
Abstract
Mesenchymal stromal/stem cells (MSCs) are a population of stromal cells present in the bone marrow and most connective tissues, capable of differentiation into mesenchymal tissues such as bone and cartilage. MSCs are attractive candidates for biological cell-based tissue repair approaches because of their extensive proliferative ability in culture while retaining their mesenchymal multilineage differentiation potential. In addition to its undoubted scientific interest, the prospect of monitoring and controlling MSC differentiation is a crucial regulatory and clinical requirement. Hence, the molecular regulation of MSC differentiation has been extensively studied. Most of the studies are in vitro, because the identity of MSCs in their tissues of origin in vivo remains undefined. This review addresses the current knowledge of the molecular basis of differentiation of cultured MSCs, with a particular focus on chondrogenesis and osteogenesis. Building on the information coming from developmental biology studies of embryonic skeletogenesis, several signaling pathways and transcription factors have been investigated and shown to play critical roles in MSC differentiation. In particular, the Wnt and transforming growth factor-β/bone morphogenetic protein signaling pathways are well known to modulate in MSCs the molecular differentiation into cartilage and bone. Relevant to the emerging concept of stem cell niches is the demonstration that physical factors can also participate in the regulation of MSC differentiation. Knowledge of the regulation of MSC differentiation will be critical in the design of three-dimensional culture systems and bioreactors for automated bioprocessing through mathematical models applied to systems biology and network science.
Mesenchymal Stem Cells: A Brief Overview
Mesenchymal stromal/stem cells (MSCs), a subset of stromal cells present at low frequency in most adult connective tissues, have been extensively studied for their multiple differentiation capabilities. Friedenstein and colleagues (1970) demonstrated that the bone marrow contains a rare population of plastic-adherent cells (approximately 1 in 10,000 nucleated cells) that were able to form single-cell-derived colonies. The adherent cell clones expanded into round-shaped colonies composed of fibroblastoid cells, thus leading to the term colony-forming unit—fibroblasts (CFU-f); after proliferation some of the colonies could differentiate into aggregates resembling small areas of bone or cartilage (Friedenstein et al., 1970, 1976). These initial observations were then extended by the study of CFU-f proliferative abilities and phenotypic characteristics (Castro-Malaspina et al., 1980; Prockop, 1997; Caplan and Bruder, 2001). These cells were eventually defined as multipotent and able to differentiate into osteoblasts, chondrocytes, adipocytes, and even myoblasts.
MSC Differentiation: The Caveat of In Vitro Studies
MSCs have been studied in several species, especially humans (Castro-Malaspina et al., 1980; Haynesworth et al., 1992; Bruno et al., 2009; Yoo et al., 2009) and mice (Gindraux et al., 2007; Sung et al., 2008). MSCs are classically derived from bone marrow (Pittenger et al., 1999), but they have been isolated as well from most connective tissues (da Silva Meirelles et al., 2006), including, to mention a few, adipose tissue (Zuk et al., 2002), periosteum (Nakahara et al., 1991; De Bari et al., 2001a, 2006), and synovial membrane (De Bari et al., 2001b, 2003). From the accumulating studies in the literature, it is now apparent that MSCs, whichever their tissue source, have ability to respond to a variety of physiological or pathogenic stimuli, producing responses that in each specific context require thorough investigation in order to assess their biological significance and exploit any potential clinical impact.
The differentiation of MSCs has been extensively studied, using mainly well-established in vitro assays with culture-expanded MSCs. Findings, therefore, have the caveat that they may not always be reliable and fully reproducible because of the vast heterogeneity of in vitro culture conditions and MSC types. Indeed, MSCs are known to undergo phenotypic rearrangements during ex vivo manipulations, losing expression of some markers while acquiring new ones (Jones et al., 2002). In addition, the possibility exists that the MSC phenotype and abilities vary between in vivo and in vitro settings because of the removal from their natural environment and the use of chemical and physical growth conditions that might alter their characteristics. The in vitro-obtained data are overly dependent on culture conditions for derivation and expansion of MSC populations and, therefore, are unlikely to be extrapolated to the native cells. Data may also not be entirely valid across species and intraspecies. As an example, murine MSCs differ not only from the human MSCs, but also between strains in marker expression and behavior in culture (Peister et al., 2004; Sung et al., 2008; Fiorina et al., 2009). Furthermore, data showed that MSCs from different tissues present phenotypic heterogeneity and different growth abilities (Baksh et al., 2007; Rebelatto et al., 2008), reflecting a tissue specificity that could be correlated to marked biological and functional differences (De Bari et al., 2008). Such heterogeneity could also underpin the regulation of MSCs and their responses to external stimuli, and it could be a main source of variation in the biological properties of MSCs. To complicate things further, studies on MSCs have not always been performed with primary cells but with cell lines such as C3H10T1/2 (Shea et al., 2003; Wang et al., 2010). This makes the reported findings not directly extrapolatable to primary human MSCs and of complex interpretation within the full biological picture.
Despite biological differences, MSCs from multiple tissue sources share common features such as the potential to differentiate into mesenchymal lineages and the expression of common surface markers (Baksh et al., 2007), although even within each tissue source, single-cell-derived clonal MSC populations are highly heterogeneous in their proliferative and differentiation potentials (Phinney and Prockop, 2007; De Bari et al., 2008).
Differentiation Potency of Culture-Expanded MSCs
One of the criteria to define MSCs is their ability to differentiate into the osteogenic, chondrogenic, and adipogenic lineages (Dominici et al., 2006). Classically, osteogenic differentiation of human MSCs (Jaiswal et al., 1997; Pittenger et al., 1999) requires incubation in fetal bovine serum (FBS)-containing medium supplemented with ascorbic acid, β-glycerophosphate, and dexamethasone, resulting in an increase in alkaline phosphatase activity and calcium deposition. The chondrogenic differentiation is performed with a high cell-density pellet or micromass culture treated with transforming growth factor (TGF)-β in serum-free medium; this results in production of cartilage-specific, highly sulfated proteoglycans and type II collagen. For adipogenic differentiation, MSCs are treated with dexamethasone, insulin, isobutyl methyl xanthine, and indomethacin (added to medium containing FBS), and the differentiation is revealed by the detection of lipid vacuoles with oil red O staining. At the clonal level, however, not all clonal populations are able to differentiate into all three lineages, as some MSC clones may lack differentiation into at least one lineage (Pittenger et al., 1999). Subsequent studies indicated the possible existence of a hierarchical model of differentiation, with human bone marrow clonal MSC populations readily differentiating into the three lineages but undergoing a sequential loss of lineage potential with the osteogenic precursors as residual cells (Muraglia et al., 2000).
Increasing evidence, indeed, indicates that MSC populations are heterogeneous with coexisting subsets having varying potency; this applies to bone marrow MSCs as well as to MSCs from other tissues. As an example, we reported that human synovium-derived clonal MSCs were all capable of osteogenic and chondrogenic differentiation although with varying potency, but only 30% of the clonal populations tested were able to differentiate into adipocytes (Karystinou et al., 2009).
Under appropriate conditions, MSCs can also differentiate into other mesenchymal lineages such as skeletal myocytes and tenocytes (Wakitani et al., 1995; De Bari et al., 2003; Hoffmann et al., 2006). MSCs have also been reported to differentiate into nonmesenchymal lineages such as neurons (Black and Woodbury, 2001). The clinical relevance of the presumptive nonmesenchymal potency of MSCs is, however, questioned because, for example, MSC-derived neuron-like cells were unable to generate action potentials and, therefore, to function as neurons (Hofstetter et al., 2002).
MSC-derived cartilage and bone
The “natural” mesenchymal propensity of MSCs has prompted researchers to devote attention to the chondrogenic and osteogenic abilities of MSCs with the clinical prospects of developing MSC-based biological approaches to the repair of articular cartilage and bone. The osteogenic potential of culture-expanded MSCs (Friedenstein et al., 1976; Ashton et al., 1985) has been studied extensively in vitro and in vivo. The first in vivo experiments with MSCs were performed with diffusion chambers loaded with culture-expanded cells (Ashton et al., 1980). Later, the adoption of bioscaffolds such as hydroxyapatite (HA) implanted in immunocompromised mice proved useful in studying MSC osteogenic differentiation in vivo (Ohgushi and Okumura, 1990). It was possible to obtain donor MSC-derived bone by subcutaneous implantation of HA scaffolds seeded with human MSCs (Muraglia et al., 1998; Bluteau et al., 2008; De Bari and Dell'Accio, 2008; De Bari et al., 2008; Dell'Accio et al., 2008). Using HA-based bioscaffolds, it then became possible to repair segmental bone defects in vivo by using autologous MSCs, under loaded conditions, both in large animals (Kon et al., 2000) as well as in humans in proof-of-concept studies (Quarto et al., 2001).
The use of MSCs in clinical practice remains challenging for issues such as the plethora of tissue sources and culture conditions, with resulting biological variability. For instance, human periosteum contains cells that after enzymatic release and culture expansion display the MSC phenotype and capacity at the single-cell level to differentiate into multiple skeletal lineages including bone (De Bari et al., 2006). Notably, in a proof-of-concept study we quantified the bone-forming potency of matched human MSCs from synovium and periosteum and analyzed the sources of variability in osteogenic outcome. We identified the tissue of origin of MSCs as the main source of variability, because MSCs from periosteum had significantly greater osteogenic potency than MSCs from synovium. A second source of variability was related to the individual donor, within each tissue. We measured the basal expression levels of osteoblast-lineage genes in clonal MSCs before osteogenic treatment, identified biomarkers that correlated with osteogenic outcome, and developed a mathematical model that predicts bone-forming potency of clonal MSC preparations, independent of donor and tissue source (De Bari et al., 2008). The development of a biomarker-based model that predicts the bone-forming potency of human MSC preparations is of considerable clinical relevance. A similar approach is likely to increase the consistency of therapies that employ MSCs for bone repair. It may also facilitate the selection of individuals that qualify for MSC-based bone repair and help identify the best source of and preparation protocol for human MSCs. It remains to be investigated whether the same formula can be applied successfully to MSC-based orthotopic bone repair in a preclinical model, where in addition to the properties intrinsic to the cell preparation, other factors such as inflammation and biomechanics will influence bone formation.
Although bone formation in vivo is relatively straightforward when MSCs are loaded onto matrices and then implanted subcutaneously in mice, the formation in vivo of stable hyaline-like cartilage resembling articular cartilage appears to be challenging when using MSCs. The chondrogenic potential of MSCs is well known in vitro in high-cell-density pellets or micromass cultures but the key question as to whether the resulting cartilage-like tissue is stable cartilage or a transient cartilage template destined to be replaced with bone in a process of endochondral ossification is unresolved. Using a nude mouse assay of ectopic cartilage formation validated with intramuscular injection of adult human articular chondrocytes (Dell'Accio et al., 2001), we demonstrated that the in vitro chondrogenic potential of synovial membrane-derived MSCs is not sufficient to predict the in vivo outcome at least in this nude mouse model, because the synovial MSCs induced in vitro into a chondrocyte-like phenotype failed to form stable cartilage when implanted in vivo (De Bari et al., 2004). Of note, Pelttari and colleagues reported that bone marrow MSC-derived cartilage pellets transplanted into ectopic sites in severe combined immunodeficiency (SCID) mice underwent endochondral ossification, via premature induction of chondrocyte hypertrophy-related molecules such as type X collagen (Pelttari et al., 2006). More recently, Scotti and colleagues (2010) reported that human bone marrow MSCs, implanted subcutaneously into nude mice at various stages of chondrogenic differentiation, formed bone only when they had developed in vitro hypertrophic cartilage traits. The underlying morphogenetic process was similar to limb bone development in embryos, thus revealing the capacity of human MSCs to generate bone tissue via an endochondral program.
Altogether, these studies suggest that the in vitro MSC-derived neoformed cartilage-like tissue is not stable. Nonetheless, they do not rule out the possibility that, as opposed to an ectopic site, the joint environment of a cartilage defect may instead be sufficient either to induce a stable cartilage phenotype or stabilize the chondrocyte-like phenotype of in vitro precommitted MSC populations. Uplift of the bone front at the expense of the overlying articular cartilage has, however, been observed in osteochondral repair by bone marrow cells (Qiu et al., 2003).
Molecular Regulation of MSC Differentiation
Several molecules have been reported to be involved in the regulation of MSC differentiation. In this review we focus on the main signaling pathways involved in the modulation of MSC differentiation but will also sketch out a concise overview of the transcription factors and the physical parameters influencing the fate of MSCs. Because studies of developmental biology have inspired a great deal of investigation in the MSC field, we think it will be helpful to report key findings in embryonic skeletogenesis before discussing the relevant pathways in MSC differentiation. Our attention revolves mainly around the Wnt canonical pathway and the TGF-β superfamily pathways.
Wnt signaling and MSC differentiation
Particularly studied is the Wnt family of secreted proteins, reported to modulate bone mass in vivo and shown as acting directly on MSCs (Liu et al., 2009; Takada et al., 2009). Wnt genes, a family of 19 genes in humans and mice, produce secreted proteins that, for their involvement in cell proliferation, differentiation, and apoptosis, are crucial in embryonic tissue development and in regeneration of adult tissues including bone (Westendorf et al., 2004). Figure 1 shows a schematic diagram of the canonical β-catenin-dependent Wnt signaling pathway (for a comprehensive review see Gordon and Nusse, 2006).
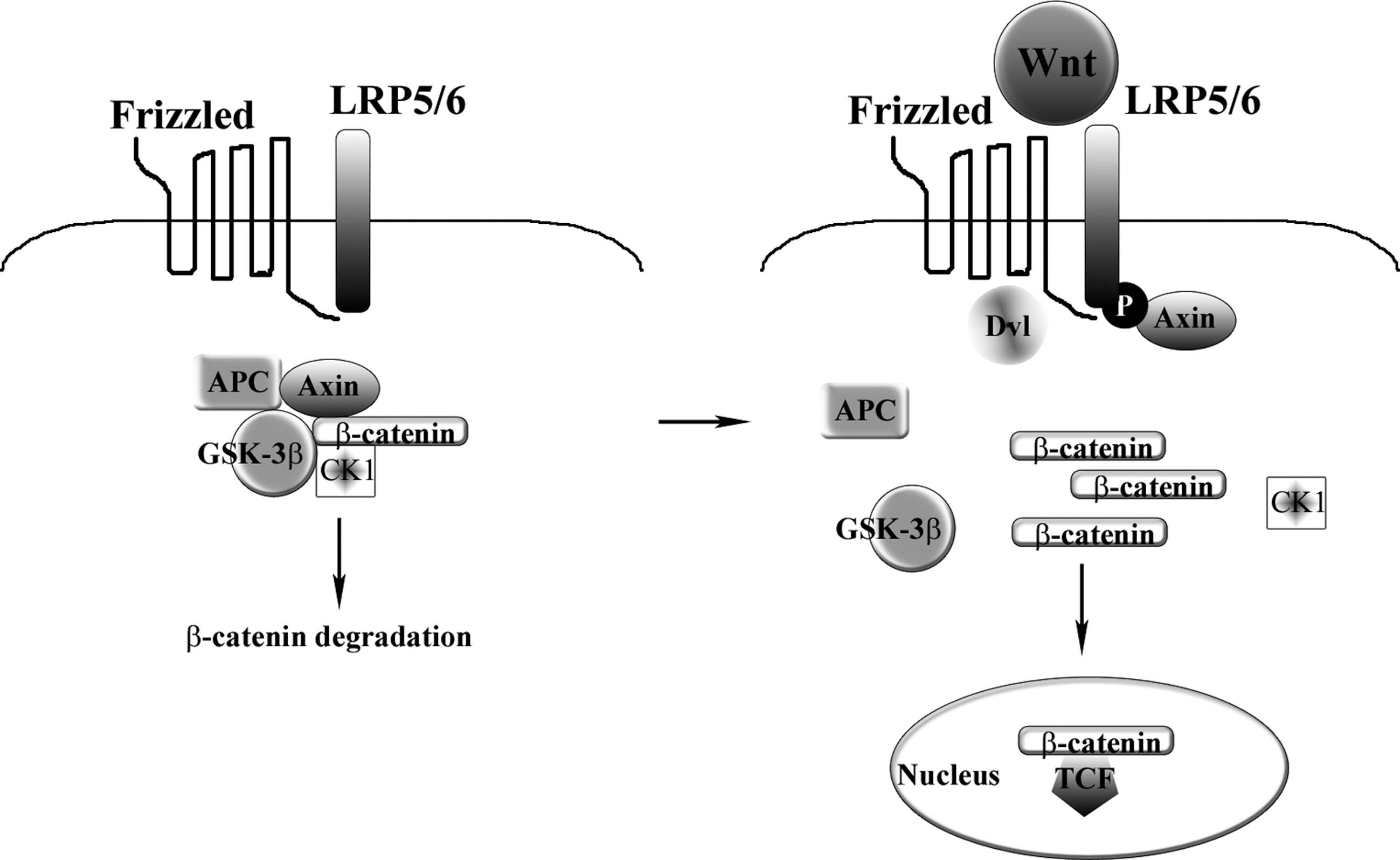
Schematic diagram of the canonical Wnt signaling pathway. In the absence of Wnt ligand, β-catenin is phosphorylated by GSK-3β and is degraded. On engagement of Wnt ligands with their receptors, the cytoplasmic degradation complex of β-catenin is disrupted; β-catenin accumulates and translocates into the nucleus, where it interacts with TCF/LEF and activates transcription of target genes. APC, adenomatous polyposis coli protein; CK1, casein kinase-1; Dv1, disheveled protein; GSK-3β, glycogen synthase kinase-3β; LRP5/6, low-density lipoprotein receptor-related protein-5/6; P, phosphorylation; TCF/LEF, T-cell-specific transcription factor/lymphoid enhancer-binding factor.
Several studies of developmental biology using mouse genetics have revealed a critical role for Wnt in skeletogenesis, and particularly in the formation of cartilage and bone. We briefly review those studies that, in our opinion, are relevant for an understanding of the molecular regulation of MSC differentiation.
Wnt and bone
Wnt proteins have a critical role in bone development and homeostasis. Gong and colleagues (2001) demonstrated that the osteoporosis-pseudoglioma syndrome (OPPG), a disorder characterized by low bone mass, ocular defects, and predisposition to fractures, was caused by a loss-of-function mutation in the LRP5 (low-density lipoprotein receptor-related protein-5) coreceptor. The production of Lrp5–/– mice (Kato et al., 2002) and Lrp6–/– mice (Holmen et al., 2004) allowed recapitulaton of these findings, with phenotypes similar to that of human OPPG. When the mutation of Lrp5 was involved, the disease was attributed to decreased osteoblast proliferation. Interestingly, other mutations in the same gene can have different effects. As an example, some individuals with high bone mass density bear a gain-of-function mutation in Lrp5 (Boyden et al., 2002; Little et al., 2002). Many studies followed these initial reports, indicating that loss-of-function mutations in the Lrp5 gene could also affect bone formation via indirect mechanisms, such as Htr1b (5-hydroxytryptamine [serotonin] receptor-1B)–CREB (cAMP response element-binding protein) (Yadav et al., 2008), but also that the gain-of-function mutation in Lrp5 may not have an effect on bone density when expressed in mature osteoblasts (Yadav et al., 2008). It appears therefore that Lrp5 could affect bone mass in a Wnt-independent way and that Lrp6 could be more relevant when considering Wnt signaling in osteoblasts. When analyzing the Wnt pathway involved in bone development and homeostasis, it is apparent that each molecule involved can affect different features of bone mass regulation (for a comprehensive list of Wnt-related factors and the in vivo effects caused by them, see the review by Hoeppner and colleagues, 2009).
Wnt and cartilage
Wnt genes and proteins also act on cartilage differentiation, by interacting with cartilage-related transcription factors. Day and colleagues (2005) showed that ectopic canonical Wnt signaling led to enhanced ossification and suppression of chondrocyte formation during skeletogenesis. On the other hand, during both intramembranous and endochondral ossification, by genetic inactivation of β-catenin they induced ectopic chondrocyte formation in place of osteoblast differentiation.
In the same year, Hill and colleagues (2005) demonstrated that canonical Wnt signaling was essential for skeletal lineage differentiation, preventing transdifferentiation of osteoblastic cells into chondrocytes, and also that β-catenin was crucial in determining whether mesenchymal progenitors would become osteoblasts in the developing embryo.
Dong and colleagues (2006) investigated the molecular mechanisms underlying canonical Wnt-mediated regulation of chondrocyte hypertrophy, using chick sternal chondrocytes, and provided evidence that in chick upper sternal chondrocytes activation of the canonical β-catenin Wnt signaling pathway induces chondrocyte hypertrophy and maturation and that Wnt/β-catenin signaling is regulated by TGF-β and bone morphogenetic protein (BMP)-2, and mediates chondrocyte hypertrophy at least partly through activation of Runx2, a transcription factor required for bone formation (Schroeder et al., 2005; Komori, 2010), which in turn may induce expression of collagen type X.
Maruyama and colleagues (2010) observed endochondral ossification in vivo in the skull of mice via knockout of Axin2, a negative regulator of the WNT/β-catenin pathway. The activation of β-catenin cooperated with fibroblast growth factor receptor-1 (FGFR1) to alter the lineage commitment of MSCs to differentiate into chondrocytes. In this way, instead of the expected intramembranous ossification, the switch in the fate of MSCs to chondrocytes resulted in endochondral ossification, abnormal suture morphogenesis, and fusion with premature cranial gaps closure and a phenotype of craniosynostosis. These findings suggest that the WNT/β-catenin pathway is involved in controlling the stem cell population by regulating its renewal and proliferation, and in modulating lineage specification, in part by setting the balance of the FGF and BMP pathways (Maruyama et al., 2010).
Wnt and MSCs
Attention has moved from osteoblasts to the cells that likely originate them, the MSCs. Etheridge and colleagues (2004) demonstrated that MSCs express a number of Wnt ligands, including Wnt2, Wnt4, Wnt5a, Wnt11, and Wnt16, and several Wnt receptors, including FZD2, 3, 4, 5, and 6 as well as various coreceptors and Wnt inhibitors. Boland and colleagues (2004) showed that during osteogenic differentiation in vitro, MSCs upregulate a number of Wnt-related molecules while downregulating others. Moreover, the same authors demonstrated that MSCs could respond to exogenous Wnt3, resulting in transient repression of osteogenesis. Similar results were obtained by Cho and colleagues (2006) using human adipose-derived MSCs. Using an in vitro differentiation culture system with human MSCs and performing global gene expression profiling on undifferentiated and differentiated MSCs, as well as on dedifferentiated cells derived from mesenchymal lineages, Song and colleagues demonstrated that differentiated cells could dedifferentiate into a primitive stem cell-like stage before transdifferentiating into another cell type (Song et al., 2006). In this study, the authors identified a list of genes that were candidate markers of MSCs and may function to maintain stem cells in an uncommitted state or to initiate their differentiation process. Prominent among them were the genes associated with the Wnt pathway.
Quarto and colleagues (2010) reported that Wnt3a has differential effects when using different in vitro models and an in vivo model of bone regeneration; the effects were dependent on the dose as well as the differentiation state of the recipient cell. When added to undifferentiated MSCs, Wnt3a inhibited osteogenic differentiation. By contrast, when added to calvarial osteoblasts, Wnt3a at high doses had an inhibitory effect in cells from juvenile mice but induced bone production in cells from adult animals, as assessed by alkaline phosphatase activity and alizarin red mineralization assay. The defect repair was influenced once again both by the Wnt3a dose and by the age of the animal, mimicking the in vitro results. These findings are in accordance with previous investigations (Kahler and Westendorf, 2003; Kahler et al., 2006, 2008; Eijken et al., 2008), showing that the effect of canonical Wnt signaling on osteogenesis is influenced by the differentiation stage of target cells. Overall, canonical Wnt signaling appears to stimulate the differentiation of MSCs committed to osteogenic lineage, while it inhibits the terminal differentiation of mature osteoblasts.
In a study on the effects of Wnt inhibitors on MSC chondrogenesis, Im and Quan (2010) found that in pellet cultures of human MSCs, inhibitors of the Wnt pathway promoted early chondrogenesis of MSCs, but they lacked a synergistic effect with TGF-β in the longer term culture, and therefore did not provide an ultimately enhancing role in the cartilage tissue engineering of MSCs.
The Wnt family and their regulation by microRNAs
Increasing attention has been devoted to the interaction between microRNAs and the classical differentiation pathways. Although many of such studies are on osteoblasts and not on undifferentiated MSCs, the results are relevant as they could be translated to primary MSC cultures and, possibly, to in vivo scenarios. It was reported that the microRNA miR-29a is induced by canonical Wnt signaling and can potentiate Wnt signaling (Kapinas et al., 2010). The microRNA miR-125b inhibited osteoblastic differentiation and proliferation (Mizuno et al., 2008). After it was demonstrated that miR-27 could influence adipogenesis and myogenesis (Feng et al., 2009; McDaneld et al., 2009), Wang and Xu (2010) reported that the miR-27 microRNA is also able to interfere with the differentiation of a fetal osteoblastic cell line through Wnt signaling modulation, by accumulation of β-catenin and repression of adenomatous polyposis coli protein (APC) expression.
TGF-β superfamily signaling pathways and MSC differentiation
The TGF-β superfamily consists of many growth factors and morphogens that have roles in developmental skeletogenesis and postnatal skeletal homeostasis. The TGF-β superfamily of ligands includes bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs), anti-müllerian hormone (AMH), Activin, Nodal, and TGF-β (Piek et al., 1999; Derynck and Miyazono, 2008). Here, we concentrate on the TGF-β and BMP families, and on the effects that they exert on the skeletal system and on MSCs (see Fig. 2 for a schematic representation of the TGF-β and BMP signaling pathways).
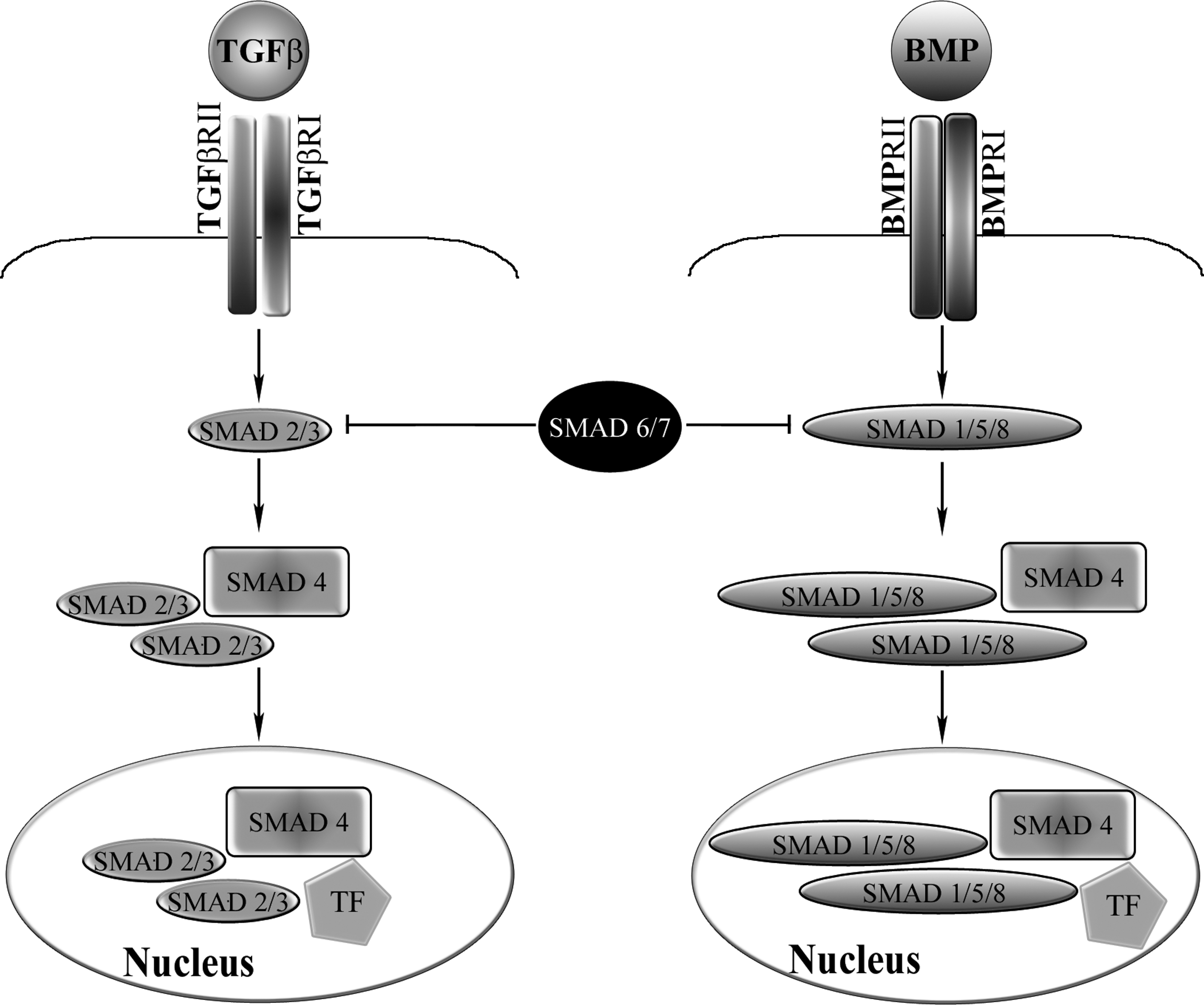
Schematic diagram of the transforming growth factor (TGF)-β and bone morphogenetic protein (BMP) signaling pathways. On engagement of the ligands with their corresponding receptors, receptor SMADs are phosphorylated and form a complex with SMAD4, which enters the nucleus and interacts with transcription factors (TFs) to modulate gene transcription. SMAD2 and SMAD3 mediate TGF-β signaling, whereas SMAD1, SMAD5, and SMAD8 mediate BMP signaling. The inhibitory SMAD6 and SMAD7 are also shown. BMPRI/II, BMP receptors I and II; TGFβRI/II, TGF-β receptors I and II.
TGF-β family
The expression of growth factors of the TGF-β superfamily has been described in embryonic bone and cartilage development as well as during adult bone repair (Hogan, 1996; Horner et al., 1998), and the chondro/osteo-stimulatory/modulatory effects of the TGF-βs as well as the BMPs have been well established in embryonic and adult mesenchymal cells (Johnstone et al., 1998; Mackay et al., 1998; Denker et al., 1999; Barry et al., 2001; Majumdar et al., 2001; Noth et al., 2002; Sekiya et al., 2002). In particular, TGF-β has been used for years in in vitro assays of cartilage micromasses or pellets starting from culture-expanded MSCs (Mackay et al., 1998; Pittenger et al., 1999), and has become a requirement for these chondrogenesis assays. TGF-β promotes cartilage-specific gene expression through intracellular signaling cascades involving SMAD proteins, the mitogen-activated protein (MAP) kinases, p38, extracellular-signal regulated kinase (ERK)-1, and c-Jun N-terminal kinase (JNK) (Lutz and Knaus, 2002; Schmierer and Hill, 2007).
After the demonstration of the crucial role of the cell adhesion protein, N-cadherin, and Wnt signaling in mesenchymal condensation and chondrogenesis (Fischer et al., 2002), Tuli and colleagues (2003) demonstrated that TGF-β1 treatment initiates and maintains chondrogenesis of MSCs through the differential chondrostimulatory activities of p38, ERK-1, and to a lesser extent, JNK. This resulted in the modulation of N-cadherin expression levels, WNT-7A gene expression, and the consequent Wnt-mediated signaling through the intracellular β-catenin pathway, with control of the condensation and early steps of MSC chondrogenesis.
BMP family
BMPs are involved in the developmental and homeostatic processes of the bone and cartilage, as well as of other tissues (Katagiri and Miyazono, 2008). BMPs are characterized by a complex transduction pathway, based on their signaling through SMAD proteins 1, 5, 8, and 4 (reviewed in Xiao et al. [2007] and in Miyazono et al. [2010]). BMPs are capable of inducing, in adult rodents, ectopic cartilage and bone formation, mimicking embryonic endochondral bone formation.
BMPs are important morphogens regulating chondrogenesis and skeletogenesis during normal embryonic development (Hogan, 1996). Individual BMPs exhibit distinct expression patterns in skeletal elements. Bmp-2 is expressed in areas surrounding the initial cartilage condensations, periosteal and osteogenic zones, whereas Bmp-4 is expressed in perichondrium. Bmp-6 is expressed in prehypertrophic chondrocytes. High levels of Bmp-7 mRNA have been observed in the perichondrium, but its expression is absent in the zones of joint formation (Zhao et al., 2002; Sekiya et al., 2005).
The effects of BMPs on MSCs have been investigated in several studies. Rickard and colleagues (1994) demonstrated that rat bone marrow MSCs cultured in the presence of BMP-2 increased osteoblastic markers such as alkaline phosphatase activity and expression of osteocalcin. This effect was strengthened when culturing MSCs with BMP-2 and dexamethasone together. Several other studies analyzed the effect of BMP-2, along with other factors, on the proliferative and osteogenic abilities of primary or immortalized MSCs and/or osteoblasts (Wang et al., 1993; Hanada et al., 1997; Lecanda et al., 1997; Jorgensen et al., 2004). In 1999, Denker and colleagues observed that the addition of BMP-2 to C3H10T1/2 mesenchymal cells in micromass cultures enhanced the appearance of chondrocytes. The effect of BMP-2 was apparently modulated by N-cadherin, a Ca2+-dependent adhesion molecule (Haas and Tuan, 1999). The addition of BMP-6 to micromass cultures of human bone marrow MSCs enhanced cartilage formation compared with controls in a time- and dose-dependent fashion (Sekiya et al., 2001).
In a study in micromass culture, Sekiya and colleagues (2005) compared the chondrogenic effect of BMP-2, BMP-4, and BMP-6 on human bone marrow MSCs and observed that the most potent chondroinductor was BMP-2. Analyzing microarray data from the micromasses cultured with the various BMPs, the authors found that only the MSCs cultured with BMP-2 expressed the cartilage synthesis-related genes with the correct pattern and time sequence.
Using human MSCs from periosteum, we detected expression of BMP receptors and confirmed the chondrogenic effect of BMPs in micromass. However, BMP-2, BMP-4, BMP-7, and GDF-5/CDMP-1 (cartilage-derived morphogenetic protein
BMP2 signaling can influence the Wnt pathway through interaction between β-catenin and N-cadherin. Treatment of the mesenchymal cell line C3H10T1/2, in micromass culture with lithium chloride, a mimetic of canonical Wnts that acts by inhibiting glycogen synthase kinase (GSK)-3β serine/threonine phosphorylation activity, significantly inhibited BMP-2 stimulation of chondrogenesis (Fischer et al., 2002), and decreased the levels of N-cadherin protein and mRNA, probably affecting mesenchymal condensation (see also Tuan, 2003), a process characterized by increased cell density and cell–cell adhesion and critical in the initiation of chondrogenic differentiation. In 2005, Modarresi and colleagues (2005) found that normal levels of N-cadherin expression were essential, in micromass pellet cultures, to obtain proper temporal MAP kinase and BMP-2 regulation of chondrogenic genes such as type II collagen, aggrecan, and Sox9.
Eyckmans and colleagues (2010) highlighted a requirement for BMP and Wnt signaling in bone formation when using human periosteum-derived MSCs seeded onto calcium phosphate carriers and implanted ectopically in immune-deficient mice. The inhibition of endogenous BMP and Wnt signaling by overexpression of the secreted antagonists Noggin and Frzb, respectively, abrogated osteoinduction.
Other signaling molecules in MSC differentiation
Other factors are known to influence the differentiation of MSCs. Many of them also interact, at different levels, with the Wnt and/or TGF-β/BMP pathways. One of those factors, fibroblast growth factor (FGF)-2, has been shown to promote cell proliferation and to maintain the MSC population in a prolonged undifferentiated state (Martin et al., 1997). Ng and colleagues (2008) studied the transcriptional profiling of MSCs and of MSC-derived differentiated cells in order to identify the factors implied in the differentiation processes in vitro. From their analysis, the authors highlighted three pathways, centered on TGF-β, FGF-2, and platelet-derived growth factor (PDGF), which proved to be important in the growth and essential in the differentiation of MSCs. Furthermore, the authors were able to grow MSCs using a combination of these growth factors in culture, under serum-free conditions.
PDGF has been shown to inhibit osteogenic differentiation (Gruber et al., 2004; Kratchmarova et al., 2005). In an elegant study, Kratchmarova and colleagues found that the osteogenic differentiation of human MSCs is stimulated by epidermal growth factor (EGF) but not PDGF. They used mass spectrometry-based proteomics to compare proteins that were tyrosine phosphorylated in response to EGF and PDGF. More than 90% of these signaling proteins were used by both ligands, whereas the phosphatidylinositol-3-kinase (PI3K) pathway was activated exclusively by PDGF, implicating it as a possible control point. Indeed, chemical inhibition of PI3K in PDGF-stimulated cells removed the differential effect of the two growth factors, conferring full differentiation effect onto PDGF.
Ng and colleagues (2008) showed that inhibition of PDGF signaling resulted, instead, in fewer osteocytes and the absence of mineralized bone nodules, implying that under specific conditions PDGF may be required for correct osteogenic differentiation. Tokunaga and colleagues (2008) studied the effects of the β receptor for PDGF, PDGFR-β, by specific PDGFR-β gene deletion using Cre–loxP technology. Depletion of PDGFR-β in MSCs decreased the mitogenic and migratory responses and enhanced osteogenic differentiation. In a mouse model of bone fracture, depletion of PDGFR-β significantly increased the ratio of woven bone to callus. The effects of PDGFR-α on osteogenic differentiation were instead very subtle. PDGFR-β could therefore represent an important target for bone tissue engineering.
MSCs express EGF receptor (EGFR)/ErbB-1, and EGF exerts mitogenic activity on MSCs (Krampera et al., 2005; Tamama et al., 2006). The role of EGFR signaling in bone is not well understood, with contrasting results having been discerned. Loss of EGFR signaling has been reported to accelerate chondrocyte and osteoblast differentiation in mice, suggesting that EGFR signaling negatively regulates bone cell differentiation (Sibilia et al., 2003). Satomura and colleagues reported that after transplantation of human MSC-loaded HA scaffolds, the MSCs able to form bone were found to have consistently low expression of EGFR compared with those MSCs not forming bone, although individual variability could not be disregarded (Satomura et al., 1998). Activation of EGFR is also associated with enhanced proliferation of the stem/progenitor cell compartment with no impairment of differentiation (Krampera et al., 2005; Tamama et al., 2006), or even enhancement of differentiation (Kratchmarova et al., 2005). A reversible EGF-driven enhancement of in vitro osteogenic differentiation was described when incubating MSCs with tethered EGF (Platt et al., 2009), probably due to a consistent and continuous interaction between EGF and its receptor. This finding in particular could lead to the development of novel bioscaffold-based tissue-engineering approaches exploiting the tethering of active substances, such as EGF, that are never released from the bioscaffold itself and are therefore constantly available to induce an otherwise reversible differentiation process.
Other signaling molecules are involved in the chondro/osteogenic differentiation pathways. Scotti and colleagues (2010) linked the in vivo endochondral ossification obtained in ectopic implants in nude mice of in vitro-engineered human MSCs to the regulation of Indian hedgehog (IHH) as an upstream signal through its receptor Patched1 (PTCH1), and GLI1 as mediator of IHH signal transduction. All these genes were either not expressed or expressed at low levels in culture-expanded MSCs, whereas their expression levels were markedly increased in the early hypertrophic and even more in the late hypertrophic constructs, indicating that the processes leading to endochondral ossification are equally regulated by the activation and/or upregulation of the signaling pathways involved in endochondral bone formation during embryonic limb skeletogenesis (Karsenty, 2008).
Transcription factors in MSC differentiation
Many transcription factors (TFs) intervene in the control and regulation of the differentiation pathways of MSCs. Once again, the developmental studies have provided an educational platform for investigations of the roles in MSC differentiation of those transcription factors that have been reported to be key regulators of embryonic skeletogenesis. In particular, the main transcription factors intervening in osteogenic differentiation are CBFA-1/Runx2 and Osterix (Osx), known to be required for bone formation during development (Schroeder et al., 2005; Komori, 2010), whereas Sox9 is a transcription factor required for cartilage formation (Akiyama et al., 2004; Quintana et al., 2009). It must be said, however, that developmental studies have addressed in great detail the formation of cartilage that undergoes hypertrophy and is replaced with bone during endochondral ossification of limb development, whereas the molecular machinery underlying the formation of stable articular cartilage remains to be defined.
During development, chondrogenesis is controlled by interactions between Sox9 and the Wnt/β-catenin signaling pathway. Either overexpression of Sox9 or inactivation of β-catenin in chondrocytes of mouse embryos in vivo produced a similar phenotype of dwarfism with decreased chondrocyte proliferation, delayed hypertrophic chondrocyte differentiation, and endochondral bone formation (Akiyama et al., 2004). Furthermore, either inactivation of Sox9 or stabilization of β-catenin in chondrocytes also resulted in a similar phenotype of severe chondrodysplasia. Sox9 markedly inhibited activation of β-catenin-dependent promoters and stimulated degradation of β-catenin. Of note, a physical interaction between β-catenin and the C-terminal trans-activation domain of Sox9 was demonstrated.
Dong and colleagues (2006) observed upregulation of type X collagen (col10a1) and Runx2 mRNA by viral overexpression of Wnt8c and Wnt9a, thereby inducing chondrocyte hypertrophy. Wnt8c and Wnt9a strongly inhibited mRNA levels of Sox9 and type II collagen (col2a1). They also found that Wnt8c further enhanced BMP-2-induced expression of Runx2 and col10a1, whereas Wnt8c and Wnt9a inhibited TGF-β-induced expression of Sox9 and col2a1. By overexpressing β-catenin, the authors upregulated Runx2, col10a1, and alkaline phosphatase (AP) mRNA levels while they inhibited col2a1 transcription; Wnt8c and β-catenin also acted at protein levels, inducing production of Runx2 in chondrocytes. These results link activation of the canonical Wnt signaling pathway with chondrocyte hypertrophy and maturation through the transcription factors Runx2 and Sox9.
Klees and colleagues (2005) found that adhesion of cultured MSCs to laminin-5 activated ERK, leading to phosphorylation of Runx2/CBFA-1. Majumdar and colleagues (2001) observed that in MSCs placed in alginate beads and cultured in serum-free medium with BMP-2 and BMP-9 there was increased expression of Sox9 accompanied by an increase in expression of mature chondrocyte markers such as type II collagen.
It has been reported that RUNX2 and SOX9 physically interact in MSCs and that SOX9 can inhibit the trans-activation of RUNX2. In addition, RUNX2 exerts reciprocal inhibition on SOX9 trans-activity. SOX9 induced degradation of RUNX2, which was proteasome independent but phosphorylation dependent, and required the presence of the RUNX2 C-terminal domain, which contains a nuclear matrix-targeting sequence. Furthermore, SOX9 was able to decrease the level of ubiquitinated RUNX2 and direct RUNX2 to the lysosome for degradation. SOX9 also directed β-catenin for lysosomal breakdown (Cheng and Genever, 2010). Thus, the reciprocal regulation between SOX9 and RUNX2, downstream of signaling pathways, will have obvious consequences on MSC fate.
Physical factors in MSC differentiation
MSCs also interact physically with the surrounding environment. Examples are numerous; here we cite but a few. The first line of interaction for MSCs is with the extracellular environment, be it plastic, a resorbable bioscaffold, or a more rigid structure. Seminal in this sense is the study by Engler and colleagues (2006), showing that the extracellular matrix can control stem cell fate, inducing seeded MSCs toward osteogenesis or chondrogenesis depending on its physical properties. More recently, it was reported that manipulation of the membrane potential of cultured MSCs can influence their fate and differentiation (Sundelacruz et al., 2008, 2009). These findings open up unprecedented avenues for the regulation of MSC differentiation in regenerative medicine using physical factors, without the use of exogenous growth factors.
Conclusion
There is an increasing amount of data on the molecular regulation of MSC differentiation, and this review has addressed only some of them (for a schematic overview see Fig. 3). As already discussed previously, a caveat of the in vitro studies relates to the known variability that is at least in part due to culture conditions and the MSC type used. Therefore, a priority in the MSC field is the standardization of isolation and culture expansion. This will minimize variability of outcome and will also allow proper comparison of studies. Nonetheless, the availability of standardized in vitro assays employing primary human MSCs has huge scientific and clinical potentials. The use of reporter assays for assessment of net signaling activation, three-dimensional culture systems, and bioreactors with sensors monitoring MSC activity will allow rapid progression from basic science to automated, highly controlled cell-bioprocessing methods monitored via mathematical models through technological advances in systems biology and network science. With this view, the investigation of MSC “omics” as well as of the influence of the surrounding niches and contained signaling molecules is of paramount importance. A deep knowledge of the mechanisms underlying MSC differentiation will be paramount for the successful development of consistent MSC-based products in tissue engineering and regenerative medicine.

Schematic overview of signaling molecules and transcription factors involved in the regulation of chondro-osteogenic differentiation of MSCs. BMP-2, bone morphogenetic protein-2; EGF, epidermal growth factor; FGF-2, fibroblast growth factor-2; LRP5/6, low-density lipoprotein receptor-related protein-5/6; Osx, Osterix; PDGF, platelet-derived growth factor; RUNX2, runt-related transcription factor-2; TGF-β, transforming growth factor-β.
Footnotes
Acknowledgments
Supported by MRC grant G108/620. Professor De Bari is a Fellow of the Medical Research Council, UK.
Author Disclosure Statement
The authors have nothing to disclose.