
Abstract
Lentiviral vectors are now widely considered one of the safest and most efficient tools for gene delivery and stable gene expression. Even though inducible gene expression cassettes are mandatory for many genetic engineering strategies, most current systems suffer from various issues, such as the requirement of two vectors, which decreases the overall efficiency of the transduction, leakiness and/or insufficient levels of activation of the inducible promoter, lack of selectable marker, low titers, or general issues associated with the cloning of large plasmids. In this article, we describe the design and functional characterization of a set of “all-in-one” multicistronic autoinducible lentivectors. They combine: (1) an optimized drug-inducible promoter; (2) a multicistronic strategy to express living color, selectable marker, and transactivator; and (3) acceptor sites for easy recombination cloning of genes of interest. These polyswitch lentivectors have good titers, very low basal activity, and reversible high induced activity, and can accept a growing number of genes already cloned in entry plasmids. These combined features make them a novel, powerful, and versatile tool for current and future genetic engineering approaches.
Introduction
Lentivectors containing inducible systems have been described by several laboratories. If the most commonly used system remains the tetracycline-inducible gene switch (TET) (Gossen and Bujard, 1992; Gossen et al., 1995), many other systems exist, starting from TET-derivative systems such as the TET-regulated KRAB system (Szulc et al., 2006), but also hormone-modulated systems (Wang et al., 1994; Delort and Capecchi, 1996; No et al., 1996), or small molecule-modulated systems such as the rapamycin system (Rivera et al., 1996) or the cumate gene switch (Mullick et al., 2006). When applied to lentivectors, the TET systems described never combined all the desirable features to make them powerful and versatile genetic engineering tools. They would come in either of two separate vectors (Reiser et al., 2000; Haack et al., 2004; Pluta et al., 2005; Vutskits et al., 2006; Vieyra and Goodell, 2007): the TET-promoter would have high basal activity (Kafri et al., 2000; Reiser et al., 2000) or low induced activity (Reiser et al., 2000; Haack et al., 2004), or the vector could not allow for selection of transduced cells before activation or did not provide easy cloning of the gene of interest (Vogel et al., 2004; Markusic et al., 2005; Vigna et al., 2005; Barde et al., 2006; Gascon et al., 2008; Hioki et al., 2009).
Recently, advanced “all-in-one” systems have been developed in retroviral vectors and transposons (Heinz et al., 2011) or lentiviral vectors (Tian et al., 2009), but although regulation and/or inducibility is improved, no system allows for easy cloning of the gene of interest or selection of the transduced cells.
Another crucial aspect of lentivector development is the expression of multiple genes from a single vector. One possibility is to use several promoters, hence several transcripts, with the disadvantage of sacrificing precious space and the possibility of leading to promoter interference, i.e., unspecific expression or quenching (Curtin et al., 2008). From a single internal transcript, another option is to express a fusion protein, which may severely alter the functionality of individual units. The last possibility is to use viral strategies to encode for several proteins from a single RNA molecule. The most widely used viral strategy is derived from internal ribosome entry site (IRES) sequences of picornaviruses (Pelletier and Sonenberg, 1988), such as the encephalomyocarditis virus. IRES sequences are able to make ribosomes initiate translation at internal sites within the mRNA, enabling the translation of several proteins from a single mRNA. IRES sequences have, however, several limitations: (1) their relatively large size (∼600 bp); (2) they can recombine if present in more than one copy on a single RNA, especially in retroviruses, leading to loss of entire cistrons; and (3) different IRESs can compete with each other (Douin et al., 2004). In addition, the level of expression of the protein encoded by the open reading frame (ORF) downstream of the IRES element is strongly affected by the nature of the ORF upstream, as well as the cell type, making the IRES element unreliable. Another viral element that has been exploited more recently is the 2A peptide (Robertson et al., 1985). 2A peptides are short peptides (18–30 amino acids) that impair the protein assembly by ribosome skipping (Donnelly et al., 2001) and allow for nonenzymatic, site-specific cleavage of 2A peptide-containing nascent polyproteins into protein subunits. This system has already been successfully used to express all four CD3 chains in one retroviral vector (Szymczak et al., 2004).
Easy cloning in large lentivector plasmids is also a recurrent issue, due to the already large size of “loaded” lentivectors, as well as the limited choice in available restriction sites. One alternative to classical cloning using restriction enzymes is the site-specific recombination derived from the phage λ recombination machinery. It allows for fast, efficient, and directional cloning of multiple DNA fragments (Hartley et al., 2000; Sasaki et al., 2004; Suter et al., 2006). More and more genes are now available in entry plasmids coming from academic researchers (available through, e.g., Addgene, Cambridge, MA), cloning companies (such as imaGenes, Nottingham, UK), or commercial kits (Gateway, Invitrogen, Carlsbad, CA).
In this work, we describe a set of “all-in-one” multicistronic autoinducible lentivectors. They combine all the desirable features described above, i.e., gene switch, polycistronic cassettes, selectable marker, living color, and easy cloning of genes of interest. The gene switch is an optimized TET-ON system providing very low basal activity and reversible high induced activity. The polycistronic cassettes are based on 2A peptide strategy. Although 2A peptides allow for simultaneous expression of living color, selectable marker, and transactivator, we show that they decrease protein expression. The insertion of genes of interest is performed using the fast, easy, and expanding recombination cloning strategy. Even though each individual element has been described previously, the combination of all these components in a single lentivector makes our system novel and particularly useful. Once transduced by such lentivectors, the cells can be selected and expanded, and then the gene of interest can be reversibly induced when desired. To our knowledge, these polyswitch lentivectors are the first all-in-one lentiviral vectors allowing for selection of mammalian cells transduced with an optimized drug-controlled gene switch.
Materials and Methods
Bacterial strains and plasmid isolation
For the construction of DEST lentivector plasmids containing the CCDB killer gene (Bernard et al., 1993), Library Efficiency DB3.1 Competent Cells (Invitrogen) were used. For construction of other vectors and clones without the CCDB gene, Top10 Competent cells (Invitrogen) were used. Plasmid DNA was prepared using the Miniprep Kit or Maxiprep Kit (GenoMed, Aventura, FL).
Lentivector design
General design of lentivectors with dual transcription cassettes
The backbone of our all-in-one autoinducible multicistronic lentivectors was obtained by digestion of a Rix lentiviral plasmid (Dayer et al., 2007) with HpaI and MluI and filled in using Klenow fragment. Detailed maps and sequences of the plasmids described in this article can be downloaded from our institutional Web site (

Principle and construction strategy of the polyswitch “all-in-one” lentiviral vectors: schematic diagram of the Rix-pTF-DEST-EX multicistronic autoinducible lentivector. The common features of the bacterial plasmid and lentivector are abbreviated as follows: Amp, ampicillin resistance gene; 5’ LTR, chimeric 5’ long terminal repeat (LTR) as described (Dull et al., 1998); ψ, packaging signal; RRE, rev-responsive element; cppt, central polypurine tract; 3’ LTR SIN, self-inactivating 3’ LTR as described (Zufferey et al., 1998). The GENE box represents the module ready to clone the gene of interest to be expressed under the control of a TET-inducible promoter. This cassette is composed of the following element: pTF, modified TET-responsive promoter as described in Materials and Methods. The attR1-CAT-ccdB-attR2 (CAT, chloramphenicol acetyltransferase; ccdB, ccdB lethal gene targeting E. coli DNA gyrase) represents the modules of the recombination cloning destination (DEST) cassette (Hartley et al., 2000). The gene of interest is cloned in place of the DEST cassette using a pENTR-L1-GENE-L2 plasmid (see Materials and Methods) and LR recombinase as described in the Gateway manual (Invitrogen). The SWITCH box represents the transcription cassette that expresses the rtTA reverse TET-transactivator. The four different SWITCH cassettes are ER, EBR, EGR, and EBGR, where the modules are abbreviated as follows: E, EF 1α short promoter; R, reverse TET-transactivator; B, blasticidin resistance gene; G, GFP or green fluorescent protein. Upon addition of DOX, the rtTA will bind the pTF promoter and activate expression of the gene of interest. Each SWITCH cassette is an assembly of PCR fragments amplified with compatible ends (see Supplementary Fig. S1). Arrows numbered from 1a to 6 indicate the positions of primers used to clone modules as detailed for the EBGR cassette in Fig. S1. The sequences of these primers are described in Table 1. The modules are assembled as described in Fig. S1 prior to incorporation into the final Rix-DEST-EX lentivector.
Inducible cassette with recombination cloning acceptor sites
The TET-inducible promoter pTF (Leuchtenberger et al., 2001) was cut with EcoRI and BamHI and inserted into pUC18 cut with the same enzymes, generating the pUC-pTF intermediate. The Gateway vector destination (DEST) cassette (Reading frame A, Conversion reagent system; Invitrogen) was inserted into pUC-pTF cut with SalI and blunted. The resulting plasmid pUC-pTF-DEST was cut with HindIII and blunted to insert the various ubiquitous expression cassettes described below, to yield pUC-pTF-DEST-EFs-X. Finally, the double cassette pTF-DEST-EFs-X was cut with SacI and BglII, blunted, and inserted into the Rix lentiviral backbone.
Multicistronic cassette using 2A peptides
The EF1α short promoter (EFs) together with the central polypurine tract were extracted from pWPTS (Kostic et al., 2003) and subcloned into pBluescript (Stratagene, La Jolla, CA). The resulting intermediate, pBS-EFs, was then digested with BamHI and SacI, and the various (multi)protein coding sequences, digested with the same restriction enzymes, were introduced behind the EFs promoter. We used the redundancy of the genetic code to design the sequences coding for the 2A peptides (Szymczak et al., 2004; Holst et al., 2006) with restriction sites in the central region. We then designed our primers with half a 2A peptide coding sequence in the N-terminal end, and the sequence of the gene to amplify in the C-terminal end. Primers are listed in Table 1. The blasticidin resistance gene (BSD) was PCR-amplified using primers BSD-for-BamHI and E2A-BSD-HindIII-rev. The green fluorescent protein (GFP) coding sequence was PCR-amplified using primers E2A-GFP-HindIII-for and F2A-GFP-NheI-rev for the cassette “EBGR,” or with primers GFP-BamHI-for and F2A-NheI-GFP-rev for the cassette “EGR.” The reverse transactivator rtTAs-M2-p65 (hereinafter referred to as rtTA) (Knott et al., 2002) was provided by Dr. Wolfgang Hillen in the plasmid pWHE1009, and PCR-amplified using primers rtTA-BamHI-for and rtTA-SacI/BglII-rev for the cassette “ER,” or primers F2A-NheI-rtTA-for and rtTA-SacI/BglII-rev for the cassettes “EBGR,” or E2A-HindIII-rtTA-for and rtTA-SacI/BglII-rev for the cassettes “EGR” and “EBR.” Sequence-verified pBS-EFs-X clones were then inserted into the final DEST backbone as EFs-X cassette using the pTF-DEST-EFs-X intermediate described above. These lentivectors have the common backbone called Rix-pTF-DEST-EX, in which DEST refers to the acceptor site for final recombination cloning and X refers to the polycistronic peptide. For the construction of the NGN3-PDX1-MAFA (NPM) multicistronic cassette, a similar strategy was applied. Each ORF was PCR-amplified with 2A flanking moieties containing compatible restriction sites. Human NGN3 cDNA was obtained from Imagenes (pENTR-Ngn3), human MAFA cDNA was generously provided by Benoit Gauthier, and human PDX1 cDNA was from pWPI-Pdx1 (Ritz-Laser et al., 2003). The NPM sequence (2,680 bp) was first cloned in pUC19 and then transferred in pENTR2b (Invitrogen).
Other lentivectors and pENTR plasmids
For this study, we used pENTR4-eGFP provided by David Suter (Suter et al., 2006). Other lentivector plasmids were pWPTs (Kostic et al., 2003), coding for GFP only, and Rix-EBG, containing the EFs promoter driving BSD-F2A-GFP only.
Production and titration of lentivectors
Production of HIV-derived vectors pseudotyped with the VSV-G envelope protein was achieved by transient cotransfection of three plasmids into 293T/17 cells as previously described (Salmon and Trono, 2007). Titers of lentivector stocks were determined using HT-1080 as target cells, followed by flow cytometry (direct measure of living color) or real-time quantitative PCR (RT-qPCR) as described elsewhere (Salmon and Trono, 2007). Calculated titers of unconcentrated stocks varied between 105 transducing units (TU)/ml and 2×106 TU/ml. Transduction of target cells was performed using multiplicity of infection (MOI; i.e., transducing units per target cells) lower than 0.2 in order to have a majority of single transduction events.
BSD selection and gene expression assay
BSD selection was performed on HT-1080 and on human neonatal foreskin fibroblast (HNF; Cambrex Corp., East Rutherford, NJ; catalog no. CC-2509) cells transduced with vectors at MOI 0.1. Three days after transduction, cells were selected using various doses of BSD (0, 1, 5, 10, 50, 100 μg/ml). Cells were rinsed with phosphate-buffered saline (PBS), and fresh medium containing BSD was added with the same doses every other day. After 7 days of selection, TET-modified pTF promoter was induced by adding doxycycline (DOX) to a final concentration of 1 μg/ml, which was found to be the optimal concentration for rtTA2s-M2 in our constructs, as previously reported by others (Koponen et al., 2003; Sheng et al., 2010). Expression of the living color was analyzed by flow cytometry on a BD FACScan Flow Cytometer. For Fig. 3, the lentivectors used were pWPTs (EG), Rix-EFs-BSD-E2A-GFP (EBG), Rix-pTF-DEST-EFs-GFP-F2A-rtTA (EGR), and Rix-pTF-DEST-EFs-BSD-E2A-GFP-F2A-rtTA (EBGR). For Figs. 4 and 5, lentivector is Rix-pTF-GFP-EFs-BSD-E2A-rtTA (G-EBR).
Western blot
Protein extracts of stably transduced and control HT-1080 were prepared in PBS supplemented with Complete Mini EDTA-free protease inhibitor (Roche Diagnostics, Mannheim, Germany) through one freeze/thaw cycle. Equal amounts of total protein (∼20 μg/lane), as assayed by the Bradford quantification (Sigma-Aldrich, St. Louis, MO), were separated in a 12% sodium dodecyl sulfate–polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Bio-Rad, Watford, Herts, UK). The eGFP was detected using goat polyclonal anti-GFP antibodies (Novus Biologicals, Littleton, CO; catalog no. NB100-1770). Detection of α-tubulin was performed as a positive control using mouse monoclonal anti-α-tubulin (Sigma-Aldrich product no. T6074). Neurogenin-3 (NGN3) was detected using mouse monoclonal antibodies from the hybridoma bank (DSHB F25A1B3). PDX1 was detected using goat polyclonal antibodies provided by Chris Wright. MAFA was detected using rabbit polyclonal antibodies (Bethyl Laboratories, Montgomery, TX; catalog no. A300-611A). Secondary antibodies for the detection of α-tubulin and NGN3 were the peroxidase-conjugated anti-mouse IgG (Bio-Rad catalog no. 170-6516). Secondary antibodies for the detection of eGFP and PDX1 were the peroxidase-conjugated anti-goat IgG (Sigma-Aldrich product no. A5420). Secondary antibodies for the detection of MAFA were the peroxidase-conjugated anti-rabbit IgG (Bio-Rad catalog no. 170-6515).
RT-qPCR
RNA extracts were obtained with the RNeasy mini kit (Qiagen, Hilden, Germany). A DNase treatment (RNase-Free DNase Set, Qiagen) was included in each RNA extraction. Reverse transcription was performed using the SuperScript II Reverse Transcriptase (RT, Invitrogen) according to the manufacturer's instructions. qPCRs were performed on an ABI PRISM 7900HT Real-Time PCR System using a FastStart Universal SYBR Green Master+Rox (Roche). Primers used for the detection of the GFP and NGN3 transcripts are listed in Table 1 as qGFPfor/qGFPrev and qNgn3for/qNgn3rev, respectively. Primers used as internal control are binding to the cyclophilin A coding sequence and are listed in Table 1 as hcyclo5’ and hcyclo3’. qPCRs were set up as follows: one cycle of 94°C/10 min (activation of FastStart Taq DNA Polymerase), then 50 cycles of 95°C/15 sec, followed by 60°C/1 min. Results were analyzed with the software SDS 2.2.2. When applicable (Figs. 5 and 6), baseline levels were calculated from gene versus cyclophilin A ΔCt values obtained in untransduced cells and set to 1.
Results
General design and titers of polyswitch lentivectors
Our goal was to generate lentivectors that would provide in one integration event the following: (1) a selectable marker to eliminate nontransduced cells; (2) a gene switch to induce expression of genes before or after the cells have been selected; and (3) a living color to allow for live tracking and/or sorting of transduced cells in complex tissue environments. For simultaneous expression of the selectable marker, the living color, and the gene switch transactivator (rtTA), we chose the 2A strategy. These various combinations of the three proteins are expressed from a single mRNA transcribed from the ubiquitous EFs promoter (Kostic et al., 2003). The pTF drug-induced promoter (Leuchtenberger et al., 2001) is a modified version of the classical TET promoter (Gossen et al., 1995) in which the core promoter is derived from a plant virus and the six tet operators are separated by five different spacers. This ingenious design provides very low basal activity and high inducibility. The inducible transcription cassette was cloned upstream of the ubiquitous cassette to minimize expression of the inducible gene in the OFF-state, due to possible read-through of long overlapping transcripts. Finally, we incorporated a recombination cloning destination cassette (DEST recombination cassette; Fig. 1) to facilitate cloning of inducible genes. Considering the overall length of the empty vector, the cloning cassette has a theoretical capacity of 4 kb, a size compatible with most cDNAs. Details on design of these polyswitch lentivectors are given in Fig. 1, Supplementary Fig. S1, and Materials and Methods.
Depending on the vector backbone and on the transgene cloned in place of the DEST cassette, the titers varied in this study from 4×105 to 2×106 TU/ml. The vectors GFP-EBR and Ngn3-EBR were produced at 2×106 TU/ml and NPM-EBR at 7×105 TU/ml. When tested, the FACS and qPCR titers were equivalent.
Efficient selection and gene induction from polyswitch lentivectors
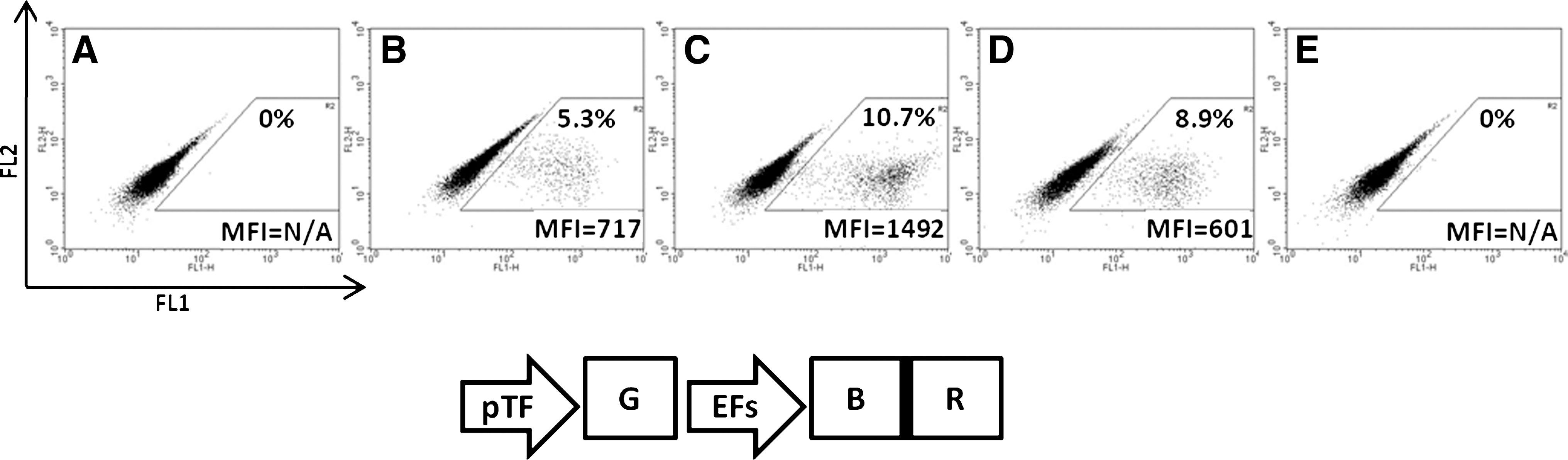
We first tested the functionality of the selectable marker as well as of the transactivator in the context of a 2A peptide-dependent expression. To analyze the three critical aspects of our vectors, i.e., the basal activity of the inducible promoter, its induction level, and the selection efficacy provided by the BSD gene (BSD), we chose to express GFP in our inducible cassette. Consequently, we used vector EBR (EFs promoter, BSD, rtTA) as the model, as EGR and EBGR already express GFP. HT-1080 cells were thus transduced with the lentivector G-EBR (pTF promoter, GFP; EBR) at an MOI of 0.1 in order to have only one copy of lentivector per genome, as well as to test the enrichment of the transduced cells using BSD.
As shown in Fig. 2B, unselected/DOX-induced cells showed a GFP-positive population of 9%. This indicates that 9% of the cells contain a transgene capable of expressing a functional rtTA together with pTF-inducible GFP. This 9% is in accordance with the calculated MOI initially applied to the cells. Neither unselected/uninduced cells (Fig. 2A) nor selected/uninduced cells (Fig. 2C) showed significant GFP expression. The mean fluorescence intensity (MFI) differences between these (Fig. 2A and C), and the untransduced cells (Fig. 2E) are within the usual variations of autofluorescence, and thus indicate that pTF promoter has an extremely low basal activity that can barely be detected using the very sensitive GFP/FACS system.

Drug-controlled induced expression of GFP in cells transduced with the G-EBR lentivector. HT-1080 cells were transduced with Rix-pTF-GFP-EFs-BSD-2A-rtTA lentivirus (abbreviated G-EBR) at an MOI of 0.1 and expanded with or without BSD selection. After selection, bulk and selected cells were treated with DOX for GFP induction. Cells were then analyzed by FACS, and GFP expression was displayed as histograms of GFP expression (FL1, x-axis, 4-decade log scale) versus cell number (y-axis, linear scale). The mean fluorescence intensity (MFI) and respective percentage were determined for both GFP-negative cells (M1 marker) and GFP-positive cells (M2 marker).
The MFI ratio between GFP-positive cells (in Fig. 2B and D, MFI≥1,200) and GFP-negative cells (in Fig. 2A and C, MFI≤12) indicates that the transcriptional activity of the pTF promoter is enhanced at least 100-fold upon DOX activation. When GFP-positive cells are compared with GFP-negative cells within the same FACS plot (in Fig. 2D), the MFI ratio is 84. This lower level of induction results from the counting of induced GFP-positive cells in the M1 marker. The MFI of GFP-positive-cells in Fig. 2B and D is similar, suggesting that selected cells have the same copy number and induced activity as unselected cells. It also suggests that selection with BSD does not introduce any enrichment for clones with higher expression levels. The lentivector G-ER (pTF promoter, GFP; EFs promoter, rtTA) in which rtTA is expressed independently of any 2A peptide provided the same level of induction (data not shown). This indicates that rtTA expression and function are not affected by our 2A peptide strategy. The level of GFP expressed from the activated pTF promoter is more than threefold higher than that from the EFs promoter (F), an intermediate/strong ubiquitous promoter (Kostic et al., 2003).
In preliminary experiments, we observed that all our BSD-containing lentivectors provided similar resistance to BSD at concentrations ranging from 1 μg/ml to 100 μg/ml. However, lower concentrations needed more time for efficient selection, and higher concentrations resulted in slower growth of cells. We thus used a concentration of 5 μg/ml for all further experiments. By using this concentration, cells could be fully selected after 1 week. As shown in Fig. 2D, selected/induced cells show a vast majority (92%) of GFP-positive cells with a high MFI. The 10-fold increase in percentage between Fig. 2B (<10%) and 2D (>90%) indicates that culture with BSD has selected against untransduced cells, leaving a majority of transgene-positive cells. The remaining percentage may be the result of integration of proviruses that have recombined leaving the BSD gene intact and inactivating either the pTF-GFP cassette or the rtTA gene. Such recombination events are not unusual in retroviruses (Negroni and Buc, 2001).
Taken together, these data show that, in our lentivector system, (1) the 2A-based strategy provides fully functional gene switch and selectable marker, and (2) the level of induced expression is compatible with most gain-of-function systems.
Expression of the living color is dependent on 2A peptide sequence
Living color selection by cell sorting can be crucial to extract and/or track specific cells from a complex tissue, independently of drug selection that can kill rare precious cells by the bystander effect. We thus next tested the expression of the living color from cells transduced with various polyswitch lentivectors. As shown in Fig. 3A, HT-1080 cells were transduced with all vectors at MOI 0.1 in order to compare levels of expression originating from a single copy of transgene. Cells transduced with pWPTS (GFP only, EG) displayed 10% of GFP-positive cells with an MFI of 270. For practical reasons, i.e., relative amounts of GFP in western-blot analysis (Fig. 3B), these control cells were not enriched by FACS. Cells transduced with other forms of GFP (2A constructs) displayed a lower MFI, partially overlapping with GFP-negative cells. We thus enriched these GFP-positive cells either by cell sorting (EGR, no selectable marker) or by BSD selection as described above (EBG and EBGR). This resulted in enrichment to at least 80%, allowing for a more significant MFI analysis of the transduced cells. Here again, as for Fig. 2, the remaining percentage of GFP-negative cells (3% to 20%, depending on the construct) may be the result of integration of proviruses that have recombined, excising GFP and leaving the BSD gene intact. Although the recombination rate in lentiviruses is described to be ∼1 in 4,000 nucleotides (Negroni and Buc, 2001), this cannot account for up to 20% of mutations affecting GFP expression. One hypothesis would be that some lentiviral constructs have recombination “hot spots” affecting GFP and/or rtTA expression. Detailed analysis of such recombination events is beyond the scope of this study.
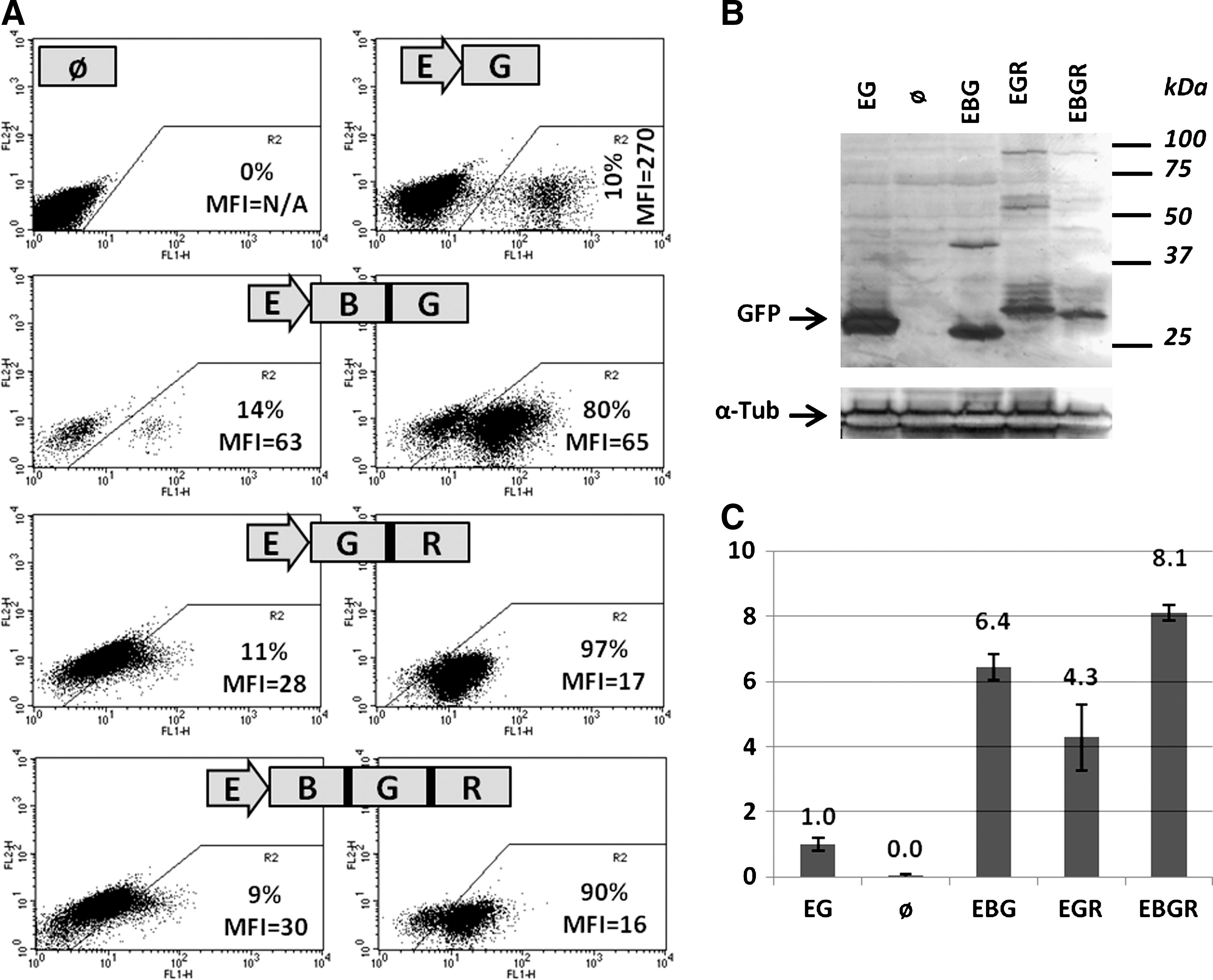
GFP expression from various 2A-based polypeptidic precursors. HT-1080 cells were transduced with lentivectors containing the EFs promoter driving GFP only (EG), BSD-E2A-GFP (EBG), GFP-F2A-rtTA (EGR), or BSD-E2A-GFP-F2A-rtTA (EBGR), cultured for 5 days, and analyzed for GFP expression by FACS (
We could observe that the MFI of cells transduced with EBG was approximately fourfold lower than GFP alone (65 versus 270). We also observed that when GFP was coupled to rtTA, whether BSD was upstream or not, the result was a 16-fold decrease in GFP signal (17 or 16 versus 270). We thus performed western-blot analysis on extracts of these cells to investigate further the mechanism underlying this decrease in GFP signal. Again, to be able to compare the amounts of GFP expressed by control pWPTS vector, on the one hand, and by 2A-based constructs, on the other, we had to use unsorted cells transduced with pWPTS (10% of GFP-positive cells). Sorted control GFP-positive cells would give, in chemiluminescence, a signal too strong for direct comparison with other constructs. As shown in Fig. 3B, the 2A-based polyproteins are correctly processed as the majority of the GFP products have expected sizes. The approximate expected sizes are 28 kDa for GFP only, due to extra amino acids at the C-terminal end in the pWPTs vector (EG), 27 kDa for BSD-GFP (EBG), and 29.5 kDa for GFP-rtTA (EGR) and BSD-GFP-rtTA (EBGR). When comparing quantities, the ratio of immunoreactive GFP protein is approximately threefold higher with native GFP (pWPTS, unsorted cells) than with BSD-E2A-GFP, and five- to 10-fold higher than with GFP-F2A-rtTA or BSD-E2A-GFP-F2A-rtTA. Thus, as for GFP fluorescence, total GFP is also significantly lower in 2A-based constructs than in GFP alone. Considering that the extract for native GFP originates from cells that are only 10% GFP-positive, as opposed to other extracts that originate from cells that are enriched to at least 80% GFP-positive, these ratios should be considered as underestimated by a 10-fold factor. This indicates that the decrease in total GFP protein is even higher than the decrease in GFP fluorescence. As shown in Fig. 3C, quantitative RT-PCR shows that these decreased levels in GFP protein are not correlated to decreased levels in mRNA coding for GFP. Here again, one must consider that the extract for native GFP originates from cells that are only 10% GFP-positive. This means that steady-state levels of transcripts containing the various sequences for our 2A-based constructs are similar to simple GFP transcript, all originating from the same EFs promoter. Still, the protein signals for EBG, EGR, and EBGR are lower than the protein signal for native GFP, despite mRNA levels that are 6.4, 4.3, and 8.1 higher than native GFP transcript, respectively. We thus show that amounts of GFP protein expressed from 2A peptide precursors are decreased compared with GFP alone. These levels of GFP expression cannot allow for robust and reliable cell sorting. We thus plan on optimizing 2A sequences to improve GFP expression in our next generation of lentivectors (see Discussion). Taken together, these results show that 2A peptide-based expression of multiple proteins is efficient, but can, at least in some cases, account for a significant decrease in the amount of proteins synthesized.
Drug-induced transgene expression is reversible
Several developmental programs require transient expression of specific genes. A useful genetic engineering tool must thus allow for transient drug-induced expression of the transgene. To test this, we analyzed the inducible cassette in a time course experiment. As shown in Fig. 4B, GFP expression could be detected as soon as 2 days after transduction/induction, with a relatively high MFI in a clearly distinct population of 5% of total cells. After 5 days, GFP expression peaked, in terms of both percentage and MFI, reaching a high value of nearly 1,500 (Fig. 4C). The percentage of GFP-positive cells is in accordance with the MOI applied to the cells, and the MFI corresponds to levels of expression 50-fold higher than uninduced levels (MFI in Fig. 4A is 30). Also, GFP can stay at this level without noticeable changes, as long as DOX is added, for at least 2 weeks. GFP expression starts to decrease as soon as 1 day after DOX removal (Fig. 4D), the percentage of GFP-positive cells remaining close to the maximum, and after 10 days, no GFP could be detected (Fig. 4E). The relatively slow GFP disappearance can be due to residual intracellular DOX, putative mRNA stability, and/or long half-life of GFP (∼26 hr; Corish and Tyler-Smith, 1999). Taken together, these data show that our polyswitch lentivector system can provide a robust drug-controlled transgene expression, with fast ON-OFF reaction and no detectable residual activity either before or after gene induction.
Reversible DOX-inducible GFP expression in cells transduced with G-EBR lentivector. HT-1080 cells were transduced with G-EBR at MOI 0.1. DOX was added 1 day after transduction, and cells were analyzed by FACS after 2 days
Regulated expression of the EBR vector in human primary cells
To demonstrate the potential of our construct, HNFs were chosen to express either GFP, NGN3, a master gene in pancreas development, or a multigene construct comprising NGN3 and two other pancreatic master genes PDX1 and MAFA, separated by 2A peptides sequences. As EGR and EBGR vectors, containing the F2A peptide coding sequence, show poor expression of GFP and seem prone to significant recombination, we decided to focus on the EBR vector (Rix-pTF-DEST-EBR). Even though our system is meant to be used at low MOI, to ensure optimal basal versus induced levels, we tested this vector at various MOIs to investigate the effect of copy number on the responsiveness of our system. HNFs were transduced with different amounts of Rix-pTF-GFP-EBR, induced (or not) and analyzed after 5 days for GFP expression (FACS and RT-qPCR) as well as lentiviral copy number (Fig. 5). When HNFs are transduced at MOI 0.05 or 0.2, i.e., statistically carry one copy of the lentiviral vector, the level of uninduced GFP is very low: uninduced MFI as determined by FACS does not vary significantly (Fig. 5A, –DOX), and RNA quantitation shows that GFP transcripts levels are very close to the background level found in nontransduced HNFs (Fig. 5B). When determined by FACS, the induction index is significantly higher than in HT1080 cells (∼200 versus ∼100). This observation could be related to the slower growth of HNF cells, allowing for increased GFP accumulation. However, as both HT1080 and HNFs are harvested at confluence, it is likely that the higher inducibility is a characteristic of HNF cells.
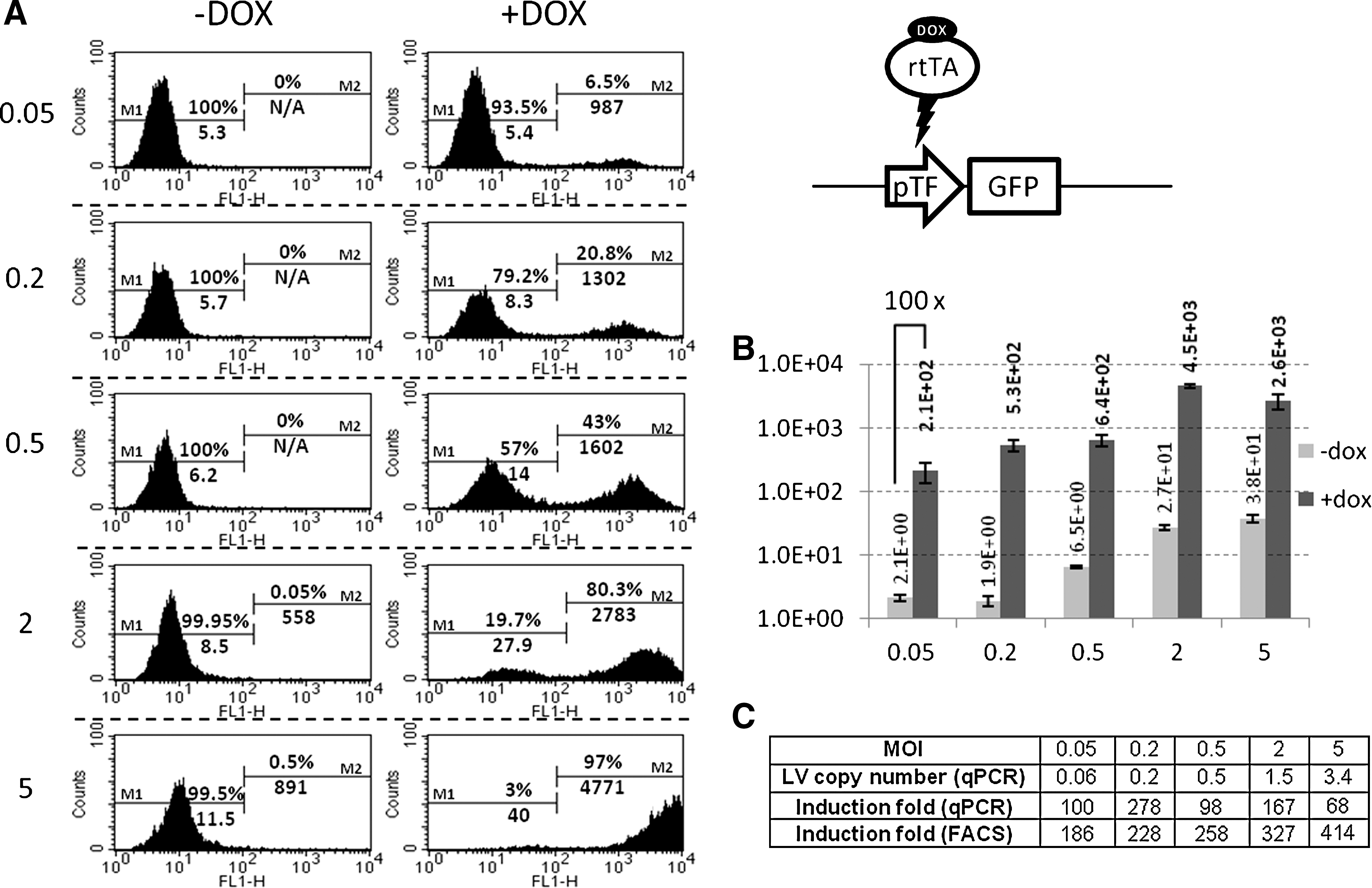
Inducible expression of GFP in HNFs.
At MOI>0.2, lentiviral vectors start accumulating into transduced cells. Consequently, the level of uninduced GFP rises up gradually. Although hardly detectable by FACS, RNA quantification shows that at MOI 5, uninduced GFP transcripts are 19-fold more abundant than at MOI 0.2 (Fig. 5B; MOI 5, –dox). This is in accordance with the theoretical 25-fold ratio of copy number between MOI 5 and MOI 0.2. FACS analysis can show evidence of a very small fraction of GFP-high cells (MOI 2: 0.05%; and MOI 5: 0.5%) that may correspond to lentivectors in which the TET promoter is influenced by enhancers close to the integration site. When transduced at MOI 20, HNFs showed a marked decrease in growth rate when uninduced, together with massive cell death upon induction (data not shown).
Quantitation of induction by qPCR shows that GFP transcripts are induced ∼100-fold independently of the MOI applied (68 to 278 without correlation with copy numbers; Fig. 5C). However, when determined by FACS, GFP induction is ∼200-fold and progressively increases up to ∼400-fold when the MOI increases. This observation can be explained by the inability of the FACS to detect low levels of expression, low amounts of GFP being comparable to cell autofluorescence. FACS analysis is thus less accurate than qPCR for determining the induction index of our system. However, it must be noted that, even at MOI 5, the level of uninduced GFP is barely detectable by FACS, showing that our TET promoter has an extremely low basal activity.
We then analyzed the induction properties of vector EBR-expressing genes implicated in specific developmental programs. HNFs were transduced with Ngn3-EBR or NPM-EBR at MOI<0.2 and selected with BSD. Note that HNFs and HT1080 can be transduced and selected with BSD with equivalent efficiencies (data not shown). Transduced BSD-resistant HNFs were cultured in the presence of DOX for 10 days, and extraction of DNA and RNA was performed. The number of integrated lentiviral copies was confirmed to be one (data not shown). We chose to measure NGN3 RNA copy numbers in a qPCR assay to determine and compare the level of induction between Ngn3-EBR and NPM-EBR vectors. As for GFP, when not induced, the NGN3 transcription level is very close to the background level (Fig. 6A; d0). In 24 hr, the level of mRNA reaches a peak and stays stable for at least 10 days. There is no significant difference in terms of transcription levels between NGN3 and NPM (Fig. 6A; d5), suggesting again that 2A peptide coding sequences do not interfere with transcription. Interestingly, the level of induction for both NGN3 and NPM exceeds 1,000-fold, whereas it is ∼100-fold for GFP. This is in accordance with reports showing that readouts other than GFP display higher induction rates (Haack et al., 2004; Heinz et al., 2011), a good omen for other gene candidates. Another interesting finding is that full shutdown requires daily washes of DOX for 3 days. Indeed, when cells are kept for 5 days after a unique DOX wash, they still express NGN3 RNA amounts comparable to those at day 1 after DOX wash (Fig. 6; d1-DOX; and data not shown). This indicates that traces of DOX can account for significant residual transcription from the TET promoter.
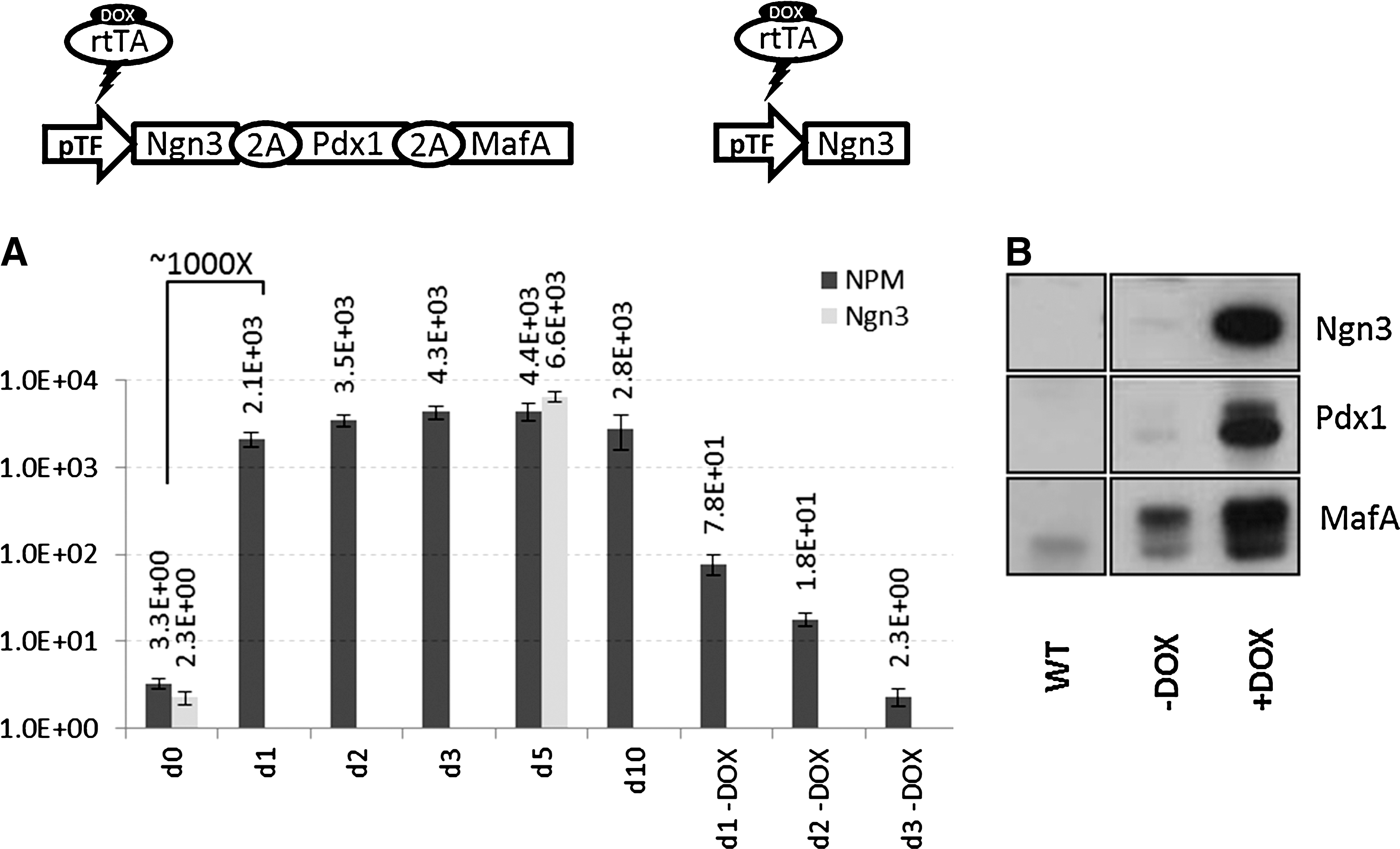
Inducible expression of Ngn3 and Ngn3-Pdx1-MafA (NPM) in HNFs.
Finally, we analyzed the processing of the NPM construct. As shown in Fig. 6B, individual NGN3, PDX1, and MAFA proteins could be detected at high levels after DOX induction, although they were barely visible before induction. The slightly denser uninduced MAFA band could be the result of higher sensitivity and/or lack of specificity of our anti-MAFA antibody, although there was no significant detection in untransduced cell extracts. This shows that the 2A peptide strategy allows for robust expression of numerous individual proteins from a single transcript.
Discussion
In this work, we describe a set of novel lentivectors that combine several of the features desired for modern genetic engineering approaches. First, they are autoinducible, i.e., they contain in the same genome both the inducible transcription cassette and the transactivating gene. Second, they allow for live selection of transduced cells prior to gene induction. Third, they can be readily used to clone hundreds of genes that are already available in the entry plasmids. And last but not least, they can be produced at titers that are compatible with most genetic engineering approaches, including transgenesis.
All-in-one autoinducible vectors are crucial when one wants to transduce cells in complex tissue context or cells that are hard to transduce. In these cases, simultaneous transduction of individual cells with two separate vectors, one for the inducible gene and one for the transactivator (Reiser et al., 2000; Koponen et al., 2003; Pluta et al., 2005; Vutskits et al., 2006; Vieyra and Goodell, 2007), would be too rare to be practically applicable. Moreover, identification of doubly-transduced cells prior to gene induction would require two independent living colors or selectable markers, one in each lentivector. Of note, such lentivector must also prove to be functional when present as a single copy, for accumulation of integrated lentiviral copies in primary cells may be either difficult, due to poor transducibility of the cells, or toxic, or both.
As biological research is moving into more and more elaborate systems, it appears that gain-of-function in primary cells often leads to dramatic changes, such as differentiation, growth arrest, or apoptosis. These changes are not compatible with cell expansion. Thus, when proliferation is needed to reach sufficient cell numbers for functional assay or prospective cell therapy, uncontrolled gain-of-function will either prevent cell expansion or lead to expansion of escape-mutants.
One thus needs an optimal inducibility index. On the one hand, the inducible promoter must have an undetectable or very low activity. One can easily imagine that even low amounts of a master gene can trigger dramatic and irreversible changes while one is trying to expand cell populations. It is in this particular context that we have chosen to test the NPM multicistronic construct. The ectopic expression of NGN3, PDX1, and MAFA, provided by three separate adenoviral vectors, was shown to reprogram mouse adult pancreatic exocrine cells in vivo into endocrine-β-like cells (Zhou et al., 2008). In order to develop this strategy further, it would be crucial to have a single vector expressing these genes with tight control. In this regard, our vectors offer levels of uninduced expression barely detectable, very close to that of untransduced cells and identical for all genes tested.
On the other hand, the activity of the induced promoter must be high enough to reach the threshold required for the function of the induced protein. This is in itself a special challenge. Our constructs offer high levels of induced expression. Although the level of induction may vary depending on the gene of interest (GFP≈100, NGN3≥1,000), we believe that it is compatible with most gain-of-function experiments. Our preliminary data indicate that the NPM construct provides amounts of each of the three proteins compatible with biological effects, i.e., induction of insulin production in transduced target cells.
Several articles have described autoinducible lentivectors that have either non-negligible uninduced activity (Kafri et al., 2000) or low levels of induced activity (Haack et al., 2004; Barde et al., 2006). Other reports have shown satisfactory basal versus induced transcription levels, but do not provide genes for selection of uninduced transduced cells (Vogel et al., 2004; Markusic et al., 2005; Vigna et al., 2005; Gascon et al., 2008; Hioki et al., 2009) or require transduction with two separate lentivectors (Suter et al., 2007). Our polyswitch lentivectors thus represent a yet unavailable tool for this application.
The very low basal and high induced activities we observe may be the result of several mechanisms. First, they may be due to the special "assemblage" of the pTF promoter, combining TET-operators with a plant virus core promoter. This pTF promoter has already been described to have low basal activity and high inducibility index (Leuchtenberger et al., 2001), but has not yet been used in lentivectors. Second, these activities may be due to the use of a third-generation rtTAsM2-p65 activator, combining a synthetic sequence providing low basal activity and high response to DOX together with lower cell toxicity by replacing the viral VP16 transactivator by nuclear factor-κB p65 (Knott et al., 2002). Third, the low level of uninduced pTF promoter may be due to the presence of 250-bp insulators present in our Rix backbone (Dayer et al., 2007), protecting it from enhancers present in the cell DNA around the vector integration site. We do not favor this hypothesis because, in early pTF-containing lentivector backbones devoid of insulators, GFP signals were as low as in Fig. 2 or 4.
A useful lentivector must also have a selectable marker or a living color expressed independently of the gene of interest, in order to select and/or visualize transduced cells independently of gene activation. Our polyswitch lentivectors described here were designed to provide both, expressed from the ubiquitous EFs promoter. We show that the BSD selectable marker is working within a large range of BSD concentrations. However, we encountered some limitations (recombination and low level of expression) when using the 2A-based expression system together with GFP. Vectors EGR and EBGR are thus not recommended in their present version, and we are currently addressing these issues in our next generation. However, we believe that vector EBR (Rix-pTF-DEST-EBR) is a state-of-the-art vector. Vector ER (Rix-pTF-DEST-ER) is also a useful vector, similar to vector EBR, but not allowing for selection of the transduced cells.
We demonstrate that the 2A approach for polycistronic vectors is efficient, because the various proteins in the multicistron are correctly cleaved. Indeed, GFP, BSD, and rtTA proteins originating from 2A-based constructs were shown to be functional, and we show that NGN3, PDX1, and MAFA proteins are efficiently processed from their 2A-based precursor. Moreover, preliminary data indicate that our NPM lentivector is biologically active, because it can activate the rat insulin promoter in several human cells. The autocleavage of 2A peptides is, however, not optimal in some cases and certainly not as straightforward as originally claimed (Szymczak et al., 2004). An observation similar to ours was recently reported (Ibrahimi et al., 2009). In that study, Ibrahimi and colleagues showed that different 2A peptides are not equivalent in terms of processing, with F2A being poorly processed. Interestingly, in our constructs, the lowest levels of GFP are observed when GFP is fused to F2A (EGR and EBGR). This phenomenon seems to occur at the translational level, as demonstrated by the combination of FACS analysis, western-blot analysis, and RT-qPCR. Although, we cannot conclude on the posttranscriptional mechanism responsible for such a decrease, this observation is valuable, because it provides clues for future 2A peptide optimization. It is also possible that additional amino acid residues resulting from the cleaved 2A peptide, on either end of the protein, contribute to the apparent lower GFP expression level, but we prefer another hypothesis. Indeed, in construct EGR, GFP is expressed without N-terminal extra residues, but gives the same MFI as in construct EBGR. In construct EBG, GFP has no additional amino residues in the C-terminal end and gives an MFI four times higher than EGR. Amino acids in the C-terminal end may be responsible for this decrease of GFP fluorescence. On the other hand, the MFI obtained with the EBG construct is still four times lower than that with the EG construct. As extra residues in the N-terminal end have no effect, the apparent decrease of expression of GFP is clearly related to the translation process. These data confirm observations made by Ibrahimi and co-workers (Ibrahimi et al., 2009).
Our polyswitch lentivectors also address an important issue for scientists working with lentivectors, namely, “easy” cloning. It is well known that cloning of genes of interest into lentivector backbones that are already big may be, at best, very time- and energy-consuming and, at worst, impossible. The presence of a recombination cloning DEST cassette in our polyswitch lentivectors makes further clonings fast and easy by just using LR enzyme mix and pENTR-Gene plasmids chosen from already existing collections, or custom-ordered from commercial sites and delivered in a few weeks.
Taken together, the polyswitch lentivectors described here represent a yet unavailable tool for genetic engineering. The combination in a single transduction unit of optimized autoinducible gene switch, selectable marker, and recombination cloning makes them novel, powerful, and versatile tools for current and future genetic engineering approaches. As an example of a straightforward application, these polyswitch lentivectors could be used in primary precursors in which, after sufficient cell expansion, one can transiently express a master gene that is crucial for terminal differentiation of cells, such as pancreatic β-cells or neurons.
Footnotes
Acknowledgments
The authors thank Dominique Wohlwend, Laureline Piriou, and Suzanne Bissat for excellence in FACS assistance; Patrizia Arboit for western-blot analysis; David Suter and Karl-Heinz Krause for sharing recombination cloning starting material; Tuan Nguyen, Karl-Heinz Krause, and Olivier Menzel for critical reading of the manuscript; Christiane Gatz for the pTF plasmid; Wolfgang Hillen for the rtTAs-M2-p65 plasmid; Jozsef Kiss for personal, scientific, and logistic support; and members of the Kiss laboratory for critical comments. This work was supported by a grant from the Chicago Diabetes Project (
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.