
Abstract
To generate sufficient clinical-grade vector to support a phase I/II clinical trial of adeno-associated virus serotype 8 (AAV8)-mediated factor IX (FIX) gene transfer for hemophilia B, we have developed a large-scale, good manufacturing practice (GMP)-compatible method for vector production and purification. We used a 293T-based two-plasmid transient transfection system coupled with a three-column chromatography purification process to produce high-quality self-complementary AAV2/8 FIX clinical-grade vector. Two consecutive production campaigns using a total of 432 independent 10-stack culture chambers produced a total of ∼2 × 1015 vector genomes (VG) by dot-blot hybridization. Benzonase-treated microfluidized lysates generated from pellets of transfected cells were purified by group separation on Sepharose beads followed by anion-exchange chromatography. The virus-containing fractions were further processed by gel filtration and ultrafiltration, using a 100-kDa membrane. The vector was formulated in phosphate-buffered saline plus 0.25% human serum albumin. Spectrophotometric analysis suggested ∼20% full particles, with only low quantities of nonviral proteins were visible on silver-stained sodium dodecyl sulfate–polyacrylamide gels. A sensitive assay for the detection of replication-competent AAV was developed, which did reveal trace quantities of such contaminants in the final product. Additional studies have confirmed the long-term stability of the vector at −80°C for at least 24 months and for at least 24 hr formulated in the clinical diluent and stored at room temperature within intravenous bags. This material has been approved for use in clinical trials in the United States and the United Kingdom.
Introduction
Numerous different serotypes of AAV have been identified (Gao et al., 2002; Rabinowitz et al., 2002), distinguished, in part, by the surface capsid protein, which is responsible for viral tropism, infection kinetics, and immunogenicity. Because its genome was the first to be cloned, AAV2 has been the serotype most extensively studied in preclinical and clinical trials. We and others have established in a number of different in vivo settings that AAV2 vectors are particularly efficient at transducing terminally differentiated cells such as neurons (Kaplitt et al., 1994; Mandel et al., 1997), myocytes (Fisher et al., 1997; Herzog et al., 1999), retinal photoreceptor cells (Flannery et al., 1997; Ali et al., 2000), bronchial epithelium (Wagner et al., 1999a; Duan et al., 2000; Halbert et al., 2000), and hepatocytes (Snyder et al., 1999; Nathwani et al., 2001, 2002), resulting in long-term transgene expression in animal models. Correction of the bleeding diathesis after a single administration of AAV2 into either muscle or liver has been consistently observed in murine (Herzog et al., 1997; Wang et al., 2000; Nathwani et al., 2001) and canine (Herzog et al., 1999; Snyder et al., 1999; Wang et al., 2000; Mount et al., 2002) models of hemophilia without significant toxicity. These preclinical studies have supported clinical evaluation of AAV2 vectors in more than 100 patients with hemophilia B (Manno et al., 2003, 2006), cystic fibrosis (Flotte et al., 1996; Wagner et al., 1998, 1999a,b), Canavan disease (Janson et al., 2002), limb-girdle muscular dystrophy (Stedman et al., 2000), α1-antitrypsin deficiency (Flotte et al., 2004), and Leber's congenital amaurosis (Bainbridge et al., 2008; Maguire et al., 2008). Collectively, these early studies suggest that AAV2 vectors are safe in humans, although efficacy has been limited, except in trials for Leber's congenital amaurosis, in which significant clinical benefit was realized.
More recently, we have focused on the use of recombinant AAV vectors pseudotyped with capsid protein of an alternative serotype, serotype 8. Although serotypes AAV2, AAV5, and AAV8 were generally similar in their ability to target and transduce rhesus hepatocytes (Davidoff et al., 2005), we have found that the prevalence of immunity to AAV8, resulting from prior wild-type viral infection, is lower in humans than for the other two serotypes (Parks et al., 1970; Gao et al., 2002; Nathwani et al., 2009). This result is of critical importance as we have shown that serotype-specific neutralizing antibodies preclude successful hepatocyte transduction (Davidoff et al., 2005; Hurlbut et al., 2010). Clearance of AAV8 from the systemic circulation in macaques was also significantly quicker than with other serotypes (Nathwani et al., 2007), and AAV8 was shown to have reduced heparin binding, which reduced cytotoxic capsid-specific T cell activation (Vandenberghe et al., 2006). Last, AAV8 vectors have distinct biological properties that enable them to uncoat and release their genome more rapidly than other AAV serotypes (Thomas et al., 2004).
Our vector development efforts for hemophilia B treatments were significantly enhanced by the adoption in our vectors of a self-complementary design, wherein one of the vector inverted terminal repeat (ITR) terminal resolution sites is mutated to force packaging of dimer genomes (McCarty et al., 2003; Wang et al., 2003). These “self-complementary” dimer vectors provide enhanced human factor IX expression (Nathwani et al., 2006), most likely because the tail-to-tail orientation of the dimer allows rapid annealing in vivo, creating a double-stranded genome that is competent for RNA transcription (McCarty et al., 2001). Although there are numerous examples of the superiority of self-complementary AAV vector designs, little information is available regarding large-scale clinical production of such vectors.
We have established a protocol for a phase I/II clinical trial of AAV-mediated FIX gene transfer in subjects with hemophilia B, using a self-complementary vector pseudotyped with serotype 8 capsid. Although an increasing number of scalable methods for purification of recombinant AAV vectors has been described for a number of different serotypes (Brument et al., 2002; Kaludov et al., 2002), including serotype 8 (Davidoff et al., 2004; Okada et al., 2009; Ayuso et al., 2010), in order to generate sufficient clinical-grade vector to support this clinical trial we had to develop a large-scale, GMP-compatible method for production and purification of recombinant AAV8 vector. This paper describes the methodology for producing and testing U.S. Food and Drug Administration (FDA)-approved clinical-grade vector for this trial.
Materials and Methods
Plasmids
The two plasmids transfected to produce vector were designated CR21-LTAAVhelp2-8 and scAAV-LP1-hFIXco+helpv3. pCR21-LTAAVhelp2-8 is the kanamycin resistance-bearing plasmid pCR2.1 (Invitrogen, Carlsbad, CA) with an inserted AAV genomic fragment expressing serotype 2 rep and serotype 8 cap genes. The fragment begins 133 bp upstream of the Rep78 start codon, which was modified from GCCATGCCG to
Vector production and purification
Phase A
293T cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) (CRL-11268) and expanded into a master cell bank that was extensively characterized for identity and microbiological contamination. These 293T cells were grown as seed cultures to seed, at 1-week intervals, eight to sixteen 10-stack CellSTACKs (referred to hereafter as “cell factories” or CFs; Corning Life Sciences, Acton, MA) that were used for AAV vector production. Calcium phosphate-mediated transfection of the two AAV plasmids was performed when the cells were 50–70% confluent (approximately 18–30 hr after seeding the cell factories). The cells were harvested approximately 40–48 hr after transfection, using 5 μM EDTA in Ca2+- and Mg2+-free phosphate-buffered saline (PBS) and were concentrated by low-speed centrifugation. The cell pellet was resuspended in TD buffer (0.71 mM K2HPO4, 25 mM Tris, 5.0 mM KCl, 140 mM NaCl, and 6.4 mM MgCl2) and then lysed via one freeze–thaw cycle and microfluidization. The lysate was cleared by low-speed centrifugation and the resulting virus-containing supernatant was frozen and stored at −80°C.
Phase B
Eight cell factory lysates were thawed rapidly at 37°C and Benzonase nuclease (25 units/ml; Merck, Darmstadt, Germany) was added to the cell lysate and incubated at 37°C. Debris was pelleted by low-speed centrifugation and the resultant supernatant was passed through a 0.22-μm pore size filter. Sephacryl S-300 high-resolution (HR) resin (GE Healthcare Life Sciences, Piscataway, NJ) was equilibrated in 10 mM Tris, 10 mM Bis-Tris propane (pH 9.0) plus 150 mM NaCl before loading the Benzonase-treated filtrate at 15 cm/hr. The virus peak was collected and then diluted to 10 mM Tris, 10 mM Bis-Tris propane (pH 9.0) plus 40 mM NaCl in preparation for anion-exchange chromatography. POROS HQ50 resin (Applied Biosystems/Invitrogen, Foster City, CA) was equilibrated in 10 mM Tris, 10 mM Bis-Tris propane (pH 9.0) plus 40 mM NaCl before loading the diluted eluate from the previous step at 311 cm/hr. The loaded column was washed with 10 mM Tris, 10 mM Bis-Tris propane (pH 9.0) plus 40 mM NaCl and eluted, using a gradient of 10 mM Tris, 10 mM Bis-Tris propane (pH 9.0) and 10 mM Tris, 10 mM Bis-Tris propane (pH 9.0) plus 1 M NaCl, and collected as fractions. The main virus-containing fraction elutes at approximately ∼140 mM NaCl. Fractions were screened at this stage by quantitative PCR (qPCR) and held at 4°C for no more than 2 weeks.
Phase C
Virus-containing anion-exchange fractions from 24 cell factories (3 × 8 CF POROS runs) were loaded onto Sephacryl S-300 HR resin, equilibrated in PBS, at 15 cm/hr and the virus peak was collected. This material was concentrated approximately 15- to 20-fold by tangential flow filtration (TFF) with a 100-kDa molecular mass cutoff Biomax filter (Millipore, Billerica, MA). The concentrated material was then sterile filtered through a 0.2-μm pore size filter, formulated with human serum albumin (HSA) to 0.25% (1:100 dilution of a 25% stock solution; Talecris, Research Triangle Park, NC), and then frozen and stored at −80°C.
Phase D
For final vialing the individual 24 cell factory purifications were thawed, pooled, mixed thoroughly, and passed through a 0.22-μm pore size sterilizing-grade filter. The virus was aseptically vialed at 2.1 ml in glass 5-ml, 20-mm type I borosilicate clear glass vials, closed with rubber butyl stoppers, and sealed with aluminum seals. The vials were placed into a −80°C freezer and stored frozen.
Vector testing
Residual bovine serum albumin analysis
Residual bovine serum albumin (BSA) was analyzed with a commercially available ELISA kit (Alpha Diagnostic International, San Antonio, TX). The assay range is from 40 to 0.15 μg/ml. Samples spiked with BSA were also analyzed to preclude interference by inhibitors/enhancers. This assay was performed on bulk purified material pooled equally from the 24 cell factory preparations before the addition of HSA.
qPCR titer
Vector genome quantitation used SYBR green dye real-time qPCR with vector-specific primers SJVL-679 (ATTTTATGTTTCAGGTTCAGGGGGAGGTG) and SJVL-680 (GCGCAGAGAGGGAGTGGACTAGT). A standard curve was generated with linearized plasmid pscAAV-LP1-hFIXco DNA (Nathwani et al., 2006). Vector preparations and standards were diluted directly in H2O plus 0.01% (w/v) Pluronic F-68. A sample of a reference vector of known titer was run with each assay as a reference control. To calculate a predicted capsid protein concentration from the qPCR titer on pre-HSA formulated material, the average qPCR titer (8.1 × 1011 VG/ml) was converted to its molar concentration, multiplied by the ultraviolet (UV) calculated value for capsids per vector genome (5.3; see below), and converted to a mass concentration, using a predicted molecular mass for AAV8 capsids of 3.74 MDa.
Dot-blot hybridization
Two microliters of sample was first incubated with 300 units of DNase I in 25 μl of New England BioLabs (Ipswich, MA) restriction enzyme buffer #3 for 1 hr at 37°C, and then supplemented with 1 μl of 0.5 M EDTA, pH 8.0, and incubated for 10 min at 95°C. Subsequently, 1 μl of proteinase K cocktail (4 μl of 10% sodium dodecyl sulfate [SDS], 4 μl or 5 units of proteinase K, 6 μl of H2O) was added and samples were incubated for 1 hr at 55°C. The proteinase K reactions were then diluted with H2O to 100 μl, and 5 μl of diluted sample was further diluted to 300 μl with H2O before processing with a 30-kDa molecular weight cutoff Ultra-0.5 ml centrifugal device (Millipore) as per the manufacturer's instructions. The recovered samples and serial dilutions of linearized hFIXco standard were diluted 1:20 in 0.4 M NaOH before transferral to Zeta-Probe GT blotting membrane (Bio-Rad, Hercules, CA). After cross-linking, the membrane was probed with an EcoRI–PstI fragment from the vector plasmid in Hybrisol I (Millipore) and washed according to standard methods. After exposure to storage phosphor screens, images were quantitated with image analysis software.
UV absorbance
Eighteen microliters of sample was mixed with 2 μl of 1% SDS, heated to 75°C for 10 min, and slowly cooled to room temperature before measurement of absorbance at 260 and 280 nm in a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA), which reports the 1-cm path length equivalent value for low-volume samples. To calculate vector genomes per milliliter, Eq. (6) from Sommer and colleagues (2003) was solved, using a value of 1.42 × 106 g/mol as the MWDNA, and the measured absorbances. To calculate capsids per vector genome, Eq. (4) in the same publication was used similarly. Capsid protein concentrations were derived as the product of vector genomes per milliliter and capsids per vector genome, using 3.74 MDa as the molecular mass of empty capsid particles.
Sterility
Sterility was performed as per the U.S. Pharmacopeia sterility test (USP Section <71>;
Residual Benzonase
Testing was performed with a quantitative Benzonase endonuclease ELISA kit II (EMD Chemicals, Gibbstown, NJ), which contains plates coated with polyclonal antibodies to Benzonase endonuclease. Known quantities of Benzonase were run along with the sample and quantitated with secondary antibody tagged with horseradish peroxidase (HRP). A sample spiked with a known amount of Benzonase was included to show inhibition/enhancement. The limit of detection was 0.2 ng/ml and the limit of quantitation was 1 ng/ml.
Viral genomic DNA analysis
DNA was purified from 1 to 2 ml of the final vector product, using a QIAamp DNA isolation mini kit (Qiagen, Hilden, Germany), and resuspended in TE. The concentration was determined spectrophotometrically before use for contaminant PCR, direct DNA sequencing, and agarose gel analysis as follows: Real-time qPCR was done to determine the concentration of hFIX vector genome (primers listed previously), AAV8 capsid sequences (primers SJVL-930 [TCAGCCAAGGTGGGCCTAATACAA] and SJVL-932 [TTGCTGCTGCAAGTTATCTGCCAC]), kanamycin resistance gene (primers SJVL-990 [GGGCGCCCGGTTCTTTTTGTC] and SJVL-991 [GCCAGTCCCTTCCCGCTTCAGTG]), as well as ITR-Rep recombinants (primers SJVL-940 [ACTCCATCACTAGGGGTTCT] and SJVL-941 [GCTGGGGACCTTAATCACAA]), using SYBR green technology. The quantity of each contaminant (capsid, kanamycin, or ITR-Rep) was expressed relative to that determined for the hFIX vector as a percentage. For native agarose gel analysis, 250 ng of purified DNA, digested or not with BglI or EcoRI (New England BioLabs), was electrophoresed on an agarose gel before ethidium bromide staining, using standard methods. Alkaline gels were run with 0.03 M NaOH as running buffer, with recirculation, and stained postelectrophoresis with 4 × GelRed stain (Biotium, Hayward, CA). For direct DNA sequencing, 40 ng of viral DNA was mixed with 3.2 pmol of primer and submitted for automated sequencing at the Hartwell Center of St. Jude Children's Research Hospital (SJCRH).
Endotoxin
Endotoxin concentration was determined with a kinetic chromogenic LAL (Limulus amebocyte lysate) assay kit (Lonza, Basel, Switzerland). The sample was mixed with the LAL/substrate reagent, placed in an incubating plate reader, and monitored over time for the appearance of a yellow color.
Potency
Samples of AAV were diluted in PBS to a concentration of 1 × 109 qPCR VG/ml. Five male C57BL/6 mice were inoculated via the tail vein with 200 μl of the diluted AAV. After 7 days, blood was collected by retro-orbital bleeding into tubes containing sodium citrate and centrifuged, and the plasma was collected. The plasma was assayed for factor IX concentration with an Asserachrom IX:Ag kit (Diagnostica Stago, Parsippany, NJ).
Transmission electron microscopy
A 5- to 20-μl drop of sample was placed on a Formvar carbon-coated grid. After 30 sec, most of the sample was removed with filter paper. The grid was rinsed with three drops of distilled water and placed in 2% phosphotungstate acid solution, pH 6.4, for 30 sec. The sample was then allowed to dry before analyzing.
Bicinchoninic acid
Total protein content was determined by mixing sample and bovine serum albumin (BSA) standards with the bicinchoninic acid (BCA) reagent (Pierce Biotechnology/Thermo Fisher Scientific, Rockford, IL) and quantitating spectrophotometrically as per the manufacturer's instructions. The concentration of protein in the test article was determined from a standard curve run with each set of samples.
SDS–PAGE
Indicated volumes of samples were separated on a 4–12% Criterion (Bio-Rad) gel as per the manufacturer's instructions. Silver staining was performed with a SilverSNAP stain kit II (Pierce Biotechnology/Thermo Fisher Scientific).
Replication-competent AAV assay
A helper-dependent infectious stock of an AAV serotype 2-8 hybrid (serotype 2 rep, pseudotyped with serotype 8 capsid) was generated by transfecting plasmid pAAV2-8wt into 293T cells with adenoviral helper sequences, followed by purification by column chromatography. The vector genome titer of this preparation, aliquoted and stored in PBS plus 0.25% HSA, was determined to be 1.15 × 1012 VG/ml by qPCR using capsid-specific primers (described previously) and linearized pAAV2-8wt plasmid as a standard. For the replication-competent AAV (rcAAV) assay, 293T cells were plated (9.5 × 106 cells per plate) onto 15-cm dishes in 20 ml of Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and
Results and Discussion
In support of our clinical research efforts for the treatment of hemophilia B, we established an AAV production process based on two-plasmid transient transfection of 293T cells in 10-stack culture chambers. In this system, the self-complementary AAV vector genome is on the same plasmid as the adenoviral plasmid helper sequences, and the AAV helper sequences are expressed from an early-generation rep/cap plasmid (see Materials and Methods). The overall process, modified from that described previously (Davidoff et al., 2004), is detailed in Fig. 1, and consists of four phases: (A) transfection, wherein 293T cells in sets of four 10-stack chambers are transfected, harvested, and lysed; (B) stage I purification, in which the lysate is treated with Benzonase, filtered, and purified by size-exclusion and ion-exchange chromatography; (C) stage II purification, in which material from stage I is pooled and again purified by size-exclusion chromatography with reduced loading volumes, and formulated with human serum albumin (HSA); and (D) the finish, in which multiple 24 cell factory sublots are thawed, pooled, filtered, and vialed as the final vialed product.
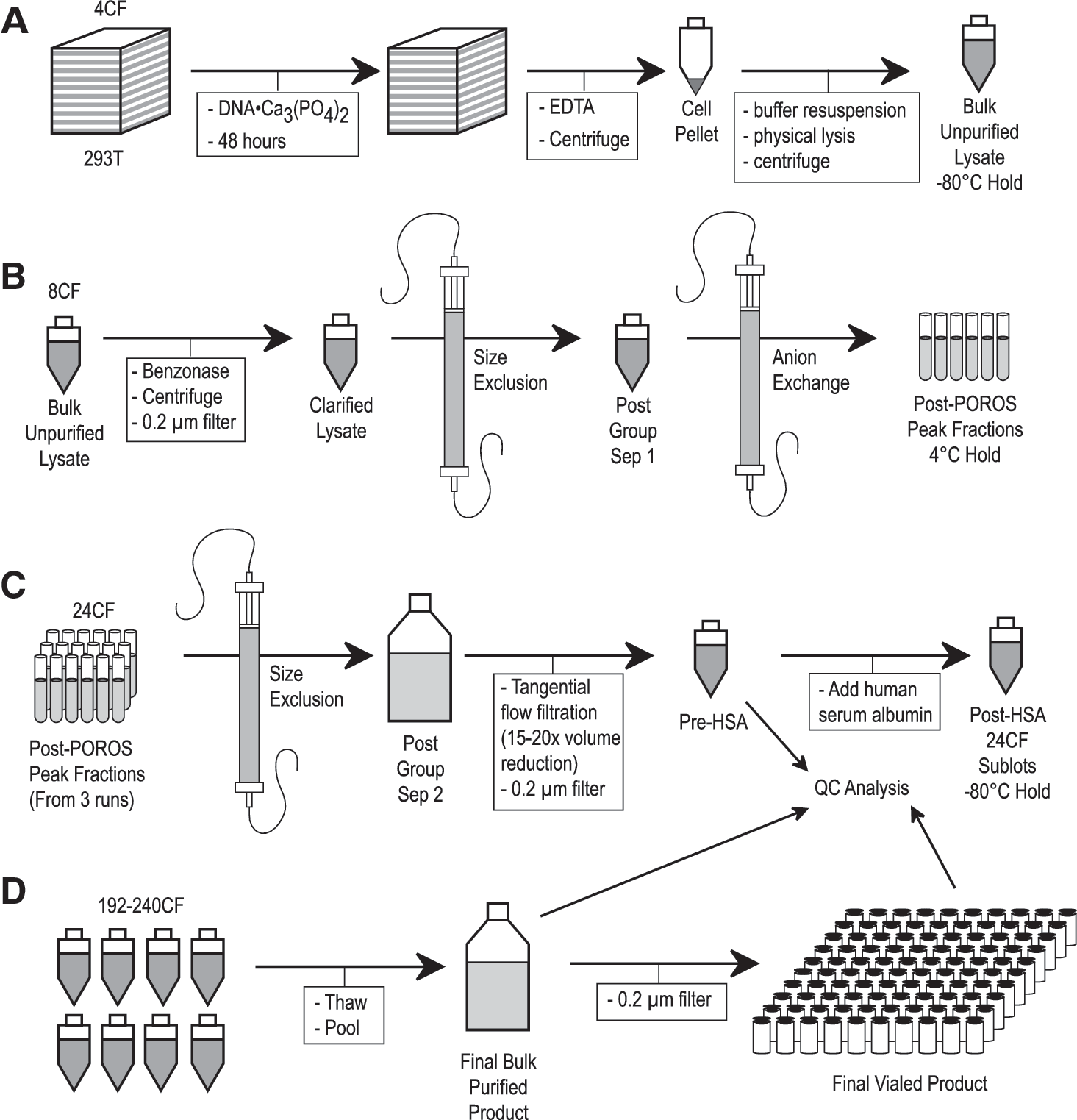
The four phases (
Product quality and yield were monitored at several stages throughout the production process. General transfection performance was evaluated by a semiquantitative Western blot of bulk crude lysates. Aliquots of this material were also retained, pooled, and tested as bulk unpurified lysate, as detailed in Table 1. Quantitative PCR estimation of vector titer (qPCR VG) was performed first after anion-exchange chromatography, and also at several stages throughout the purification and formulation process. By this assay, our yield during the last two phases of production was ∼42%. As we did not measure the vector content in crude lysates, we cannot calculate a yield for the earlier phases. Because of our selection of 0.25% HSA in the final formulation, SDS–PAGE, UV absorbance, transmission electron microscopy (TEM), and total protein (BCA method) assays were performed before addition of HSA, on each 24 cell factory sublot (see Table 2).
CFR, Code of Federal Regulations; hFIX, human factor IX; PBRT, PCR-based reverse transcription; EP, European Pharmacopeia; PTC, U.S. Food and Drug Administration Points to Consider; TEM, transmission electron microscopy; USP, U.S. Pharmacopeia.
BCA, bicinchoninic acid; CF, cell factory; HSA, human serum albumin; qPCR, quantitative PCR; UV, ultraviolet; see also Table 1.
Quantitative results, when specified, represent mean values ± standard deviation for all clinical sublots (n = 18).
Our established method for the critical vector titration assay used for subject dosing was quantitative PCR. All of our qPCR titration assays on both preclinical and clinical material used both linearized plasmid DNA as a quantitative standard as well as an in-house reference AAV preparation, for which the measured values showed relatively small variation over 3 years of analysis (standard deviation, 48% of the mean value; n = 24). We did, however, observe a significant and consistent discrepancy between the qPCR titer and that inferred by UV spectrophotometry when both assays were performed on pre-HSA 24CF sublots (Table 2). Subsequent investigation into the causes of this discrepancy demonstrated that the qPCR assay consistently underreports self-complementary AAV vector genome titers relative to dot-blot hybridization, apparently because of rapid reannealing of genome templates to exclude primer binding (data not shown). When dot-blot hybridization methods were performed on our formulated clinical material, the observed titer was nearly 10-fold higher than that determined by qPCR (see Table 4), which is significantly more consistent with UV titration results before formulation. In addition, when genomic DNA was purified from the formulated clinical material for the DNA analysis (Fig. 2C and D), and quantitated by UV spectrophotometry, our yield was similar to what would be predicted from the dot-blot titer (∼8.3 μg of DNA/ml vector ≈ 3.5 × 1012 VG equivalents/ml vector), and inconsistent with the lower, qPCR-determined titer.
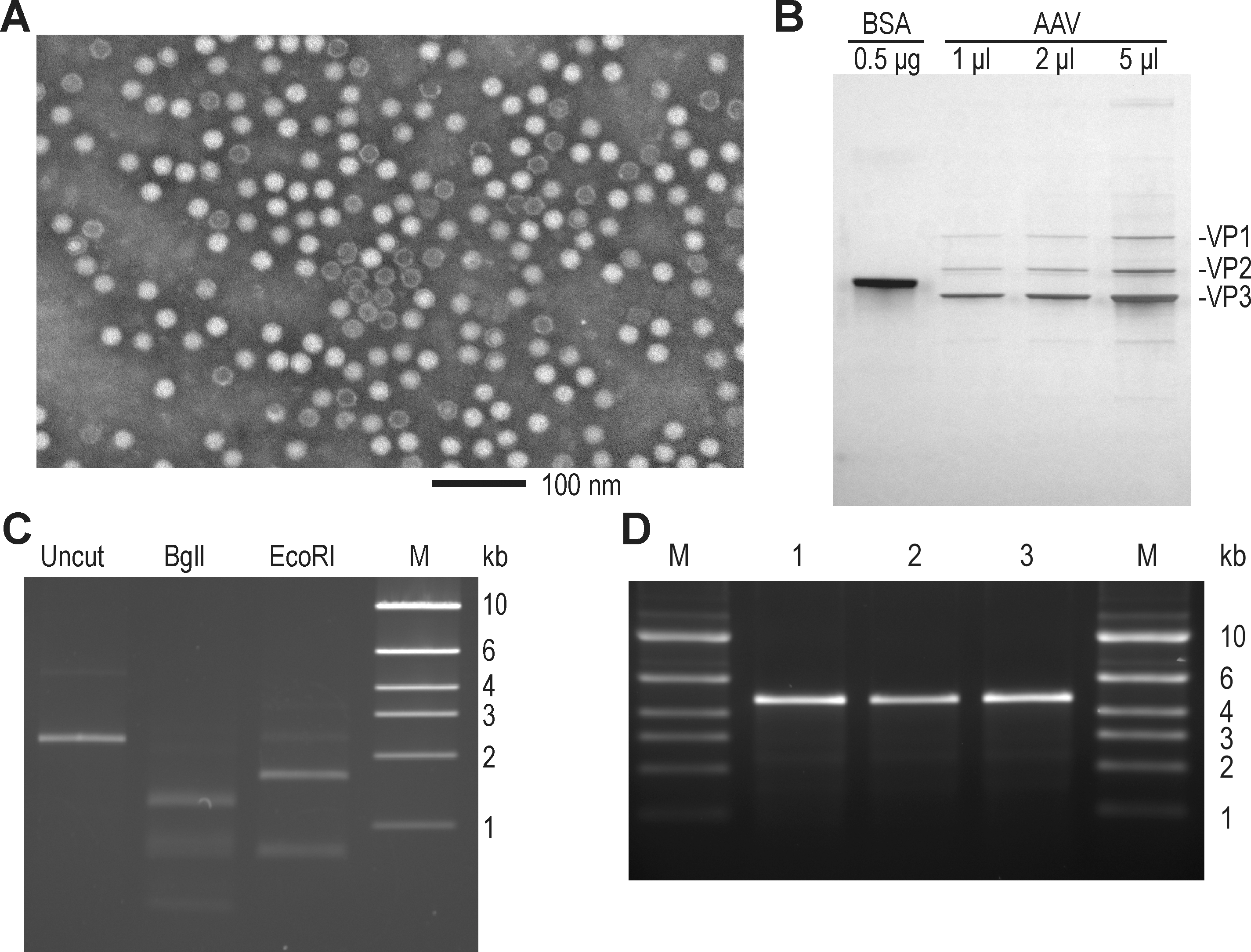
Assessment of AAV vector purity. (
Regarding the protein content of our product, silver-stained SDS–polyacrylamide gels before HSA formulation showed only minor contaminants (Fig. 2B). The average A 260nm/A 280nm ratio of 0.98 measured at this pre-HSA stage suggested that our clinical material contains ∼5.3 capsid equivalents/VG (see Materials and Methods), assuming that this parameter did not change during formulation and vialing stages. Although electron micrographs of material at this stage showed intact, icosahedral particles with lower quantities of stain-penetrated particles (Fig. 2A), in our experience quantitation of these images was not reliable and frequently produced inconsistent results. The size and intensity of viral capsid bands on the protein gel of Fig. 2B were roughly congruent with the signal expected by UV analysis of that material (Fig. 2B, see legend). The total protein content at the pre-HSA stage as determined by BCA methods (Table 2, again using bovine serum albumin as standard), when interpreted as entirely deriving from capsid protein, would suggest a 2-fold higher content of capsid protein; however, this could also be due to differential sensitivity of the assay to capsid protein relative to the BSA standard.
Numerous additional assays for vector purity and identity were performed on the final bulk purified product, and are summarized in Table 3. The DNA content of our vector preparation was analyzed by multiple methods. Because of the double-stranded nature of our self-complementary vector genome, native agarose gel electrophoresis was used to size the genome (Fig. 2C), and to confirm that after restriction digestion fragments of the proper size were generated. In addition, the genomic DNA was analyzed by alkaline gel electrophoresis (Fig. 2D), confirming little contaminating monomer genomic DNA (on the order of 2% by quantitative analysis of that image). The purified genomic DNA was also directly sequenced with an array of specific primers and confirmed the expected sequence for all but the structured ITR hairpins (data not shown). Sensitive PCR assays quantitated potential contamination by various DNAs from the 293T cell line, the backbone of transfected plasmids, and nonhomologous recombination products between the vector and helper DNAs (see Table 3 for results). Although we did detect capsid DNA, host cell DNA, and bacterial plasmid backbone DNA, the levels of these contaminants were in the range observed by others (Chadeuf et al., 2005; Hauck et al., 2009), and presumably reflect mispackaging of these DNAs in particles. We should also point out that these values were calculated relative to concurrent qPCR VG concentration, and could be as much as 10-fold lower as a result of the qPCR underestimation of vector genomes, but without confirmation that the contaminants were not similarly affected we have chosen to report these values without modification. Potentially transforming DNA sequences from the 293T cell line (E1A and simian virus 40 [SV40] large T antigen) were not detected.
Quantitative results, when specified, represent values determined for first of two clinical lots. Contaminants reported as percentages are relative to vector genome copy number in purified DNA.
Because of the inefficiency of transducing immortalized cell lines in vitro with serotype 8-pseudotyped AAV vectors, we opted to perform the critical potency assay for our vector product in mice, in which vector-derived human factor IX protein can be easily quantitated in plasma by ELISA. Figure 3A shows the dose response of this assay, which measures human factor IX levels 7 days after tail vein injection. Our standard assay uses 2 × 109 qPCR VG per animal, and was used to confirm product potency at several stages of production (see Tables 2 and 4). This assay was also used for stability testing of the final clinical trial material (CTM), in accordance with the specifications of International Conference on Harmonization (ICH) Q1A(R2) (Stability Testing of New Drug Substances and Products). Figure 3B shows a time course from one of these stability studies, confirming product stability for at least 24 months. Additional stability studies confirmed the potency of vector product during the 4°C hold step after anion-exchange purification (for 28 days), and in the dilute room temperature formulation used for patient administration (for 24 hr).
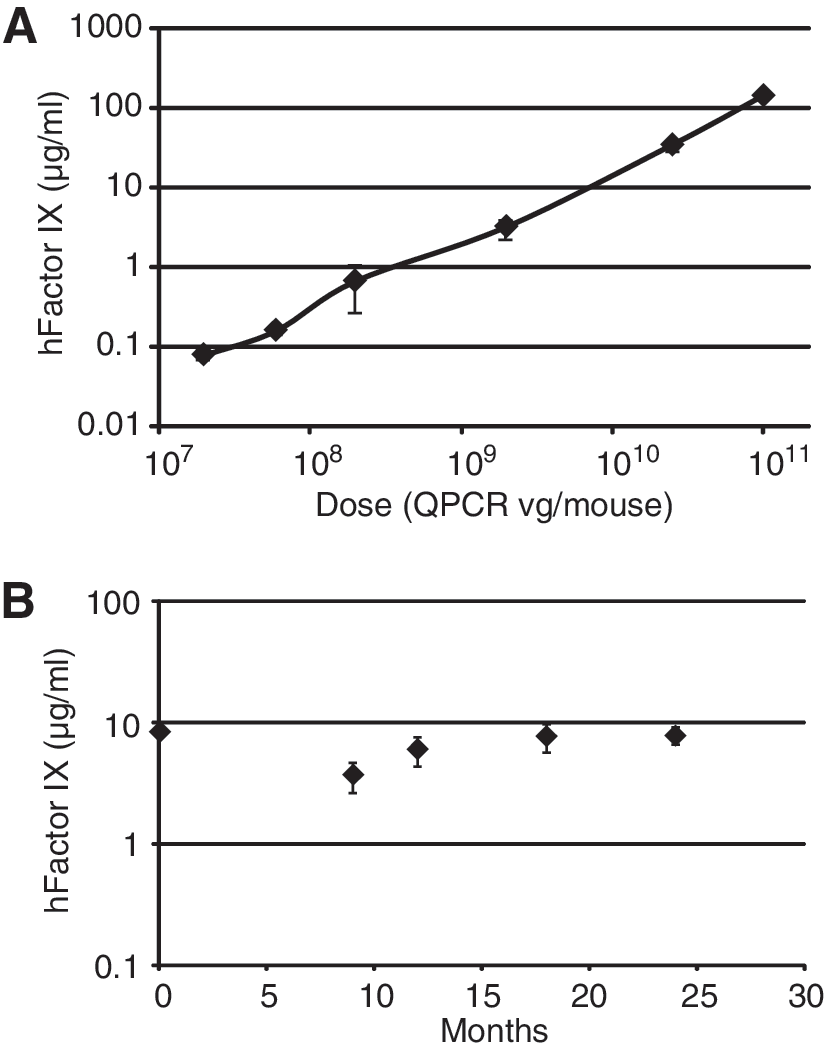
In vivo mouse potency assay. (
LAL, Limulus amebocyte lysate.
Quantitative results, when specified, represent values determined for the first of two clinical lots.
Some transient transfection systems for the production of AAV vectors have been shown to generate rare contaminants via nonhomologous recombination, and if ITR sequences are independently fused with each end of the rep/cap helper sequences then the resulting product can replicate in the presence of helper viruses such as adenovirus (Allen et al., 1997). To screen for such contaminants we developed an rcAAV assay using 293T cells, which were selected on the basis of a slightly improved ability to be transduced over other lines, and the advantage of complementing adenoviral E1A, which enabled the use of a ΔE1 adenoviral vector (Ad-GFP) as a helper virus. The assay process is outlined in Fig. 4A, and involves multiple rounds of infection in 293T cells in the presence of Ad-GFP helper, and qPCR of total DNA at each round to quantitate serotype 8 capsid DNA as an indicator of successful replication. A positive control recombinant, hybrid rcAAV virus with serotype 2 rep and serotype 8 cap genes, was generated (see Materials and Methods) and when tested in the presence of Ad-GFP helper, could be detected in a dose-dependent manner (Fig. 4B), although detection of 103 qPCR VG/plate occasionally failed and so future experiments used 104 VG/plate. With this diluted control virus, capsid sequences were not detected during any round in the absence of adenoviral helper (data not shown). When testing preclinical lots of the scAAV2/8-LP1-hFIXco vector in the presence of an rcAAV spike control, vector quantities greater than 2.25 × 1010 qPCR VG/plate or rcAAV quantities less than 104 VG/plate occasionally failed to allow rcAAV replication (data not shown), and so our standard assay used these input levels. Figure 4C shows typical results with one of our preclinical production lots, and confirms that there are low levels of adenovirus-dependent replicative AAV present in the product. On the basis of the significant increase in capsid sequence quantity after a 104 VG spike of rcAAV, we infer that the contaminant is present at less than 1 rcAAV per 2.25 × 106 qPCR VG, a result consistently observed with all preclinical and clinical production lots. Although any contamination with such recombinants is undesired, we note that AAV is a nonpathogenic virus that does not replicate in the absence of helper virus, and that at our highest proposed clinical dose of 2 × 1011 qPCR VG/kg, a 100-kg patient would receive only 107 rcAAV infectious units, an infectious dose less than that of the widely used replication-competent flu virus vaccine (FluMist). In addition, treatment with AAV vectors at the doses planned for our clinical trial has been shown to generate robust humoral immunity in both animals and humans, which should strongly protect against any further mobilization (Davidoff et al., 2005; Manno et al., 2006; Hurlbut et al., 2010).

Replication-competent AAV assay. (
It is difficult to predict what parameter of an AAV vector preparation, if any, will best predict performance in a human clinical trial. Although there are several alternative AAV production methods that have been described (Clement et al., 2009; Virag et al., 2009; Yuan et al., 2011), some of which are more easily scalable than those described here, few products produced by these alternative methods have reached the clinic. Open communication of vector production and characterization data will inevitably facilitate interpretation of clinical results from our trial and others, and hopefully lead to improved evaluation of alternative production methods and an established set of relevant quality measurements for gene therapy vectors. A clinical trial using the product described in this paper was initiated in both the United States and the United Kingdom in August 2009, and patient treatment is ongoing.
Footnotes
Acknowledgments
The authors thank Aaron Shafer, Yunyu Spence, Chris Morton, Annastasia Ouma, Cecilia Western, and Sheetal Bhatara for excellent laboratory contributions to this work, and John Coleman for outstanding management of the GMP facility. This work was supported by grants from the Katharine Dormandy Trust (UK) and the Medical Research Council (UK), a Department of Health NIHR Biomedical Research Centres funding award to UCLH/UCL (UK) and Department of Health funds for vector production, the Assisi Foundation of Memphis, the American Lebanese Syrian Associated Charities (ALSAC), the National Heart, Lung, and Blood Institute (Grant HL073838), and the National Cancer Institute (Cancer Center Support Grant CA027165).
Author Disclosure Statement
J.A.A., S.E.S., S.L., D.M.T., R.C., G.C., J.M., P.F., and A.W.N. have no competing interests to disclose. St. Jude Children's Research Hospital (SJCRH) retains intellectual property rights for certain aspects of the scAAV-LP1-hFIXco vector design, which under SJCRH policy may generate licensing and royalty revenue that would be shared with J.T.G., A.M.D., and A.C.N.