
Abstract
Viral vectors for vaccination can be optimized by targeting vector entry to antigen-presenting cells (APCs). Here we describe lentiviral vectors (LVs) that are targeted to APC using a chimeric measles virus (MV) hemagglutinin (H). The MV H protein is mutated to prevent binding to MV receptors and incorporates a single-chain antibody that recognizes murine major histocompatibility complex class II (MHC II). This targeted LV is highly efficient in transduction of freshly isolated mouse B cells and dendritic cells. MHC II–positive cells in spleen are transduced after intravenous injection, and a robust immune response to an antigen transgene is generated.
Introduction
Lentiviral vectors (LVs), based on human immunodeficiency virus type 1, are very efficient in gene delivery to skin-derived DCs in mice (He et al., 2006; Furmanov et al., 2010), and they activate the DCs that they infect (Breckpot et al., 2010). This stimulates CD8-positive and CD4-positive T-cell responses and antibodies against the LV-encoded transgene (Esslinger et al., 2003; Palmowski et al., 2004; Rowe et al., 2006; Dai et al., 2009). LVs are effective as antitumor or antiviral vaccines in mice (Coutant et al., 2008; Loisel-Meyer et al., 2009), and a similar vector based on simian immunodeficiency virus is an effective antiviral vaccine in macaques (Beignon et al., 2009).
Antibodies against cell-surface molecules can be displayed on the LV particles to direct attachment to specific cell subsets (Sandrin et al., 2003). Recently, two different strategies have been developed to trigger efficient LV and cell membrane fusion after antibody-mediated attachment, using either Sindbis virus glycoproteins (Morizono et al., 2005; Yang et al., 2006) or measles virus (MV) hemagglutinin (H) and fusion protein (F) (Funke et al., 2008, Anliker et al., 2010). Both approaches use mutated glycoproteins that no longer recognize their natural receptor. The advantage of the MV H/F strategy is that fusion occurs at the cell membrane at neutral pH so that the targeted surface receptor does not need to be endocytosed after vector binding, which is necessary for the Sindbis strategy. The MV H/F strategy relies on incorporation of a single-chain antibody (scFv) into the binding-defective H protein (Hmut). Thus, there is the potential to target any surface marker against which there is an scFV that folds efficiently in mammalian cells in the context of Hmut.
We recently isolated an scFv that recognizes a nonpolymorphic determinant on mouse major histocompatibility complex class II (MHC II) α chain, and that can be expressed at a high level in the context of a retroviral envelope (Gennari et al., 2009). We therefore decided to examine whether this scFv could be fused to Hmut in order to target LV vaccines to APCs in vivo.
Materials and Methods
The mouse MHC II complex I-Ad α and β chain plasmids (Lechler et al., 1986) were a kind gift from Prof RN Germain, NIH, USA. 293T cells were cultured in Dulbecco's modified Eagle medium with 10% fetal bovine serum (FBS) at 37°C with 10% CO2. A20, RAW264.7, and EL4 cells were cultured in RPMI 1640/10% FBS/50 μM β-mercaptoethanol at 37°C with 5% CO2. Mouse bone marrow DC cultures were prepared and transduced as previously described (Escors et al., 2008). For in vitro transduction, mouse splenocytes were cultured in RPMI 1640/10% FBS/50 μM β-mercaptoethanol at 37°C with 5% CO2 for 48 hr, and surface-marker staining was with the appropriate biotinylated antibodies and streptavidin-conjugated allophycocyanin. For in vivo transduction, mice were sacrificed 5 days after LV injection in the tail vein; then splenocytes were isolated, incubated in red blood cell lysis buffer (Sigma, St. Louis, MO), incubated in blocking buffer containing anti-Fc receptor antibody, then stained for MHC II, and analyzed by flow cytometry. To detect a CD8-positive T-cell response, mice were sacrificed 10 days after tail-vein injection, and splenocytes were isolated and used in an enzyme-linked immunospot assay, stimulated with ovalbumin257–264. The number of interferon-γ (IFN-γ)–producing cells was counted with an EliSpot reader (AID Diagnostika, Strassberg, Germany).
LV production
LVs with chimeric envelopes, Hmut with no scFv insertion, or vesicular stomatitis virus glycoprotein (VSV-G) 293T cells were produced by cotransfection of 293T cells using FuGENE 6 (Roche Applied Science, Indianapolis, IN) with envelope plasmids together with packaging plasmid pCMVRΔ8.91 and an LV plasmid expressing green fluorescent protein (GFP) (pSINCSGW), as previously described (Funke et al., 2008; Lopes et al., 2008). Producer cells were stained with an anti-H antibody (Chemicon, Temecula, CA). LV-containing supernatant was collected 24–36 hr later and centrifuged at 2,500 g for 12 hr. LVs were resuspended in PBS and left on ice for 2 hr, then aliquoted and stored at −80°C. Vector titers were determined as nanograms of reverse transcriptase (RT) activity using an RT assay kit (Roche Applied Science).
Results and Discussion
Figure 1A shows the measles Hmut (pCG-HcΔ18) and F (pCG-FcΔ30) constructs (MV strain Edmonston B) (Funke et al., 2008) with scFvs recognizing murine MHC II complex (Gennari et al., 2009) or human carcinoembryonic antigen (CEA) (Chowdhury et al., 2004). Figure 1B shows that Hmut and the two chimeric Hmut envelopes were all efficiently expressed on the LV producer cells. Specificity of targeting of the MHC II-Hmut LV was demonstrated by transfection of 293T cells with mouse MHC II complex I-Ad α and β chain plasmids, followed by LV transduction. Figure 1C shows selective transduction of the MHC II–positive cells in the transfected 293T population by MHC II-Hmut LV. VSV-G LV transduced both MHC II–negative and MHC II–positive populations, in this latter case significantly more efficiently than MHC II-Hmut LV at an equivalent dose. However, Fig. 1D shows that LVs with the MHC II-Hmut chimeric envelope are highly efficient in transduction of the MHC II–positive B cell line A20 and the MHC II–positive macrophage cell line RAW264.7, giving more transduced cells than the equivalent dose of VSV-G LV, with no transduction of the MHC II–negative T-cell line EL4. None of the cell lines was transduced by LVs with Hmut or CEA-Hmut envelopes. The MHC II-Hmut LV also transduced mouse bone marrow–derived DC cultures with approximately the same efficiency as a VSV-G LV (Fig. 1E). The titer of a Sindbis-enveloped LV targeted to DC-specific intercellular adhesion molecule (DC-SIGN) was reported as 1 × 106 TU/ml on DC-SIGN–expressing 293T cells (Yang et al., 2008). The MHC II-Hmut LV has a considerably lower titer on MHC II–expressing 293T cells, but has an equivalent titer on MHC II–positive murine cell lines or murine cell cultures.
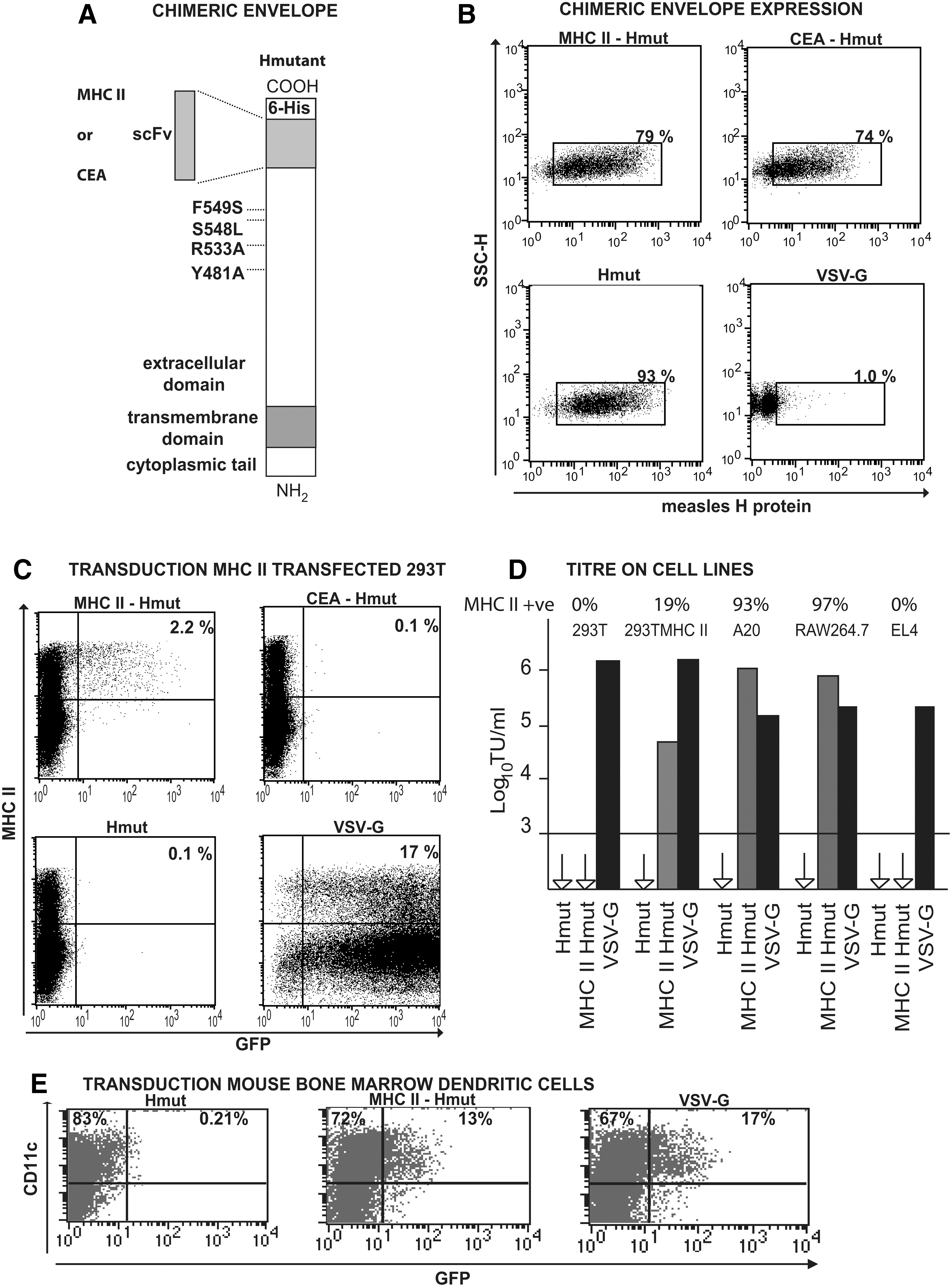
Expression and function of a chimeric measles H envelope targeted to MHC II. (
We next examined transduction of freshly isolated splenocytes by incubating them with LVs for 48 hr. Figure 2D shows efficient infection of the MHC II–positive primary cells by the MHC II-Hmut LV; a VSV-G LV is less efficient in transduction of the MHC II–positive cells and also transduces an MHC II–negative population. We also identified the subsets of these freshly isolated cells that were transduced by MHC II-Hmut LV as CD19-positive B cells (Fig. 2A), CD11c-positive DCs (Fig. 2B), and F4/80- or CD11b-positive macrophages (Fig. 2C). The gating on Fig. 2A and B is designed to remove, as far as possible, the autofluorescent population of splenic macrophages. VSV-G LV was much less efficient in the transduction of freshly isolated B cells, DCs, and macrophages, but did transduce a population of CD3-positive T cells under these conditions (Fig. 2E). The specificity and efficiency of transduction are an advance on our previous study, using the same scFv fused to an amphotropic murine leukemia virus (MLV) envelope. The amphotropic envelope itself was not binding-deficient, so it gave a background level of transduction, and the targeted envelope gave a maximum transduction of 8% of MHC II–positive cells (Gennari et al., 2009).

Transduction of mouse splenocytes ex vivo. Freshly isolated splenocytes were transduced with LVs (400 ng of RT per 1 × 106 splenocytes). After incubation for 48 hr, cells were stained for CD19 (
LVs with Edmonston strain H/F or with Hmut carrying an anti-CD20 scFv have also been reported to transduce resting human B cells very efficiently (Funke et al., 2008; Frecha et al., 2009). Our data show that efficient B-cell transduction also applies to mouse B cells and must reflect a binding receptor–independent property of MV H/F. We have also demonstrated very efficient transduction of other primary mouse cells that are MHC II–positive. To examine targeting in vivo, the MHC II-Hmut LV and controls were injected into the tail vein of C57BL/6J mice. Figure 3A shows detectable transduction of MHC II–positive cells in spleen by MHC II-Hmut LV. In fact, VSV-G LV also transduced mainly MHC II–positive splenocytes as previously reported (VandenDriessche et al., 2002; Arce et al., 2009). In vivo more cells were transduced by VSV-G LV, likely reflecting the transduction of DC precursors, which are MHC II–negative, as we previously described (Arce et al., 2009).

Transduction of mouse splenocytes in vivo and immunization. (
To examine immunization, an MHC II-Hmut LV was prepared encoding ovalbumin epitopes fused to human invariant chain (IiOVA) (Escors et al., 2008). Figure 3B shows efficient generation of a CD8–positive T-cell response by the MHC II-Hmut LV at a low vector dose of 10 ng of RT per mouse. We also examined the immune response to three high doses of LV encoding IiOVA, comparing it with the number of cells transduced with the same doses of LV encoding GFP. Despite considerably higher numbers of cells being transduced by the VSV-G LV, the CD8–positive T-cell response to MHC II-Hmut LV was equivalent (Fig. 3C). This represents a considerable advance on our previous study, using the same scFv fused to an amphotropic MLV envelope. The much more efficient transduction by MHC II-Hmut LV means that we have been able to track transduced cells in vivo for the first time. Furthermore, the amphotropic envelope itself gave a significant level of immunization, so the efficacy of the targeted MHC class II cell subset was not clearly demonstrated (Gennari et al., 2009). Finally, the mechanism of efficient transduction of primary mouse cells by the MHC II-Hmut LV will be an important topic of future investigation.
This proof-of-principle study demonstrates that scFv-targeted LVs using the MV H/F strategy are highly effective in immunization. This shows that these targeted LV particles must activate APCs to allow effective antigen presentation, as LVs with VSV-G or MLV-A envelopes do (Breckpot et al., 2010). Secondly, the wealth of new and emerging information on DC subsets, with differing efficiencies in antigen uptake, antigen presentation, and lymphocyte stimulation, begs the question as to whether rational vaccine design should harness particular DC subsets. As DC subsets are defined by surface-marker expression, a logical method is to target antigen using antibodies to these markers. Pioneering work in this case has been the use of antigen coupled to an anti-DEC205 antibody to enhance immunization with model antigens (Bonifaz et al., 2004) and to improve the efficacy of an antiviral DNA vaccine in mice (Nchinda et al., 2008). However, this method relies on the surface marker being competent for antigen internalization and processing. Also, the need to use an extraneous adjuvant means that bystander activation of other cells may enhance or inhibit the antigen-specific effect.
The use of a targeted viral vector to deliver antigen genes and activation signals to DC subsets overcomes these limitations. Only one previous study reports LV targeting to DC in vivo using Sindbis envelope (Yang et al., 2008). However, this requires modification of envelope glycosylation so that the LV binds the lectin DC-SIGN, which will be hard to achieve reproducibly on a large scale (Yang et al., 2008; Morizono et al., 2010). Our approach is more flexible as it will allow targeting of a range of surface receptors.
Footnotes
Acknowledgments
This work was supported by Cancer Research UK and UCL/UCLH Comprehensive Biomedical Research Centre and by a grant from the 7th European Framework Program “PERSIST” to CB. We thank Luciene Lopes for help with experiments.
Author Disclosure Statement
All authors have nothing to disclose.