
Abstract
Allogeneic mesenchymal stem or stromal cells (MSCs) are proposed as cell therapies for degenerative, inflammatory, and autoimmune diseases. The feasibility of allogeneic MSC therapies rests heavily on the concept that these cells avoid or actively suppress the immunological responses that cause rejection of most allogeneic cells and tissues. In this article the validity of the immune privileged status of allogeneic MSCs is explored in the context of recent literature. Current data that provide the mechanistic basis for immune modulation by MSCs are reviewed with particular attention to how MSCs modify the triggering and effector functions of innate and adaptive immunity. The ability of MSCs to induce regulatory dendritic and T-cell populations is discussed with regard to cell therapy for autoimmune disease. Finally, we examine the evidence for and against the immune privileged status of allogeneic MSCs in vivo. Allogeneic MSCs emerge as cells that are responsive to local signals and exert wide-ranging, predominantly suppressive, effects on innate and adaptive immunity. Nonetheless, these cells also retain a degree of immunogenicity in some circumstances that may limit MSC longevity and attenuate their beneficial effects. Ultimately successful allogeneic cell therapies will rely on an improved understanding of the parameters of MSC–immune system interactions in vivo.
Introduction
These insights are of particular significance to the development of MSCs as modulators of localized tissue inflammation and as therapeutic agents for immune-mediated diseases. Beginning approximately 10 years ago with in vitro co-culture experiments and progressing, more recently, to sophisticated in vivo models of immune/inflammatory disease, a clear and compelling profile of MSCs has developed as potent modifiers of a wide range of targets within the innate and adaptive arms of the immune system (Barry et al., 2005; Uccelli et al., 2008). The recognized clinical potential of MSC immunomodulatory effects now encompasses acute myocardial ischemia, stroke, kidney injury, inflammatory bowel disease, graft-versus-host disease (GVHD), multiple sclerosis, diabetes mellitus, and organ transplantation (Uccelli et al., 2008; Caplan, 2009). Initial clinical trials have been completed or are underway in several of these areas (Ankrum and Karp, 2010). One important concept that has carried through from basic and preclinical studies to human clinical trials is that of the immune privileged status of MSCs transferred into an allogeneic host (Heng et al., 2009). Stated in its simplest form, this concept implies that allogeneic MSCs (allo-MSCs) fail to activate the innate or elicit the adaptive cellular (T-cell) or humoral (B-cell/antibody) immune responses that typically result in rapid rejection of allogeneic cells and organs transplanted into a host in the absence of additional immunosuppressive therapy (Kahan, 2003). This would imply that allo-MSCs, prepared and stored in advance for “off-the-shelf” therapy, are of equal efficacy to individually prepared autologous MSC (auto-MSC) cultures and can be repeatedly administered without losing potency because of immune sensitization. A further extrapolation of some of the current literature would also suggest that allo-MSCs promote active immunological tolerance to “donor” major histocompatiblity complex (MHC) and other alloantigens—a property that could specifically enhance their therapeutic value in organ and tissue transplantation.
Although early, uncontrolled clinical case series provided exciting evidence of therapeutic benefits, the resulting optimism for off-the-shelf allo-MSC therapy has become tempered by the outcomes of recent, larger clinical trials in which allo-MSC products proved disappointing in terms of efficacy despite achieving safety endpoints (Ankrum and Karp, 2010). Thus, the time is right to ponder the route ahead for MSC-based cell therapies—particularly those based upon the large-scale expansion of cells from allogeneic donors. Most immunologists treated early descriptions of non-rejection of allo-MSCs or xenogeneic MSCs with some suspicion, but it is now the case that many thousands of patients have safely received allo-MSC therapies, and a large body of preclinical data has accumulated in support of the capacity of MSCs to modulate diverse immune processes in vivo (Uccelli et al., 2008). Despite this, the more recent clinical results indicate that a more critical analysis of the interaction between allogeneic cell therapies and the recipient immune system will be essential for the rational development of commercially attractive MSCs and MSC-like products in the future (Ankrum and Karp, 2010).
Several questions present themselves for which definitive answers remain elusive: (1) What is the hierarchy of the immune suppressive functions of MSCs? (2) How much redundancy exists among the many suppressive processes that have been identified to date? (3) What are the limits of MSC immune modulation? (4) Are cellular and humoral components of the allo-immune response equally suppressed by MSCs? (5) Can the longevity and efficacy of allo-MSC therapies be enhanced through further suppression of allo-immune responses? In this article we consider the major mechanisms that convey immune modulatory properties to MSCs and examine the evidence for and against the immune privileged status of allo-MSCs in vivo. Along the way we discuss disease targets for which MSCs may be most suitable and highlight the hurdles that remain to be overcome in translating the promising laboratory studies into mainstream clinical practice.
MSC Suppression of Innate Immunity
The first encounter for any cell therapy upon delivery is with the components of the innate immune system that provide an effective antimicrobial defense but also a barrier to allogeneic and xenogeneic transplantation. It is clear that allo-MSCs (and indeed some xenogeneic MSCs) avoid acute and hyperacute rejection mechanisms normally mediated through the complement system. This is achieved through secretion of Factor H (Tu et al., 2010) and most likely supported by MSC expression of the complement control proteins CD55, CD46, and CD59 (Komoda et al., 2010) (B.P.M., unpublished data). Thus MSCs are protected from frontline deletion mechanisms operating in other tissue and cell transplant scenarios. However, MSCs are not inert to innate immune signaling, and there is evidence that MSCs are recruited by the anaphylatoxins C3a and C5a (Schraufstatter et al., 2009), suggesting that they are attracted to and activated at sites of tissue damage rather than deleted.
The interaction of MSCs with natural killer (NK) cells has received little attention to date, particularly in the in vivo setting. Initial in vitro observations suggested that human MSCs were not susceptible to lysis by freshly isolated allogeneic NK cells and that MSCs inhibited NK cell secretion of interferon (IFN)-γ (Rasmusson et al., 2003; Aggarwal and Pittenger, 2005). Subsequently, it has been demonstrated that, although human MSCs suppress proliferation, surface receptor expression, and effector functions of NK cells via prostaglandin E2 (PGE2) and 2,3-indoleamine dioxygenase, they can be lysed by activated NK cells (Poggi et al., 2005; Sotiropoulou et al., 2006; Spaggiari et al., 2006, 2008). In the case of MSC interactions with neutrophils, the experimental evidence is even more limited. Of interest, however, is that Raffaghello et al. (2008) recently reported that human bone marrow–derived MSCs inhibited both apoptosis and the oxidative burst of resting and activated neutrophils while preserving their phagocytic and chemotactic functions. Although more work needs to be done in this area, the results fit with a model whereby MSCs modify (“reprogram”) the functional properties of innate immune mediators in a manner that can both protect the MSC from frontline deletional mechanisms and broadly suppress a range of potentially destructive inflammatory pathways.
Recent studies of the influence of ligands of Toll-like receptors (TLRs) have reinforced the concept of MSCs as cells responsive to and modulatory of innate immunity (Pevsner-Fischer et al., 2007; Liotta et al., 2008; Tomchuck et al., 2008; Opitz et al., 2009; Z.J. Wang et al., 2009). It is now clear that MSCs express a range of TLRs and that signaling via these receptors influences migration, survival, differentiation, and immunosuppressive capacity. Some studies have observed that MSC immune modulation can be downregulated by TLR3 and TLR4 ligands (Liotta et al., 2008; Romieu-Mourez et al., 2009) but enhanced by IFN-γ (English et al., 2007). This suggests that MSCs may be particularly effective in suppressing chronic inflammation seen in autoimmunity (not driven by pathogens) without impairing inflammatory responses essential to antimicrobial defense (where TLR ligands would be abundant). There is also evidence, however, that TLR ligation in MSCs results in altered patterns of induction of cytokines and other inflammatory mediators that may, under some conditions, further enhance MSC immune suppressive properties (Tomchuck et al., 2008; Lombardo et al., 2009). In the future, it will be interesting to determine whether this increased attraction of innate immune cells by TLR-activated MSCs represents a barrier to therapeutic immune modulation or, in fact, facilitates anti-inflammatory cell–cell interactions. Overall, the impact of TLR ligation on MSC functions (and immunogenicity) in vivo is incompletely understood at present and may prove to be a key modifiable factor for optimizing the clinical benefits of allo-MSCs.
Another important element of the influence of MSCs on innate immunity relates to the interaction with monocytes and monocyte-derived inflammatory cells. Evidence is accumulating that monocytes and macrophages may be “programmed” by their surrounding microenvironment either to mediate potent, locally destructive and lytic effects (perhaps appropriate for immediate clearance of dead cells and prevention of infection at a site of injury) or to produce a range of anti-inflammatory, pro-regenerative factors (indicative of a central role in the resolution and repair phase of tissue injury) (Tesar, 2008; Stout et al., 2009; Geissmann et al., 2010). Several recent studies have provided direct evidence that MSCs are involved in this programming (Ohtaki et al., 2008; Kim and Hematti, 2009; Nemeth et al., 2009). Most notably, Nemeth et al. (2009) demonstrated convincingly that both auto- and allo-MSCs reduced mortality from sepsis in a mouse model through a direct interaction with macrophages in the lung that resulted in enhanced production of interleukin (IL)-10 and was mediated by a complex monocyte/MSC cross-talk involving TLRs, tumor necrosis factor (TNF), nitric oxide, and PGE2. Taken together with the previously discussed literature on MSC interactions with the complement system, NK cells, neutrophils, and ligands for pattern recognition receptors or TLRs, these studies paint a striking picture of the complexity of MSC cross-talk with the innate immune system and of the rich potential for harnessing these effects for therapeutic benefits.
MSC Influence on Dendritic Cells
Dendritic cells (DCs) play a critical role in adaptive immunity acting as the primary antigen-presenting cell to initiate antigen-specific CD4+ helper T cells. This function has been extensively reviewed elsewhere (Steinman and Banchereau, 2007), but a simplified summary is useful to convey the importance of the MSC–DC interaction. There are a variety of specialized DC subsets with considerable flexibility in development such that precursors with myeloid or lymphoid characteristics can be identified. Some lymphoid tissues generate conventional but non-migratory DC locally from precursors also found in bone marrow (Naik et al., 2007), whereas conventional migratory DCs are generated in the bone marrow from hematopoietic pro-DC precursors often via a monocytic intermediate (Fig. 1). On differentiation, conventional bone marrow–derived DCs expressing the α x β2 integrin (CD11c:CD18) and C-C chemokine receptor (CCR) 6 migrate to peripheral tissues (Fig. 1) (Cook et al., 2000; Kucharzik et al., 2002; Osterholzer et al., 2005). Such immature DCs (iDCs) can be found within skin (Langerhans cells), interstitial and epithelial tissues where they express tissue-anchoring E-cadherin and perform sentinel functions. iDCs have the capacity to take up antigen through phagocytosis and macropinocytosis and to process antigen for loading onto MHC class II molecules; thus iDCs can be considered to perform antigen acquisition functions in the periphery (Steinman and Nussenzweig, 2002; Steinman and Banchereau, 2007). On antigen encounter, iDCs undergo a process termed maturation that sees a remarkable alteration in biological activity, to become mature DCs (mDCs). mDCs downregulate CCR6 and E-cadherin but express CCR7, resulting in chemotaxis to local secondary lymphoid tissues such as the lymph nodes to fulfill their role in antigen display to the adaptive immune system (Iwasaki and Kelsall, 2000). Maturation is accompanied by the expression of the naive T-cell chemoattractant C-C chemokine ligand (CCL) 18 and upregulation of MHC class II and the co-stimulatory molecules CD80 and CD86, among others (Masten et al., 1997). Thus the mDC in the lymph node is in “antigen presentation mode” and ideally placed to initiate and expand antigen-specific CD4+ helper T cells (Fig. 1). This process is essential to initiate adaptive immunity against foreign antigen, but of course if DCs derive from a non-MHC identical transplanted organ, graft, or transfusion, then conventional DCs will promote T-cell-mediated alloreactivity and consequent rejection (Morelli and Thomson, 2003). Conversely, antigen presentation in the absence of costimulation (CD80/CD86, etc.) can result in T-cell non-responsiveness or anergy, and DCs with immature or semi-mature phenotypes are now thought to play a role in peripheral tolerance induction (Turnquist and Thomson, 2008). These processes are thus essential to the initiation of alloreactivity, autoimmunity, and tolerance.

MSCs modulate DC function. DCs are the primary initiators of adaptive T-cell immunity by acting as professional antigen-presenting cells; consequently they contribute to allogeneic cell rejection. In vitro studies suggest that allogeneic MSCs interfere with immature DC generation and maturation through contact-dependent and -independent processes resulting in a tolerogenic phenotype. Color images available online at
A distinct lineage of plasmacytoid DCs (pDCs) can also be derived from a common proDC precursor (Naik et al., 2007; Shortman and Naik, 2007) and are identified by expression of CXCR3 and BDCA-2 (human) or Siglec-H (mouse). Although pDCs appear to be less effective in supporting the expansion of naive antigen-specific T cells, they play an important role in sustaining conventional DC production of IL-12 and in the detection and amplification of antiviral responses through TLR7 and TLR9, leading to type I IFN production and pro-inflammatory cytokine release (Liu, 2005; Zucchini et al., 2008).
Given the central role of DCs in allogeneic rejection and the powerful allosuppressive influence of MSCs, it is not surprising that the interaction of MSCs with DCs has been the focus of much attention. Clearly MSCs modulate different aspects of DC function in vitro (Zhang et al., 2004; Djouad et al., 2007; English et al., 2008; Zhang et al., 2009), and this has a functional counterpart in vivo (H. Li et al., 2008; Popp et al., 2008; Rossignol et al., 2009; Li et al., 2010). It is worthwhile separating the functions related to DC generation and DC maturation as these events may occur in different anatomical locations and concern distinct biological functions. First, it is clear that MSCs influence DC development. This is not surprising as both MSCs and many DC precursors are bone marrow residents and MSCs play a role in conditioning the niche for hematopoiesis (Dazzi et al., 2006; Sacchetti et al., 2007; Morikawa et al., 2009). At the level of development, MSC co-culture strongly inhibits the initial differentiation of monocytes to iDCs in vitro (Beyth et al., 2005; Nauta et al., 2006a). This effect is reversible (Beyth et al., 2005) and can be replicated by MSC-derived soluble factors, including PGE2 and IL-6 (Djouad et al., 2007). It is interesting that MSCs seem to have differential effects on the generation of conventional DCs (suppression) and pDCs (no suppression) (Chen et al., 2007). The implications of these observations require careful interpretation, but it may indicate that MSCs have suppressive effects that can be bypassed when antiviral responses are required for protection—an interpretation supported by recent work (Karlsson et al., 2008).
The major function of DCs in the epithelial tissues is to act as sentinels and, upon maturation, to initiate cell-mediated immunity. There are now consistent data from several sources showing that MSCs modulate or interfere with DC maturation in both the mouse and the human (Zhang et al., 2004; Djouad et al., 2007; Jung et al., 2007; English et al., 2008; Magatti et al., 2009; van den Berk et al., 2009; Zhang et al., 2009). DCs exposed to maturation factors such as lipopolysaccharide or TNF-α co-cultured with MSCs failed to show regular upregulation of maturation markers such as MHC class II, CD40, or CD86 costimulatory molecules (Djouad et al., 2007; English et al., 2008). Similar effects have been seen with MSCs from amniotic, umbilical cord, or adipose sources (Wang et al., 2008; Magatti et al., 2009; van den Berk et al., 2009; M. Wang et al., 2009).
The encounter of DCs with MSCs abrogates the capacity of antigen-pulsed DCs to support cognate CD4+ T-cell proliferation (English et al., 2008). This extends to allo-recognition as well. Allo-MSCs suppress DCs from presenting (MHC-derived) allo-antigen, thus suppressing a major pathway of allo-recognition (English et al., 2008). MSCs also prevent loss of iDC E-cadherin expression, prevent upregulation of CCR7, and inhibit chemotactic ability of DCs (English et al., 2008). Thus MSCs suppress maturation marker expression, antigen presentation capability, and capacity to respond to lymph node–derived chemotactic signals—the three cardinal features of conventional DC maturation. Unlike suppression of T-cell proliferation in mixed lymphocyte reaction (MLR), both contact-dependent and soluble factors contribute to this immunomodulatory phenomenon. The contact-dependent signal appears to involve members of the Notch-Jagged signaling pathway (Y.P. Li et al., 2008; Zhang et al., 2009) (L. Tobin, personal communication), whereas MSC-derived IL-6 contributes to the soluble signal (Djouad et al., 2007; English et al., 2008) (Fig. 1).
The consequence of this immune modulation is that DCs may display an altered profile of cytokine expression (Aggarwal and Pittenger, 2005) with reduced IL-12 (Zhang et al., 2004; Jiang et al., 2005) or increased IL-10 (Aggarwal and Pittenger, 2005) production, adopt a tolerogenic capacity (H. Li et al., 2008; Y.P. Li et al., 2008; Popp et al., 2008), and become capable of an indirect suppression through induction of regulatory T (Treg) cells (Beyth et al., 2005). The functional significance of MSC modulation of DCs in vivo is difficult to assess, and there is an urgent need for more focus on this topic. However, MSCs can alter the migratory property of DCs to delay the development of murine lethal acute GVHD (H. Li et al., 2008) and can suppress DC function during allogeneic islet transplant in a diabetic model (Kim et al., 1997). These characterizations of MSC function have considerably extended our understanding of MSC-mediated immune suppression beyond the limited understanding achievable from MLR studies to suggest that DC modulation is a major pathway of MSC immune suppression.
MSC Induction of Treg Cells
There are numerous mechanisms by which the adaptive immune system achieves tolerance to self-antigen, and understanding these offers an opportunity to develop new interventions against autoimmunity and to prevent rejection of allografts. The central mechanism of tolerance is the deletion of self-reactive lymphocytes in the thymus (T cells) and the bone marrow (B cells) (Peterson et al., 2008; Irla et al., 2010). The mechanisms of peripheral tolerance support central tolerance, and among these are suppressor actions mediated by a group of cells loosely termed Treg cells. This term encompasses a variety of cells of different phenotypes and lineages, including Tr1 cells, T-helper (Th) 3 cells, CD8+ suppressor cells, NK-like cells, and some γδT cell populations (Tang and Bluestone, 2008). The two principal suppressor populations are considered to be CD4+ CD25high FOXP3+ T cells that develop in the thymus (sometimes called natural Treg) and T cells that can develop from naive T cells in the periphery (termed inducible or adaptive Treg). The latter may also express the FOXP3 transcription factor. A full discussion of the role of Treg cells is not possible in the current context, but there is abundant evidence that Treg cells play a central role in suppressing a range of autoimmune disease and that loss of functional FOXP3 results in fatal multiorgan autoimmunity (Brunkow et al., 2001; Lin et al., 2005; Lahl et al., 2007).
Treg cells achieve suppression by multiple mechanisms involving specific cytokines and other factors, but the principal processes are via bystander suppression and so-called infectious tolerance (Fig. 2) (Jonuleit et al., 2002). In bystander suppression, antigen-activated Treg cells express cytokines such as IL-10, transforming growth factor (TGF)-β, and IL-35 that suppress local effector T cells irrespective of antigen specificity (Fig. 2) (Masteller et al., 2005; Babu et al., 2006; Walsh et al., 2009). In contrast, during infectious tolerance, activated Treg cells condition the host to promote further Treg cell populations of broader specificity (Shevach et al., 1998; Cobbold et al., 2009; Miao et al., 2009). Infectious tolerance can be adoptively transferred between animals and can persist beyond the lifespan of the original clone. Infectious tolerance is therefore of intense interest in the context of transplantation and for cell therapy to prevent or treat autoimmune conditions such as type 1 diabetes (Han et al., 1996; Zelenika et al., 2001; Waldmann et al., 2006).
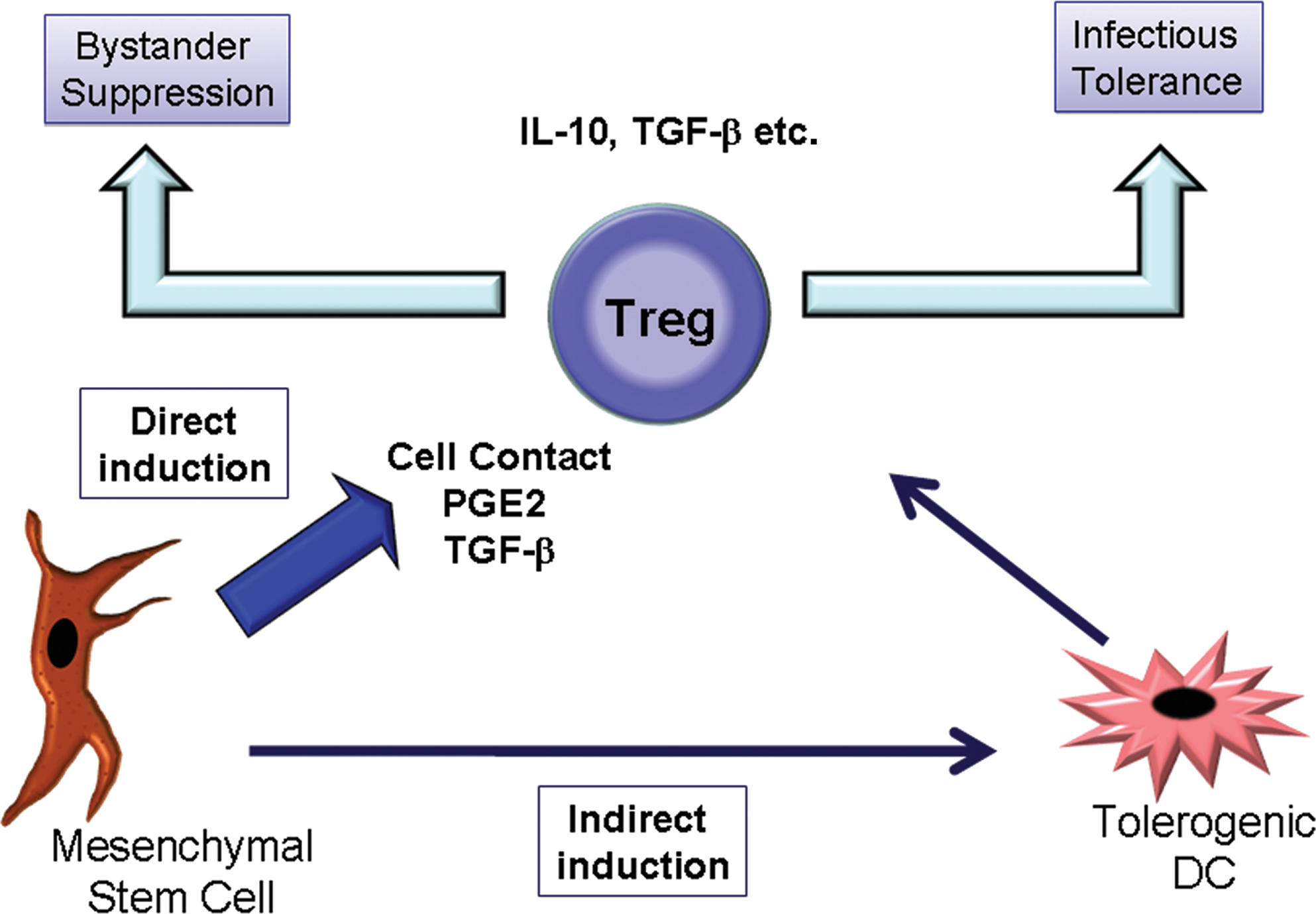
Allogeneic MSCs induce a range of regulatory T-cell populations (simplified here as Treg). This can occur via an indirect route requiring tolerogenic DCs or by a direct MSC–T-cell interaction requiring cell contact and soluble factors. Once generated, Treg may suppress allogeneic rejection through multiple pathways including creation of a suppressive microenvironment (bystander suppression) or induction of further regulatory cells (infectious tolerance). The latter effect could persist well beyond the detectable presence of MSCs. Color images available online at
MSCs can induce Treg cells indirectly via their modulating effects on DCs as described earlier (Zhang et al., 2004; Aggarwal and Pittenger, 2005; Djouad et al., 2007; English et al., 2008; H. Li et al., 2008; Wang et al., 2008). It is now clear, however, that MSCs can also directly induce Treg cells in the absence of DCs (Prevosto et al., 2007). This is clearly seen when allo-MSCs are co-cultured with purified naive CD4+ T cells from mice in which green fluorescent protein (GFP) has been “knocked in” to the locus for the FOXP3 gene. Co-culture with MSCs results in nuclear GFP expression (indicating activation of the FOXP3 gene) not seen in CD4+ cells cultured in the absence of allo-MSCs (L. Tobin, personal communication). A range of studies support the hypothesis that MSC-induced CD4+ CD25high FOXP3+ T cells actively suppress the immune effector responses to allo-antigen (Di Ianni et al., 2008; Nasef et al., 2008; Selmani et al., 2008; Gonzalez et al., 2009; Madec et al., 2009). This is further supported by the results of human clinical studies of MSCs in the prevention of allograft rejection, GVHD, chronic inflammatory disease, and autoimmunity (Koc et al., 2002; Ringden et al., 2006; van Laar and Tyndall, 2006; Uccelli et al., 2008; Caplan, 2009; Togel et al., 2009; Tyndall and Gratwohl, 2009; Zhou et al., 2010).
There are well-described roles for soluble mediators such as TGF-β1 and PGE2 in the generation and expansion of Treg cells from CD4+CD25– precursors (Horwitz et al., 2002; Yamagiwa et al., 2001; Zheng et al., 2002; Baratelli et al., 2005), with TGF-β1 signaling identified as a key regulator of the pathway that initiates and maintains FOXP3 expression and suppressor function (Fu et al., 2004). The exact mechanisms responsible for induction of Treg cells by allogeneic human MSCs have been studied by several groups. Cell contact, PGE2, and TGF-β1 appear to play complementary, non-redundant roles (English et al., 2009; Ghannam et al., 2010). Although cell contact, PGE2, and TGF-β1 contribute to Treg cell induction by MSCs, human MSCs also secrete the soluble MHC isoform HLA-G in an IL-10- and contact-dependent manner. Leukemia inhibitory factor and HLA-G contribute to the expansion of CD4+ CD25high FOXP3+ Treg cells (Nasef et al., 2008; Selmani et al., 2008), and this may also explain the observations that MSCs sustain Treg cell survival and the suppressor phenotype over time (Di Ianni et al., 2008). It is important that Treg cells generated by MSC encounter have been repurified and shown to further suppress alloreactivity in the absence of the original MSCs (English et al., 2009). The implications of these data are that MSC therapies against autoimmunity may cast a regulatory or immunosuppressive shadow long after the original stromal cells have declined, reminiscent of infectious tolerance, and thus suggest a mechanism whereby MSCs with a brief persistence in vivo may have profound long-term effects.
The biological significance of MSC induction of CD4+ Treg cells has also been studied in vivo. Pretransplant infusion of MSCs prolongs the survival of a semi-allogeneic heart transplant through the generation of Treg cells (Casiraghi et al., 2008; Popp et al., 2008; Ge et al., 2009). Recently MSCs were also shown to prevent autoimmune β-cell destruction and subsequent diabetes mellitus by inducing Treg cells in NOD mice (Madec et al., 2009) and in a similar rat model (Boumaza et al., 2009). The latter autologous study also showed that MSCs sustained Treg cell responses in the periphery, supporting studies described above. Infused MSCs and rapamycin synergize to attenuate allo-immune responses and promote cardiac allograft tolerance, a process associated with tolerogenic DC and Treg cell induction (Ge et al., 2009). Finally, in a murine model of asthma, MSC therapy had a beneficial therapeutic effect that was lost when Treg cells were chemically depleted (H. Kavanagh, personal communication). Taken together, these results extend the very clear in vitro data to show that MSC induction of Treg cells has functional relevance in vivo.
MSC-mediated immunomodulation occurs by multiple redundant pathways, of which CD4+ Treg cell induction is only one (English et al., 2007, 2008; Ryan et al., 2007). For example, MSCs also induce other regulatory T-cell populations, including CD8+ regulatory cells (Prevosto et al., 2007) (H. Kavanagh, personal communication). Djouad and colleagues have demonstrated that MSC induction of CD8+ Treg cells was responsible for at least some immunosuppressive activities of MSCs in vitro (Djouad et al., 2003), and these cells may amplify other suppressor mechanisms (Prevosto et al., 2007). The outstanding questions with regard to MSC induction of Treg cells center around the robustness of the regulatory effect, delineation of the contribution of MSCs to bystander or infectious tolerance, the precise identity of the suppressor mechanisms such as the contact-dependent signal, and the degree to which preclinical animal models translate to human disease. Answers to these questions are imminent and may well determine the ultimate utility of MSC therapy against autoimmune and other immune-mediated diseases.
MSC Suppression of T-Cell and B-Cell Effector Responses
In addition to modifying antigen presentation by DCs and promoting the expansion of Treg cell populations, MSCs may also directly influence effector functions of the adaptive immune system, including Th differentiation programs and B-cell/plasma cell activation and antibody production. These direct interactions with the effector arms of the adaptive immune system constitute an important additional element of the therapeutic effect of MSCs in diseases involving damaging inflammation, autoimmunity, or allograft rejection. In the case of Th differentiation, there has been some debate about the modulating influence of MSCs on the T-cell subsets induced by allogeneic, autoimmune, or other model antigens. Although there have been reports that MSCs preferentially reduce Th1 responses to favor Th2-like cytokine responses (Batten et al., 2006; Li et al., 2007; Wang et al., 2008; Lu et al., 2009), this is unlikely to be a universal feature. For instance, in the context of MSC-induced Treg cells, Th2 responses are effectively suppressed at a level sufficient to reduce pathology in vivo (Nemeth et al., 2010; Sun et al., 2010) (H. Kavanagh et al., personal communication). With regard to other CD4+ populations, Ghannam et al. (2010) showed that MSCs prevented the in vitro differentiation of naive CD4+ T cells into Th17 cells and inhibited the production of the effector cytokines IL-17, IL-22, IFN-γ, and TNF-α by fully differentiated Th17 cells. Furthermore, under inflammatory conditions, MSCs appear to mediate the adhesion of Th17 cells via CCR6 and exert anti-inflammatory effects through the induction of a Treg phenotype in these cells (Ghannam et al., 2010). These data support earlier studies showing that MSCs ameliorate experimental autoimmune encephalitis through suppression of CD4+ Th17 cells (Rafei et al., 2009b), again suggesting that MSC immune modulation of T-cell subsets is not merely a rebalancing of the Th1–Th2 axis. In summary, there is now a considerable body of data demonstrating that MSCs have potent direct suppressive influences on effector CD4+ T cells while promoting and sustaining Treg cells.
Studies of MSC effects on B-cell function—particularly the in vivo production of antibody during antigen-specific immune responses—have been less frequent and have produced some conflicting results (Corcione et al., 2006; Gerdoni et al., 2007; Rasmusson et al., 2007; Comoli et al., 2008; Rafei et al., 2008; Tabera et al., 2008; Traggiai et al., 2008; Asari et al., 2009; Schena et al., 2010; Youd et al., 2010). Many antibody responses are dependent on T-cell help, and so it is important to consider the distinction between direct MSC modulation of B cells and indirect B-cell effects resulting from suppression of DCs and T cells. In vitro experiments involving co-culture of human MSCs with purified B-cell populations under a variety of stimulatory conditions have predominantly shown inhibition of B-cell proliferation (via G0/G1 cell cycle arrest), differentiation, immunoglobulin production, and chemotaxis with preserved or improved cell survival (Corcione et al., 2006; Comoli et al., 2008; Tabera et al., 2008). Similar observations have been reported for purified mouse B cells and plasma cells in vitro (Rafei et al., 2008; Asari et al., 2009; Schena et al., 2010). Mediators that have been identified for MSC suppression of B-cell functions to date include alternatively cleaved CCL2 (Rafei et al., 2008), IFN-γ, and PD1/PDL1 interaction (Schena et al., 2010). In contrast, however, several groups have reported stimulatory effects of MSCs on in vitro–activated B cells or plasma cells from healthy humans (Rasmusson et al., 2007) or patients with systemic lupus erythematosis (Traggiai et al., 2008). The reasons for such apparently contradictory results are not entirely clear but may include variability in the sources and properties of MSCs as well as in the different antigen-dependent and polyclonal stimuli that have been used to activate B cells in culture.
The limited numbers of studies carried out in in vivo models of pathogenic antibody production have also yielded inconsistent outcomes. MSC-mediated inhibition of antigen-specific antibody production (including T-cell independent antibody responses) was observed in mice by three groups (Gerdoni et al., 2007; Rafei et al., 2008; Asari et al., 2009), whereas others have reported failure of in vivo MSC administration to suppress autoantibodies or increased auto-antibody titers and disease activity in a mouse model of systemic lupus erythematosis (Schena et al., 2010; Youd et al., 2010). It is interesting that there is also some experimental evidence that pre-existing antibody responses may be suppressed by MSCs through inhibition of plasma cell antibody production (Comoli et al., 2008; Rafei et al., 2008). Taken together, the existing literature regarding MSC effects on B-cell and plasma cell functions suggest a complex interaction that includes both inhibitory pathways of high clinical interest as well as the potential for stimulatory effects that could limit the benefit of MSC-based therapies for some immune/inflammatory diseases.
To What Extent Are Allo-MSCs Immunoprivileged In Vivo?
Despite numerous clinical trials with allo-MSCs, it remains unclear to what extent these cells elicit anti-donor immune responses in vivo and whether efficacy is truly equivalent for allo-MSCs compared to auto-MSCs for a given therapeutic target. A key question is whether the inherent immune suppressive properties of MSCs are sufficient to overcome the potent and diverse processes of immunologic priming, effector responses, and memory that are typically engendered by allogeneic cells in a healthy individual. As reviewed in previous sections, many aspects of the mechanistic basis for MSC-mediated immune modulation have been uncovered, and at least some of these are known to be operational in vivo. Furthermore, several studies have indicated that donor-specific MSC infusion prior to or at the time of allogeneic organ or tissue transplantation may delay rather than hasten allograft rejection (Bartholomew et al., 2002). In humans, donor MSCs have been reported to attenuate some aspects of GVHD following allogeneic hematopoietic stem cell transplantation (Lazarus et al., 2005). More recent data from animal models also suggest that MSCs of allogeneic or xenogeneic source can effectively protect from death due to sepsis (Nemeth et al., 2009), neuronal loss following cerebral ischemia (Ohtaki et al., 2008), and neurological injury in experimental autoimmune encephalomyelitis (Zappia et al., 2005; Rafei et al., 2009b) in comparable fashion to auto-MSCs.
Although such preclinical and clinical studies provide evidence in favor of a therapeutic benefit of allo-MSCs, the question of whether they enjoy complete immune privilege in vivo remains highly relevant to the true clinical and commercial benefits of allo-MSC therapies in the long term. In the majority of potential clinical applications it is not clearly known for how long MSCs need to persist in vivo in order to exert their maximal beneficial effects. For conditions in which a short-lived presence of MSCs within diseased tissue is of benefit, it is, nevertheless, likely that strong immunogenicity of allo-MSCs will have a negative influence on the potency and duration of treatment effect as well as the feasibility of subsequent dosing. For clinical applications in which permanent MSC engraftment, prolonged therapeutic effect, or subsequent allogeneic organ transplantation is anticipated, even weak in vivo immunogenicity may prove to be a formidable barrier to successful translation. It is important, therefore, that basic observations regarding the effectiveness of allo-MSCs in preclinical disease models be extended to define the extent and limits of their immune privileged state. Table 1 summarizes the results of several studies in which the in vivo immunogenicity of allo-MSCs has been specifically examined or in which the therapeutic efficacies of allo-MSCs and auto-MSCs have been directly compared in immune competent hosts. It is important that the work carried out to date has included a variety of species and administration routes. In some studies, observations regarding immunogenicity of allo-MSCs have been strengthened by re-challenging recipient animals with donor allo-antigen through strategies such as skin grafting. In others, experimental observations of allo-MSC longevity in vivo provide indirect evidence for or against immune-mediated rejection. Finally, in a smaller number of studies, the influence of allo-MSC number, exposure to inflammatory cytokines, or differentiation along one or more lineages on in vivo immunogenicity has been examined. Some significant inconsistencies remain to be resolved, but this literature (summarized in Table 1) does allow several provocative statements to be made regarding the in vivo immune responses to allo-MSCs:
EAE, experimental autoimmune encephalitis; i.m., intramuscular; i.p., intraperitoneal; i.v., intravenous; NA, not applicable; NT, not tested; post-MI, post--myocardial infarction; s.c., subcutaneous.
The majority of studies that have carefully analyzed donor-specific responses in immune competent rodents, pigs, and non-human primates following allo-MSC administration have generated evidence of immunogenicity. Notably, donor-specific antibody was observed in all studies in which allo-antibody assays were carried out. In several studies allo-specific responses were relatively weak, whereas in others allo-MSCs proved to be strongly immunogenic and sensitizing against subsequent donor antigen exposure (Table 1).
Immunogenicity and therapeutic immune modulation can co-exist in vivo. In some disease models allo-MSCs and auto-MSCs were found to be of comparable efficacy despite eliciting anti-donor immune responses, whereas in others, efficacy was lower for allo-MSCs compared with auto-MSCs (Table 1). The parallel influences of immunogenicity and suppression may be especially beneficial for proposed therapies using MSCs against autoimmune conditions. In these scenarios the ability of MSCs to induce infectious tolerance may be the critical correlate of efficacy.
Site of administration is an important modifier of allo-MSC immunogenicity. Sites for which allo-MSCs appeared to be non-immunogenic or very weakly immunogenic included intracranial, intracerebral, intra-articular, and implanted into skin wounds (Table 1). In contrast, intravenous, intraperitoneal, subcutaneous, and intramyocardial administration were sometimes associated with detectable anti-donor immunity and sometimes active rejection in the absence of other immune suppressive therapy (Table 1).
Overall, it is reasonable to state at this time that MSCs have the capacity to initiate both cellular and humoral allo-immune responses in vivo but that, in some conditions, immunogenicity may be considerably attenuated compared with other allogeneic cell types because of inherent anti-inflammatory and immune modulatory properties.
Some additional important issues are linked to the basic question of the in vivo immunogenicity of allo-MSCs and its significance for their therapeutic application. First, it remains unclear whether allo-MSC immunogenicity is altered following differentiation into chondrocytes, osteocytes, or other lineages. This consideration is of particular relevance to the use of MSCs in bone and joint disease but has been little studied to date. Nonetheless, it has been shown in rabbits that osteogenic cells differentiated from MSCs retained immunosuppressive properties in vitro and functioned as osteoblasts in allogeneic hosts for up to 28 days without precipitating primary rejection or sensitizing to a subsequent MSC-donor specific skin graft (Liu et al., 2006). Second, it is of interest to know whether immunosuppressive therapies currently prescribed in organ transplantation can be effectively used to prevent anti-donor immune responses to allo-MSCs in vivo without diminishing therapeutic efficacy. In this regard, Poncelet et al. (2008) have shown, in a miniature pig model of myocardial infarction, that the calcineurin inhibitor tacrolimus significantly attenuated the anti-donor antibody response to allo-MSCs delivered directly into the infarct. Recently, Ge et al. (2009) also demonstrated that a combination of allo-MSC infusion and low-dose sirolimus (rapamycin) therapy resulted in long-term survival of fully MHC-mismatched heart transplants in mice. As a final issue, allo-MSCs may be deployed into sites of inflammation, rich in pro-inflammatory mediators such as IL-1, TNF, and IFN-γ. The questions arise, therefore, whether allo-MSC immune modulation persists and anti-donor immune responses are enhanced in the inflamed tissue environment. Fortunately, several advances have been made in this regard. Stimulation of MSCs with IFN-γ upregulates both MHC class I and II (Le Blanc et al., 2003a,b), which may render these cells susceptible to rejection in an immune competent host especially as an elevated MHC class I level makes the cells vulnerable to cytotoxic T-cell-mediated lysis in vitro (S. Schu et al., manuscript submitted). In a mouse model of experimental autoimmune encephalomyelitis, IFN-γ increased MSC expression of CCL2, MHC I, and MHC II, leading to loss of disease suppression and allo-MSC rejection (Rafei et al., 2009a). In the pig, Cho et al. (2008) have demonstrated that both T-cell and antibody responses to allo-MSCs were enhanced in vivo by pre-exposure of MSCs to IFN-γ. Thus, there is evidence that allo-MSC immunogenicity and rejection may be more of a barrier to successful therapeutic application in the setting of localized inflammation. In contrast, it is also well established that exposure of MSCs to some inflammatory signals (e.g., high-dose IFN-γ) can enhance their suppressive effects on T cells, monocyte/macrophages, and DCs (English et al., 2007; Ryan et al., 2007; Polchert et al., 2008; Opitz et al., 2009). In models of GVHD, chronic obstructive pulmonary disease, and allergic airway disease, prestimulation of MSCs with IFN-γ improves the efficacy of cell therapy (Polchert et al., 2008) (B.P.M., unpublished data; H. Kavanagh et al., manuscript submitted). Mechanistically, these observations have been linked with IFN-γ-mediated upregulation of IL-10, TGF-β1, PGE2, and, in particular, the immune suppressive enzyme indoleamine 2,3-dioxygenase (English et al., 2007; Ryan et al., 2007; Popp et al., 2008; Opitz et al., 2009; Crop et al., 2010). Although paradoxical in some senses, the literature in this area indicates that allo-MSCs introduced into a site of existing tissue injury and inflammation engage in a complex, active cross-talk with the cells and mediators around them. On the one hand, these interactions may result in further induction of beneficial, immune suppressive features of the MSCs, whereas, on the other hand, they may render the MSCs more susceptible to lysis by cytotoxic T cells and NK cells. A better recognition that these two processes are not mutually exclusive and should be carefully studied in parallel in the future will be an essential step toward improving allo-MSC-based therapies to the point of routine clinical use for inflammatory and immune-mediated diseases.
Outlook
The field of MSC-related immune modulation has reached an exciting juncture. Despite some lingering controversy remaining regarding the degree to which MSCs truly differ from fibroblasts (Jorgensen, 2010), it has become very clear that stromal progenitor cells from multiple sites interact dynamically with almost every component of the immune system and, as reviewed here, do so with predominantly suppressive effects. The list of potential therapeutic applications continues to grow, and the use of pre-expanded allo-MSCs appears to be the most practical and commercially viable approach. In confronting the question of why recent large clinical trials produced disappointing results, therefore, it must be acknowledged that some assumptions made about the interaction between allo-MSCs and the host immune response in vivo may have been overly simplistic. Furthermore, as the specific mechanisms whereby MSCs exert their beneficial effects in a given disease remain poorly understood, there are few genuine, quantifiable, correlates of efficacy upon which to base comparisons between different sources of MSCs. From our perspective, the literature to date supports a view that therapeutic MSCs are conditionally subject to allo-immune responses in vivo. Furthermore, there is experimental evidence to suggest that cellular and humoral anti-donor responses in immune competent recipients are sufficient, in some settings, to limit MSC longevity, attenuate beneficial effects, and sensitize to subsequent allo-antigen exposure. It is important that the available literature also provides reasons to believe that allo-MSCs may be truly immune privileged at some anatomical sites or that detrimental anti-donor responses to allo-MSCs may be readily controlled or even converted to donor-specific immune tolerance. In the future, achieving the optimal benefits of allo-MSC therapy for each disease process will require further careful comparisons with autologous cells coupled with a more rigorous application of methods in transplant immunology to ongoing preclinical and clinical studies.
Footnotes
Acknowledgments
The authors are supported by a Science Foundation Ireland Centre for Science and Engineering Technology grant to the Regenerative Medicine Institute (REMEDI) (to M.D.G., T.R., and B.P.M.) and by individual grants from the Science Foundation Ireland (SFI): SFI 06/RFP/BIC056 and SFI PI 07/IN.1/B925 (to T.R.) and SFI PI 06/IN.1/B652 (to M.D.G.). Dr. H. Kavanagh and Ms. Laura Tobin are thanked for sharing unpublished data with the authors during the preparation of this article.
Author Disclosure Statement
M.G., T.R., and B.P.M each declare that no competing financial or other competing interests exist.