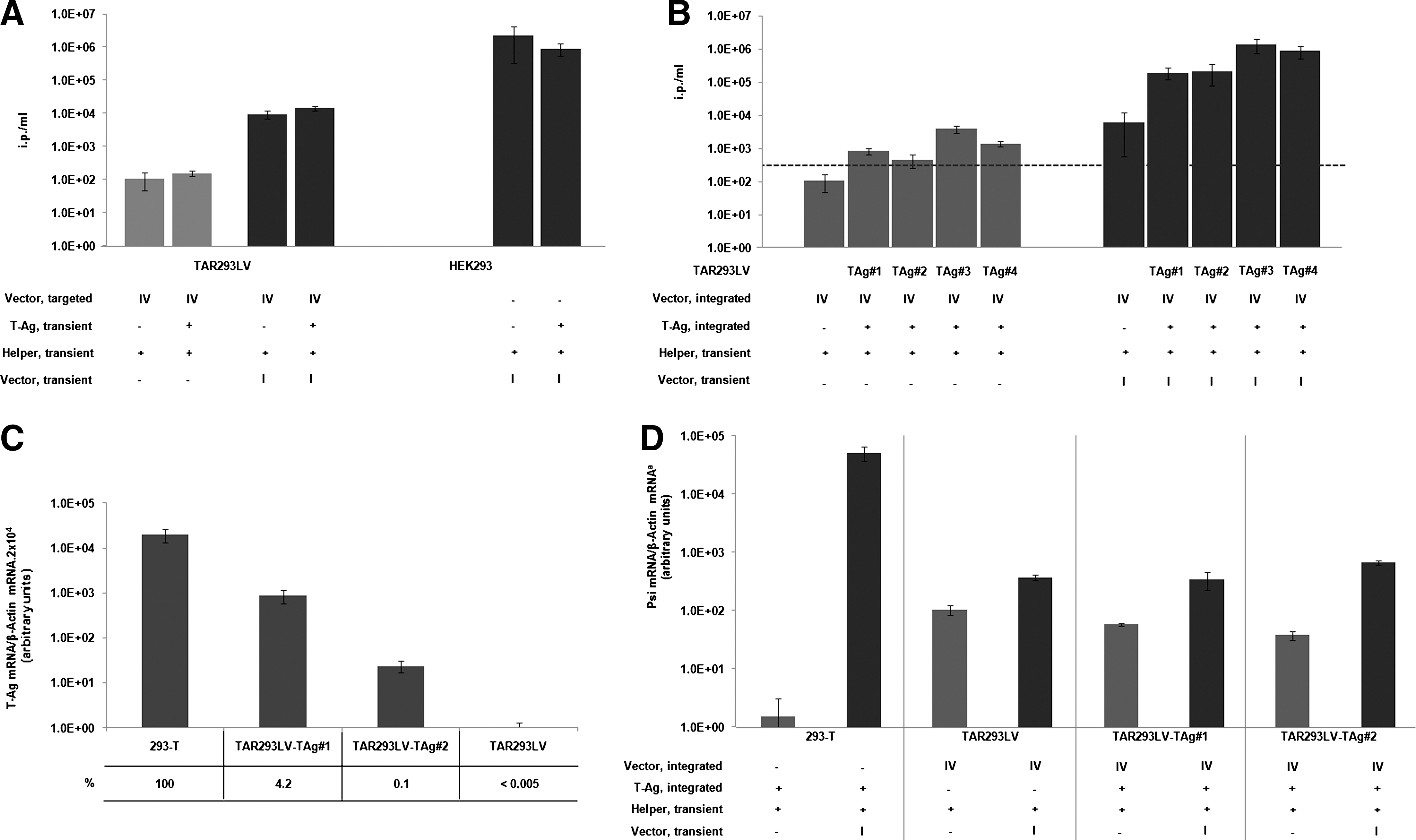
293-T cells have been shown to allow high titer production of lentiviral vectors in transient production protocols (Sinn et al., 2005; Cockrell and Kafri, 2007). We evaluated the lentiviral production upon stable vector integration in these cells. 293-T cells were tagged by infection with the non-SIN HIV-1–derived vector III (Fig. 1A) according to the previously described protocol. Transduced cell clones, harboring one or a few copies of the lentiviral vector were identified on the basis of GFP reporter gene expression (Supplementary Fig. S1E for GFP expression of two representative clones). Independent cell clones were analyzed for infectious virus production upon transient transfection of the helper functions. The results obtained for 41 cell clones are shown in Fig. 4A. Overall, production of lentiviral vectors upon chromosomal integration in 293-T is about 10-fold higher when compared to titers obtained from corresponding HEK293 clones, (Figs. 2A and 3B). This suggests that the expression of T-Ag in these cells supports lentivirus formation from integrated vector copies. Importantly, 293-T–derived clones were not prevented from producing lentiviral vectors when the viral genome was additionally provided as an episome, achieving titers up to 1×107 ip/ml, which is comparable with virus production capacity of the parental cell line (Fig. 4B).
Stable integration of SV40 large T-Ag improves lentiviral vector production from HEK293-derived cell lines
To evaluate if T-Ag expression exerts a positive effect on the production of infectious lentiviral particles from a single integrated HEK293 cell line, we complemented TAR293LV cells with SV40 T-Ag. First, the impact of transient T-Ag expression on virus production was analyzed (Fig. 5A). In this set-up T-Ag did not affect virus production with (TAR293LV) or without (HEK293) stable lentiviral integrates. We then tested if long-term expression of T-Ag would affect virus production. Four cell clones designated as TAR293LV-TAg#1–4 were arbitrarily selected after calcium phosphate precipitation mediated stable transfer of the T-Ag plasmid into TAR293LV cells. Integration of the T-Ag gene was confirmed by PCR (data not shown). Figure 5B shows that long-term T-Ag expression gene in the genome of lentivirally tagged TAR293LV cells increased transient production of lentiviral vectors up to 200-fold. Moreover, an increase from the integrated lentiviral genome was observed when compared with the same experimental setting in the absence of T-Ag expression. In order to evaluate the levels of T-Ag expression behind such an effect, we compared T-Ag mRNA production in 293-T cells and TAR293LV-TAg#1 and 2 cells (Fig. 5C). Both clones expressed significantly lower levels of T-Ag compared with 293-T cells. In clone TAR293LV-TAg#1, 4.2% of T-Ag expression was monitored, while TAR293LV-TAg#2 was related to a 40-fold lower level of T-Ag, corresponding to 0.1% of T-Ag in 293-T. This suggests that already low levels of T-Ag expression are sufficient to increase lentiviral vector production. However, since this effect is only observed after long-term expression of T-Ag, an indirect mechanism is likely to be responsible. Interestingly, a comparable increase in viral titer from integrated lentiviral vectors was observed upon stable expression of the ras oncogene (Fig. S6). This suggests that the increase of virus production is an indirect effect triggered by both ras and SV40 T-Ag.
Restoration of infectious particle production in TAR293LV by T-Ag expression is not due to increased levels of lentiviral mRNA
To define the level at which T-Ag is acting in lentiviral vector production, the amounts of full-length lentiviral mRNA in 293-T cells, TAR293LV cells, TAR293LV-TAg#1, and TAR293LV-Tag#2 cells were measured. qRT-PCR with primers that specifically bind to the packaging signal (psi) present in the leader region of the lentiviral genome was performed. The results are summarized in Figs. 5D and 6. 293-T cells expressed the highest level of full-length viral RNA. In contrast, HEK293-derived cells with single copy vector integration showed about two to three orders of magnitude less efficient expression of full-length mRNA. RNA expression from transiently transfected vectors in these cells was about 3.5 times higher compared with viral genome produced from integrated copy. Interestingly, stable integration of T-Ag had no effect on the full-length lentiviral RNA level in all examined TAR293LV cells, despite increased vector production from these clones (Fig. 5B). This indicates that T-Ag expression does not act on the level of packagable mRNA transcription or RNA stability.
We characterized the levels of viral protein produced in HEK293-derived cell lines in the presence or absence of T-Ag or ras expression. While 293T cells gave rise to highly efficient expression of lentiviral helper functions VSV-G and gag, neither T-Ag nor ras per se could significantly increase expression of helper functions in HEK293 cells or TAR293LV cells bearing a single copy lentiviral vector (see Supplementary Fig. S7). Thus, we can exclude the increased viral production capacity of HEK293-derived cells with single copy of lentiviral vector upon long-term expression of T-Ag being mediated by a substantially improved expression of lentiviral particle components such as RNA or viral helper proteins.
Discussion
Lentiviral producer cells are frequently accompanied by instability in expression and loss of titer (Klages et al., 2000; Throm et al., 2009), which might be due to genetic and/or epigenetic instability of helper functions and/or vector expression. Lentiviral production from a well-characterized stable producer cell line would be safer and might contribute to stability of production. Indeed, both of these conditions were achieved for high titer γ-retroviral producer cells by integration of retroviral vectors into single defined chromosomal loci (Coroadinha et al., 2006; Schucht et al., 2006). The modular nature of these cells further allowed the systematic evaluation and optimization of vector design in the specific loci (Gama-Norton et al., 2010). In this study we intended to identify chromosomal loci in HEK293 that support high levels of lentiviral vector expression as a prerequisite to the establishment of a stable, modular packaging cell line for lentiviral vector production. We screened more than 100 independent cell clones that had been tagged with HIV-1–derived lentiviral vectors. Different DNA transduction methods were used to exclude any bias with respect to integration sites favored by the transduction method. Although internal reporter genes were readily expressed, none of the individual chromosomal sites in these clones was capable of supporting significant infectious virus production. In addition, we exploited two defined integration sites in HEK293 cells that were previously shown to provide high expression levels of integrated cassettes. Upon complementation with the helper functions, these sites resulted in high virus titers when used for integration of γ-retroviral SIN and non-SIN vectors (Coroadinha et al., 2006; Schucht et al., 2006). These loci also support expression of nonviral cassettes, such as antibody expressing and conditionally regulated expression cassettes (data not shown). Unexpectedly, upon integration of lentiviral vectors by RMCE a reasonable titer could not be achieved, although reporter gene expression was very high (Supplementary Fig. S1D). Indeed, the full-length vector RNA production from these integrated vectors was about 500-fold lower than the expression from transiently transfected 293-T cells (Fig. 5D), and concomitantly, the level of virus produced from such cells was 10,000-fold reduced if compared with the titer achieved with the classical 293-T cell protocol (Fig. 4). Together, the results show that none of the tested chromosomal loci allowed production of lentiviral vectors from integrated viral vector genomes.
Intriguingly, once a lentiviral vector is stably integrated, cells are severely restricted from producing infectious particles from transiently transfected vector molecules, with a reduction of more than 100-fold on the level of viral particles. Currently, it is not clear how the presence of an integrated lentiviral vector copy can inhibit the production of lentiviral particles from episomal vector copies. However, the fact that all the clones tested showed comparable results excludes the vector integration having knocked out a crucial cellular factor. More likely, a general phenomenon is behind impaired lentiviral production from single copy integration events in HEK293 cells. It would be interesting to analyze if such a restrictive phenomenon could be circumvented by random changes of individual cells in a clonal population if selected on the basis of vector production capacity, rather than reporter gene expression. In this respect, subcloning of cells with single copies of lentiviral vectors could selectively identify clones with an altered genetic background that supports lentiviral production at high levels.
Moreover, the integration of helper function coding sequences such as gag-pol, rev, and envelope might contribute to achieving an ideal stoichiometry of all viral particle components necessary for high titers in HEK293 cells integrated with a single copy of the lentiviral vector.
Our study shows that the impairment is independent of the family of HIV-1–derived lentiviral vectors or on the vector promoter content used. Although the lentiviral titer was not significantly improved upon altering the promoter equipment of single integrated vectors, it was shown that the promoter constellation of the lentiviral vector should be considered with respect to the characteristics of the site of integration. Indeed, the vector with the promoter composition RSV at 5′ LTR and SFFV as the internal promoter (vector V, Fig. S2) was associated with a 10-fold increase in titer when compared with the performance of other lentiviral vectors integrated in this particular integration site. This information might be of relevance for future producer cell systems.
Finally it was shown that impaired viral production by HEK-293 cells from a single lentiviral integrant is not exclusive for HIV-1–derived vectors but is also observed in an SIV-based system. This indicates that the impairment is not a specific feature of the HIV-1–based lentiviral vectors, but it also applies to other lentiviral constructs. With respect to the effect on virus production from episomal vector copies, the impairment mediated upon chromosomal integration of HIV-1 vectors is more pronounced than that of SIV-1 vectors. In contrast, single retroviral vector integrants do not inhibit lentiviral vector production from episomal vector copies. This might indicate that this “trans” effect is particularly relevant to HIV-1–based vectors.
The titer from standard protocols for transient lentiviral vector production in 293-T cells are about 10-fold higher compared with titers achieved from HEK293 cells lacking T-Ag (Fig. 3B and 4B). The reason for this difference has not yet been elucidated. SV40-derived T-Ag has been characterized as an oncoprotein that elicits multiple functions (Ahuja et al., 2005). The pleiotropic effects exerted by T-Ag expression account for the dramatic alterations in the cellular transcriptome. This is reflected by the reversible deregulation of around 5% of cellular genes upon conditional expression of T-Ag (May et al., 2004). In addition, T-Ag is known to exert nonreversible effects, often as a result of increased genome instability but possibly also by other mechanisms.
Here, we show that expression of T-Ag is associated with higher lentiviral vector titers, both for single copy integrated vectors and for episomal state vectors. We demonstrated that this effect is not mediated by increase in helper protein expression (Fig. S7). Further, our results show that the T-Ag effect relies on its long-term expression because freshly T-Ag–transfected cells do not show this phenomenon (Fig. 5A). Importantly, stable integration of ras oncogene also promotes an increase in lentiviral production from integrated single copy proviral genomes (Fig. S6). This suggests a common pathway triggered by the T-Ag and ras oncogene that finally results in increase of titer.
We evaluated if suboptimal transcript formation of the lentiviral genome is the limiting step that impairs lentiviral vector production from a single copy integrated in HEK293 cells. Real-time PCR analysis showed that transiently transfected 293-T cells express a higher relative value of full-length lentiviral transcripts compared with the other cell clones analyzed (Figs. 5D and 6). This shows that the higher titer of lentiviral vectors in T-Ag–expressing TAR293LV cells is not due to an increase of full-length mRNA transcripts. However, we cannot exclude such an effect at very high expression levels of T-Ag as found in the 293-T cells.
The transient production of lentiviral vectors in cell lines impaired by integration of the lentiviral vector is increased up to 200-fold upon long-term expression of T-Ag. However, the capacity of HEK293 expressing T-Ag or 293-T from single integrants of lentiviral genomes was not found to be higher than 1×104 ip/ml. This indicates that lentiviral vector integration is the major restriction factor for lentiviral vector production from a single copy and this effect is uncoupled from the impaired ability of single copy HEK293-derived cells to transiently produce viral vectors. This is in line with the discussed effect of integrated HIV-1 versus SIV-1 vectors.
In summary, the reason why HEK293-derived cell clone with an integrated lentiviral cassette has a compromised ability to produce infectious particles remains obscure. However, we show that both T-Ag and ras can partly overcome this block, and we can exclude T-Ag significantly modifying the levels of viral mRNA or viral protein production. Thus, we assume that the effect of T-Ag is indirect and might promote nondefined cellular alterations that eventually result in improved virus particle production. Since the positive effect of these oncogenes on viral vector production is only observed upon long-term culture, we anticipate that expression of T-Ag renders an accumulation of cellular genetic and/or epigenetic alterations that are necessary to convert a nonproductive cell system towards one with a capacity for lentiviral production Currently, we cannot exclude that other changes in HEK293-derived cells, e.g., beneficial properties evident upon subcloning, might similarly contribute to an improved lentiviral production. Indeed, several subclones of HEK293 cells have been isolated that were shown to have improved lentiviral vector production in transient systems (Ansorge et al., 2009) and when integrated with lentiviral genomes (Kafri et al., 1999; Klages et al., 2000; Ni et al., 2005; Cockrell et al., 2006; Broussau et al., 2008). However, in all the reports published so far, the number of lentiviral genomes in stable producer cell lines has never been estimated. The notion that only multicopy integrants lead to higher lentiviral titers has been implied in several reports (Kafri et al., 1999; Klages et al., 2000; Cockrell et al., 2006; Throm et al., 2009). It remains to be elucidated if higher titers achieved upon multicopy integration are due to the increased number of vector genomes transcribed, the augmented chances of hitting a rare chromosomal locus that supports per se high levels of lentiviral genome expression, or to a different, unanticipated scenario.
The results presented here point toward the need of multiple copy integration for lentiviral vectors in order to bypass a still undefined bottleneck for lentiviral vector generation imposed by the producer cell. The observation that both T-Ag and ras expression improve this blockage opens a potential way to overcome this bottleneck. Preferably, non-oncogenic perturbations should be followed to achieve such an improvement.
Footnotes
Acknowledgments
We thank Rainer Loew and Axel Schambach for stimulating discussions and Sara Behme for experimental support. This work was supported by grants from the EU (Clinigene), the Bundesministerium für Bildung und Forschung (FKZ0313940; 0315275C; 0315271A), Deutsche Forschungsgemeinschaft (Cluster of Excellence REBIRTH), and the Helmholtz Society (SBCancer). L.G.N. acknowledges the financial support received from Fundação para a Ciência e Tecnologia, Portugal (SFRH/BD/22081/2005).
Author Disclosure Statement
No competing financial interests to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.