
Abstract
Gene therapy studies in primates can provide important information regarding vector tropism, specific cellular expression, biodistribution, and safety prior to clinical trials. In this study, we report the assessment of transduction efficiency of recombinant adeno-associated virus (rAAV) vectors using human postmortem retina. Transductions were performed using two in vitro models prepared from human tissue: dissociated cell cultures and retinal explants. These models were used to assess cellular tropism and selectivity of rAAV vectors encoding for fluorescent proteins under the control of different promoters. These promoters were a ubiquitous cytomegalovirus promoter and a cell type–specific promoter targeting expression in ON bipolar cells. The results demonstrate that this in vitro approach can limit the use of living primates for the validation of gene therapy in vision and ophthalmology.
Introduction
Successful gene therapy testing in animal models is a usual prerequisite for human clinical trials. Thus, LCA2 gene therapy was first assessed in several animal models to assess safety and efficacy of a subretinal injection with rAAV vectors encoding the functional RPE65 gene. Efficacy was demonstrated in transgenic and natural murine models for LCA2 (Dejneka et al., 2004; Lai et al., 2004; Pang et al., 2006), as well as in a natural occurring canine model of the disease, the Briard dog (Acland et al., 2001; Le Meur et al., 2007). The subretinal rAAV delivery allowed a functional expression of the correct RPE65 protein in the RPE leading to the restoration of visual function in these models (Narfström et al., 2003; Acland et al., 2005; Le Meur et al., 2007). Although the therapeutic evaluation in non-human primates is not an absolute prerequisite to proceed for gene therapy, it provided information on the vector biodistribution and the systemic toxicity of LCA2 treatment (Jacobson et al., 2006). Such primate experiments can also demonstrate the vector efficacy for new targeted cell types, new genes, new gene promoters, and new viral vectors.
To limit the use of primates for ethical and economic reasons, we propose to use cell cultures of postmortem human retinal cells and tissue for validation steps. Retinal neurons from human postmortem tissue can indeed survive in monolayer culture for extended periods, maintaining their functional properties (Picaud et al., 1998). The mammalian retina can even be kept in vitro as an ex vivo tissue or retinal explant with normal tissue architecture and intact network activity (Pinzón-Duarte et al., 2000; Vallazza-Deschamps et al., 2005; Agulhon et al., 2007). Although such in vitro experiments are not relevant for safety and biodistribution assessments, they can already provide crucial information concerning comparative efficacies in cell targeting with different gene promoters and viral vectors in human retinal tissues with vector applications from both retinal sides.
Materials and Methods
rAAV preparation
The transgene cassette in all viruses consisted of cytomegalovirus (CMV) or metabotropic glutamate receptor 6 (GRM6)-driven fluorescent protein (channelrhodopsin-2 [ChR2]–yellow fluorescent protein [YFP] or green fluorescent protein [GFP]). Adeno-associated virus (serotypes 2/5 and 2/8) was prepared by triple transfection by the Medical Genetics Vector Core Facility at the University of Pennsylvania (Philadelphia, PA) as previously described (1 × 1013 genomic copies [GC]/ml) (Auricchio et al., 2001). The vectors were purified using CsCl gradient centrifugation. Vector physical titers were assessed by real-time polymerase chain reaction, and purity of the virus was ensured by endotoxin assay and gel electrophoresis. The titer of the purified virus used was 3 × 1012–1 × 1013. Virus aliquots were stored at −80°C and thawed prior to administration to retinal explants and cell cultures.
Human ocular tissue
Human ocular tissues were obtained from the Cornea Bank of Amsterdam (Amsterdam, The Netherlands) under approved human subject protocols. Eyes were collected from six donors (ranging in age from 46 to 71 years) subsequent to death through natural causes. After removal of the cornea, eyes were stored immediately in cold phosphate-buffered saline (PBS) solution and shipped to our laboratory. Postmortem delay times were always less than 48 hr before the production of retinal explants and cell cultures.
Retinal explants and vector administration
Human retinal explants were prepared similarly as previously described for murine or rat tissues with the exception of enzymatic digestion (Pinzón-Duarte et al., 2000; Vallazza-Deschamps et al., 2005; Agulhon et al., 2007). Immediately after receipt of the eyes, anterior parts were removed, and vitreous humor with attached neural retina was transferred in CO2-independent medium (Invitrogen, Carlsbad, CA). The retinal tissue was carefully separated from the vitreous to prepare retinal fragments (∼1 cm2). With the photoreceptor face up, these retinal fragments were placed on the polycarbonate membrane of a Transwell 0.4-μm (pore size) cell culture insert (Corning, Corning, NY) with one drop of CO2-independent medium and carefully flattened with a polished Pasteur pipette. Before transduction, rAAV vector solutions were diluted in 50 μl of Neurobasal-A medium supplemented with 2 mM
Retinal cell cultures and vector administration
Monolayer cell cultures were prepared as described previously (Picaud et al., 1998). Retinas separated from the eyes were placed in CO2-independent medium and chopped into small fragments (∼1 mm2). The fragments were then washed twice in mammalian Ringer's solution without Ca2+ and incubated with 0.2% activated papain (Worthington Biochemical, Lakewood, NJ) for 25 min at 37°C. Digestion was stopped by addition of 2 volumes of Dulbecco's modified Eagle's medium containing 10% fetal calf serum (Invitrogen) and 25 μl of DNase I (Sigma-Aldrich, St. Louis, MO). Cells were dissociated by gentle trituration of retinal fragments using a plastic Pasteur pipette. After removal of the cell suspension, medium was added to the residual retinal fragments to repeat the procedure until their complete dissociation. After mild centrifugation of all cell suspensions, NBA+ medium was added on the cell pellets, and an aliquot of this new cell suspension was incubated with trypan blue vital dye and examined on a hemocytometer to count viable cells. Cells were seeded at an initial density of 4 × 105 viable cells/cm2 into 24-well culture plates containing coverslips coated with polylysine and laminin (Sigma-Aldrich). One week after seeding, culture medium was removed, and then retinal culture cells were exposed to 1 × 1012 GC of rAAV2/8-GRM6-CY diluted in 100 μl of NBA+ medium for each well. The next day, 400 μl of NBA+ medium was added to each well. Culture medium was renewed weekly.
Immunohistochemistry
Retinal explants were fixed by incubation with 4% paraformaldehyde in phosphate buffer (0.1 M, pH 7.4) for 1 hr at 4°C, cryoprotected in graded PBS-sucrose solution (10%, 20%, and 30%), and included in Shandon Cryomatrix embedding resin (Thermo Scientific, Waltham, MA), and then 10-μm-thick vertical sections were cut on a cryostat. Monolayer cell cultures were fixed with 4% paraformaldehyde for 20 min at 4°C.
Procedures for immunostaining were identical for retinal explant sections and monolayer cell cultures: Samples were permeabilized and blocked by incubation for 30 min in 0.5% Triton X-100, 1% bovine serum albumin, and 3% normal goat serum in PBS and incubated with primary antibodies for 2 hr at room temperature. Samples were then washed, incubated for 1 hr at room temperature with secondary antibodies and diamidinophenylindole (Sigma-Aldrich) at a dilution of 1:1,000, washed again, and observed. We used the following antibodies: a polyclonal rabbit anti-GFP antibody (diluted 1:200) (Molecular Probes Inc., Eugene, OR), a monoclonal anti-G0α antibody (diluted 1:500) (Chemicon International, Temecula, CA), a monoclonal anti-rhodopsin antibody (diluted 1:500; rho4D2) (Hicks and Molday, 1986), and a monoclonal anti–protein kinase Cα antibody (diluted 1:500) (Sigma-Aldrich).
Results and Discussion
Optogenetic therapy targeting ON bipolar cells was recently found to restore visual function in blind mice. To investigate if the same construction could be use in humans, the viral vector rAAV2/8-GRM6-CY encoding for this construction was applied on dissociated cultured human retinal cells (n = 4 with at least 12 coverslips in each test) (Picaud et al., 1998). This viral vector encodes for the ChR2-YFP fusion protein, composed of the algal ChR2 and YFP, under the control of the mouse GRM6 enhancer element associated with the SV40 promoter. This expression cassette has been applied on the mouse retina to target ON bipolar cells with high selectivity (Ueda et al., 1997; Lagali et al., 2008).
Monitoring the cell fluorescence showed an absence of visible YFP expression during the first 2 weeks following transduction. The YFP endogenous fluorescence appeared in scattered cells only from the third week. This fluorescence persisted until 6 weeks after transduction when the cells were fixed. The size and morphology of cells with endogenous fluorescence were consistent with a glial identity (Fig. 1A–C); and none of them exhibited a neuronal shape. To visualize all transduced ON bipolar cells, double immunostaining was performed 4 weeks after transduction using an anti-GFP antibody, recognizing YFP, and an anti-G0α antibody labeling ON bipolar cells (Vardi, 1998). Double immunolabeled cells were visible, revealing YFP immunofluorescence in the cell body and processes of human ON bipolar cells (Fig. 1D–F). However, only a few double immunolabeled cells were visible on each culture coverslips: They represented 2% of the whole ON bipolar cell population (eight double-labeled cells/437 ON bipolar cells). Double immunostainings were also performed with an anti–protein kinase Cα antibody, recognizing rod bipolar cells, a subpopulation of ON bipolar cells (Osborne et al., 1992). Again, few (2%) scattered double immunolabeled cells were detected (four double-labeled cells/222 rod bipolar cells) (Fig. 1G–L). These results demonstrated the ability of the rAAV2/8-GRM6-CY vector to transduce and drive transgene expression in human ON bipolar cells in vitro. However, the low density of YFP-positive cells and the expression of the transgene in glial cells revealed the poor tropism of rAAV2/8 vector for ON bipolar cells and a lack of selectivity of the GRM6 regulation element. Moreover, the absence of endogenous fluorescence in transduced ON bipolar cells indicated the low level of transgene expression.
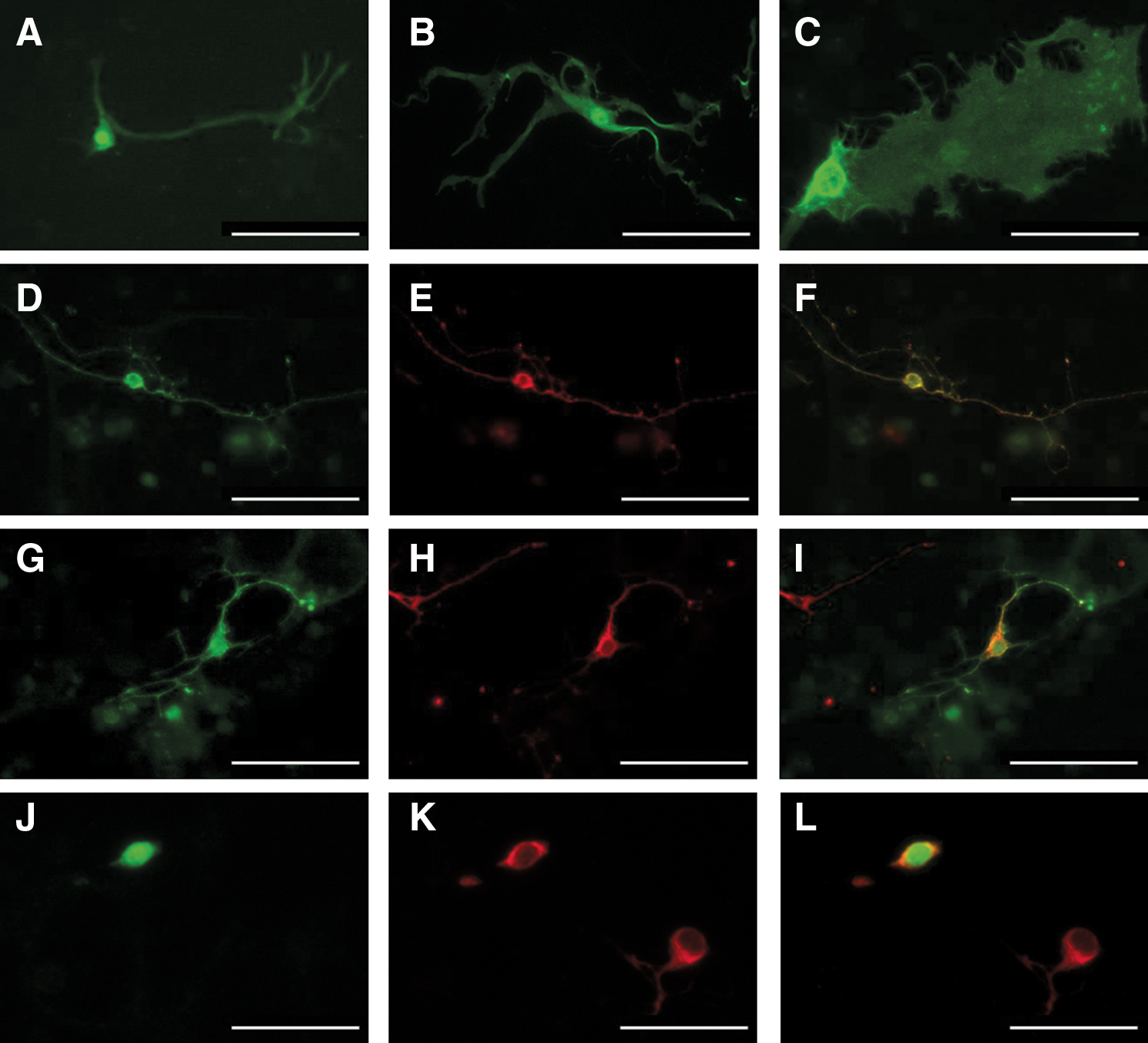
Transduction with rAAV2/8-GRM6-CY vector on monolayer cultured cells of postmortem human retina. (
To determine if a similar or higher cell transduction could be achieved in a more clinically relevant environment, transductions using rAAV2/8-GRM6-CY were also performed on human retinal explants (n = 4 with at least three explants in each test). Three weeks after transduction, observation of whole-mount retinal explants only revealed rare cells with YFP endogenous fluorescence, with size and morphology identifying them as glial cells (Fig. 2A and B). Immunolabeling with the anti-G0α antibody demonstrated the presence of numerous ON bipolar cells in the inner nuclear layer (Fig. 2C), whereas rhodopsin immunolabeling indicated rod identity of most photoreceptors in the outer nuclear layer (ONL) (Fig. 2D). However, we failed to found a specific marker for retinal ganglion cells in our model, preventing us from labeling the ganglion cell layer. Both G0α and rhodopsin immunolabeling demonstrated the preservation of the retinal laminar structure of the explant after several weeks, despite the absence of RPE, supporting the validity of our model. Even after anti-GFP immunolabeling, no transduced cells were detected in the inner nuclear layer, indicating an absence of transduced ON bipolar cells. This absence is consistent with the very low rate of ON bipolar cell transduction observed on monolayer cultures.

Transduction with ON bipolar cell–directed rAAV2/8-GRM6-CY vector on cultured retinal explants of postmortem human retina. (
To further examine if retinal explants could serve to investigate viral vector transduction in human retinal neurons, experiments were achieved with the rAAV2/5-CMV-GFP vector encoding GFP under the control of a ubiquitous CMV promoter. Fixation was carried out 3 weeks after transduction, although continuous monitoring during the incubation period showed endogenous fluorescence to be developing within 2 weeks. Examination of cryosections without immunostaining showed many cells with GFP endogenous fluorescence localized essentially in the ONL, identifying them as photoreceptors (Fig. 3A); other transduced cells had the typical morphology of Müller glial cells (Fig. 3B and C). Double immunostaining was performed to confirm the identity of the GFP-positive cells. In most regions of the explant, anti-G0α labeling failed to detect ON bipolar cells expressing GFP (Fig. 3D); only a very few double immunolabeled cells were found at occasional cuts within the explant (Fig. 3E). A more thorough analysis at retinal explant edges was not possible because peripheral cells were progressively dissociating from the explants during the long incubation periods required to achieve gene expression. However, these observations at occasional cuts suggested that the low rate of transduction for ON bipolar cells is caused by barriers formed by the ONL and the ganglion cell layer.
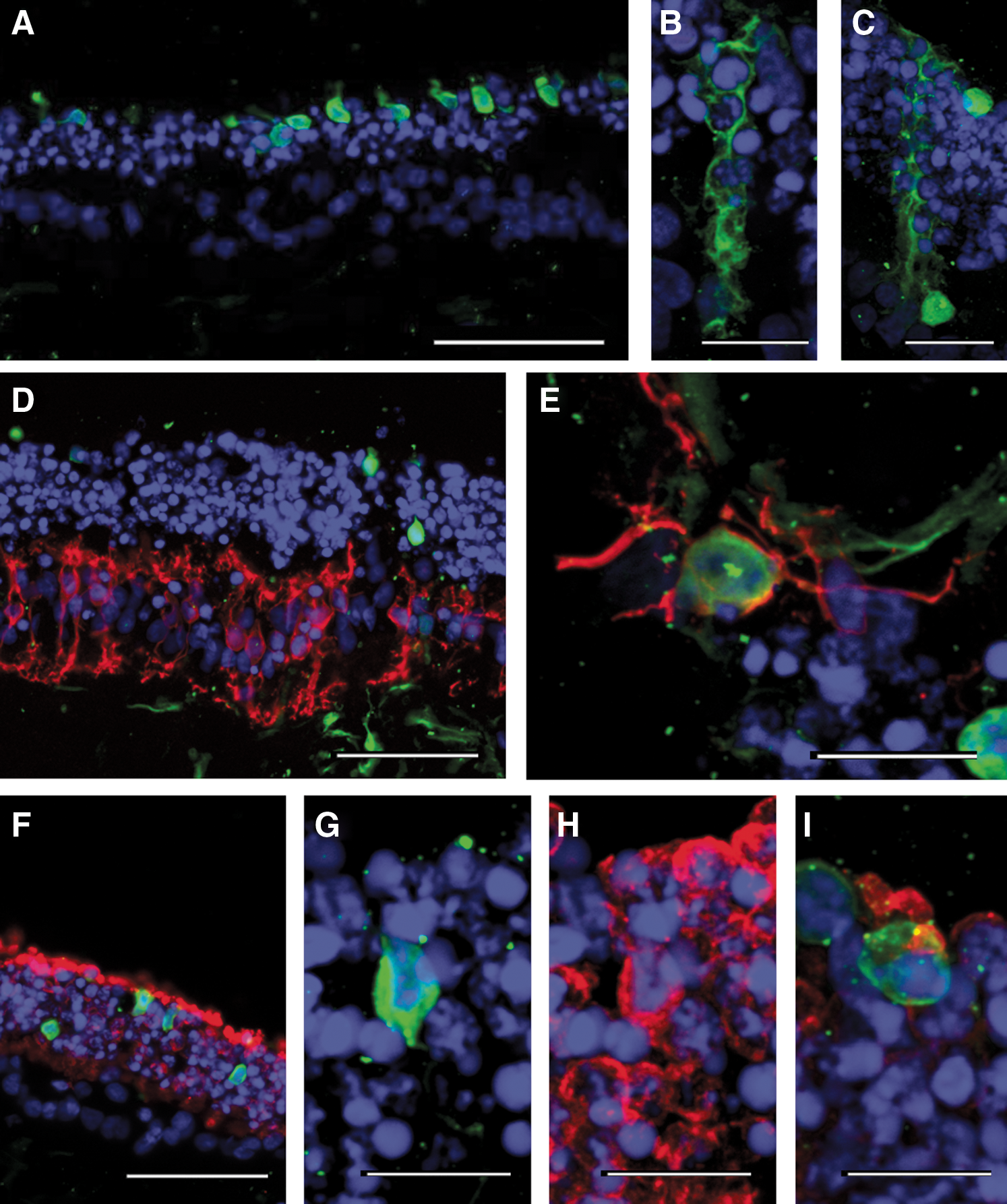
Transduction with an unspecific rAAV2/5-CMV-GFP vector on cultured retinal explants of postmortem human retina. (
In the ONL, we found that transduced cells represented 4.9 ± 1.6% (SD, n = 6) of all photoreceptors. When sections were immunolabeled for rhodopsin (Fig. 3F), confocal imaging of GFP-positive cells indicated the presence of both rhodopsin-positive (Fig. 3G and H) and rhodopsin-negative (Fig. 3I) transduced photoreceptors. Although rod photoreceptors are more numerous, most transduced photoreceptors were rhodopsin-negative; only 40% were double GFP/rhodopsin-labeled cells. Furthermore, most transduced cells were localized in the outer part of the ONL (Fig. 3A), consistent with their cone identity. This relative high rate of cone transduction could rely on either a good tropism of rAAV2/5 vector for cone photoreceptors or an easier access due to their position at the retinal outer surface. Although GFP-expressing cells were visible in the ganglion cell layer (Fig. 3D, bottom), the absence of specific markers for ganglion cells in the human retinal tissue cultured for several weeks prevented their identification.
In summary, this study demonstrates the feasibility of rAAV vector transduction in two in vitro models of postmortem human retinal cells. This approach provides information about cellular tropism of rAAV vectors and selectivity of transgene promoters. These two models enable an evaluation of vector tropism from two models of cell accessibility: in dissociated cell cultures with all cell types equally accessible to vector and in retinal explants where the native tissue diffusion constraints are partly preserved. Indeed, vector diffusion has to follow similar paths as during vitreal or subretinal injections when vectors are applied on either side of the explants. Thus, absence of or low gene expression in both monolayer cultures and explants can reveal poor vector tropism and promoter efficacy for the targeted cells, whereas lack of transduction in explants only suggests diffusion problems for the vector to reach the targeted cells. For instance, our experiments using the GRM6 sequence showed expression of transgene in glial cells in addition to the few bipolar cells, indicating that selectivity of this cell-specific enhancer is not absolute. These in vitro approaches could thus provide ways to screen for vectors and promoters with high transduction efficacies and selectivities, although these parameters could be affected in vitro with respect to the in vivo situation. In fact, intact eye cups would provide conditions closer to the in vivo situation, but the delays for obtaining postmortem human tissues are not compatible with this procedure because the retina is rapidly detaching from the RPE. Such experiments on intact eye cups or eyes may become possible on non-human primate tissues because postmortem delays can be shortened. However, in non-human primates, it is then preferable to perform in vivo experiments providing more information on immune responses and biodistribution.
Our work paves the way for a wider use of postmortem human retina in other studies, even if these models cannot fully replace in vivo testing on primates. This approach should speed up the development of new ocular gene therapy by screening and excluding vectors with low transduction rate or poor selectivity of transgene. Moreover, the use of these models would reduce experimentation on non-human primates, enabling a more ethical and less expensive validation of treatments before human clinical trials.
Footnotes
Acknowledgments
We thank the Amsterdam Cornea Bank (Amsterdam, The Netherlands) for providing postmortem human eyes. This work was carried out with the financial support of the Fondation Roland Bailly (Geneva, Switzerland), the Fondation pour la Recherche Médicale (Paris, France), the city of Paris, the regional council of Ile-de-France, and the European Community (contract RETICIRC number HEALTH-F2-2009-223 156 and contract TREATRUSH).
Author Disclosure Statement
No competing financial interests exist.