
Abstract
Mesenchymal stem cells (MSCs) possess tumor-tropic properties and consequently have been used to deliver therapeutic agents for cancer treatment. Their potential in cancer therapy highlights the need for a consistent and renewable source for the production of uniform human MSCs suitable for clinical applications. In this study, we seek to investigate whether human embryonic stem cells can be used as a cell source to fulfill this goal. We generated MSC-like cells from two human embryonic stem cell lines, HuES9 and H1, and observed that MSC-like cells derived from human embryonic stem cells were able to migrate into human glioma intracranial xenografts after being injected into the cerebral hemisphere contralateral to the tumor inoculation site. We engineered these cells with baculoviral and lentiviral vectors, respectively, for transient and stable expression of the herpes simplex virus thymidine kinase gene. In tumor-bearing mice the engineered MSC-like cells were capable of inhibiting tumor growth and prolonging survival in the presence of ganciclovir after they were injected either directly into the xenografts or into the opposite hemisphere. Our findings suggest that human embryonic stem cell-derived MSCs may be a viable and attractive alternative for large-scale derivation of targeting vehicles for cancer therapy.
Introduction
The vast therapeutic potential of MSCs highlights the need to identify an accessible and reliable source of these cells. Although adult MSCs could be reliably isolated from bone marrow and adipose tissues, the isolation procedure is invasive, with the isolated MSC population declining in proliferation capacity and differential potential with increasing age in culture (Kern et al., 2006). Another source of MSCs, umbilical cord blood, allows the derivation of MSCs with relatively greater proliferation capacity in culture. However, it still remains controversial whether cord blood can generate sufficient MSCs for subsequent studies and clinical applications (Perdikogianni et al., 2008). Human embryonic stem cells (hESCs), on the other hand, are an alternative source that enables standardization and large-scale production of downstream differentiated cells. This offers the potential for the manufacture of single batches of cell therapy products sufficient for repeated treatments in multiple patients, thus eliminating variability in the quality of cell therapeutics and facilitating reliable comparative analysis of clinical outcomes. The use of hESC-derived cells for cell therapy may also increase cost-effectiveness by reducing the laboriousness in collecting and expanding cells from individual patients and simplify the logistics by employing cryopreserved cells in ready-to-go format.
Techniques enabling the differentiation of MSCs from human embryonic stem cells have been developed (Lian et al., 2007; Arpornmaeklong et al., 2009; Liu et al., 2009). These hESC-derived MSCs are similar to adult MSCs in their gene expression profile, surface marker profile, and differential potential. They exhibit much greater proliferative capacity than their adult counterparts, and could be consistently derived in large amounts. hESC-MSCs have also been shown to possess the same immune-suppressive effects found in adult MSCs, and could potentially be tolerated by the immune systems of allogeneic recipients (Trivedi and Hematti, 2008; Yen et al., 2009). Most importantly, they are functional in in vivo disease models, conferring neuroprotective effects in rat transient focal cerebral ischemia, cardioprotective effects during porcine myocardial ischemia–reperfusion injury, as well as in repairing defective rat cartilage (Timmers et al., 2007; Hwang et al., 2008; Liu et al., 2009; Lai et al., 2010).
In view of these advantages, we set out to investigate the feasibility of using hESC-derived MSCs as prodrug delivery vectors for cancer gene therapy. We are interested in whether these cells can exhibit the tumor tropism property demonstrated in adult MSCs and whether they can produce an efficient tumor-killing effect when coupled with the herpes simplex virus thymidine kinase gene (HSVtk)–ganciclovir system. We loaded the cells with the HSVtk transgene, using either the baculovirus or lentivirus gene transfer method. Baculovirus, or Autographa californica multiple nucleopolyhedrovirus, is an insect virus that is emerging as a potentially safe class of gene delivery vectors because of its inability to replicate or to cause toxicity in mammalian cells (Wang and Balasundaram, 2010). Because of its nonintegrating nature, it mediates transient transgene expression in cells. Lentivirus, on the other hand, is an animal virus well known to preferentially integrate into transcriptionally active regions of host genomes, raising concerns over insertional mutagenesis, oncogene activation, and cellular transformation (Schroder et al., 2002; Hacein-Bey-Abina et al., 2003; Liu et al., 2006). Yet, despite the risks involved in its use, the stable transgene expression instigated by this virus has proven useful for many applications. Our study seeks to reveal the differences in tumor-killing efficacy that arise from the use of these two gene transfer systems mediating different durations of transgene expression, and most importantly, highlights the potential of hESC-MSCs in cancer therapy.
Materials and Methods
Cells
The generation and characterization of HuES9 hESC-derived MSCs were described previously (Lian et al., 2007). These cells have been sorted to obtain a homogeneous CD105+ and CD24− population and tested for purity by flow cytometry and for their ability to differentiate into osteogenic, chondrogenic, and adipogenic lineages (Lian et al., 2007). Cells are positive for CD105, CD166, CD29, and CD44 and negative for CD14, CD34, and CD45. They were obtained at passage 13 and used not later than passage 20 for the current study. Human embryonic stem cell line H1-derived MSCs were generated according to the method described by Hwang and colleagues (2008). H1 embryonic stem cells were obtained from the WiCell Research Institute (Madison, WI) and cultured in mTeSR-1 medium (StemCell Technologies, Vancouver, BC, Canada) on Matrigel (BD Biosciences, San Jose, CA). H1 cells were incubated for 7 min with Dispase (1 mg/ml), followed by two washes in unsupplemented Dulbecco's modified Eagle's medium (DMEM)–F12 (GIBCO/Invitrogen, Carlsbad, CA). Cells were scraped and cultured in suspension in DMEM–F12 supplemented with 20% knockout serum replacer, 1 mM
MSC migration in vitro and in vivo
For the in vitro migration assay, hESC-MSCs were serum-starved overnight in reduced serum Opti-MEM (Invitrogen) and collected for use. Fifteen thousand MSCs were seeded into each 96-well Corning cell culture insert (Corning Life Sciences, Lowell, MA) with a polyester (PET) membrane (pore size, 8 μm). Inserts were placed into a Corning 96-well receiver plate containing Opti-MEM, U87 cells seeded in Opti-MEM, or Opti-MEM supplemented with 15% fetal bovine serum (FBS). U87 cells were seeded at a density of 50,000 cells per well. The plates were incubated for 24 hr at 37°C in 5% CO2, after which the inserts were transferred to a new receiver plate. Migrated cells on the bottom side of the membrane were treated with a cell dissociation and staining solution consisting of 4× diluted trypsin containing 2.4 μl of Calcein-AM (BD Bioscience) (50 μg resuspended in 30 μl of dimethyl sulfoxide [DMSO]) per milliliter. Fluorescence from the detached cells was measured with a microplate reader (GENios Pro; Tecan Group, Maennedorf, Switzerland) and relative fluorescence units were plotted against a standard curve to estimate the actual number of cells that had migrated. All experiments were done with at least six replicates and values are expressed as means ± SD.
The in vivo migration assay was performed with three adult male BALB/c athymic, immuno-incompetent nude mice (weight, 20 g; age, 6–8 weeks). U87 cells and hESC-MSCs were labeled with CM-DiO and CM-DiI (Molecular Probes/Invitrogen, Eugene, OR), respectively, according to the manufacturer's protocol. The mice were inoculated with 5 × 105 CM-DiO-labeled U87 cells via the right striatum of anesthetized animals (designated as day 0). On day 7, 1 million CM-DiI-labeled hESC-derived MSCs were injected into a site contralateral to the tumor inoculation site. The brains were collected 7 days later, fixed in 4% paraformaldehyde overnight, and sectioned.
Viral vector preparation and cell transduction
Baculoviral vectors with the enhanced green fluorescent protein (eGFP) reporter gene were constructed in our previous studies (Zeng et al., 2007, 2009; Du et al., 2010). The transfer plasmid pFastBac1 (Invitrogen) was used to generate recombinant baculoviruses with different expression cassettes. To generate the recombinant baculoviral vector containing the HSVtk gene (Balani et al., 2009), the HSVtk gene with flanking EcoRI and XhoI sites was obtained through PCR amplification from the pORF9-HSV1tk plasmid (InvivoGen, San Diego, CA), using forward primer 5′-GTGAACCGTCAGATCGAATTCCTGAGATCACCGGCGAAGGA-3′ and reverse primer 5′-CCAGAGGTTGATTATCGCTCGAGTCAGTTAGCCTCCCCCATCT-3′, and used to replace the eGFP gene in our CMV-W vector. BacPAK6, a baculoviral vector without a mammalian gene expression cassette, was purchased from Clontech (Mountain View, CA). Viruses were produced and propagated in Spodoptera frugiperda 9 (Sf9) insect cells according to the Bac-to-Bac baculovirus expression system manual (Invitrogen). Budded viruses in the insect cell culture medium were centrifuged at 2000 × g for 10 min to remove cell debris, and concentrated by a second round of centrifugation at 28,000 × g for 60 min. Viral pellets were resuspended in 0.1 M phosphate-buffered saline (PBS) and their infectious titers (plaque-forming units) were determined by plaque assay on Sf9 cells.
For baculoviral transduction, cells were transduced in a minimal volume of medium at a multiplicity of infection (MOI) of 100 and left to incubate overnight at 37°C, 5% CO2. This was followed by a top-up of medium the next day, and a full change of the medium on day 2 posttransduction if transduced cells were cultivated for longer than 2 days.
Lentiviral vectors with CMV-W expression cassettes containing either the eGFP or HSVtk transgene were constructed in our previous study (Zhao and Wang, 2010). Briefly, expression cassettes were cloned into the pLenti6/v5-TOPO expression plasmid (Invitrogen), using the ViraPower lentiviral directional TOPO expression kit (Invitrogen). The pLenti6/v5-TOPO expression plasmid, containing either the expression cassette for eGFP or HSVtk, was cotransfected with pLP1, pLP2, and pLP/VSVG packaging plasmids into 293FT cells. Transfection was performed overnight, followed by a full medium change. Lentivirus was collected 48 hr posttransfection and concentrated at 75,000 × g, 4°C for 1 hr. Concentrated viral particles were resuspended in PBS and stored at −80°C or used immediately for transduction.
For lentiviral transduction, 3 million cells were suspended in 5 ml of medium and seeded into 100-mm dishes; 40 μl of concentrated virus was added to each dish while cells were still in suspension (MOI < 1). Transduction was allowed to take place overnight at 37°C and medium was topped up to 10 ml the next day. On the second day after lentiviral transduction, virus-containing medium was completely removed and cells were washed three times with copious amounts of PBS. This was followed by 2-week antibiotic selection for transduced cells, using medium containing blasticidin at 5 μg/ml (Invitrogen). Selected cells were expanded in normal medium and used for subsequent experiments.
Fluorescence-activated cell-sorting analysis and Western blotting
When fluorescence-activated cell-sorting (FACS) analysis was used to evaluate transduction efficiency, baculovirus-transduced cells were trypsinized to single cells and collected in PBS before analysis with a FACSCalibur flow cytometer (BD Biosciences). To examine the effects of viral transduction on surface marker expression, the cell pellet was resuspended in 1% bovine serum albumin (BSA)–PBS and incubated for 1 hr with phycoerythrin (PE)-conjugated primary monoclonal antibodies, including anti-CD34, anti-CD44, anti-CD73, anti-CD90, anti-CD105, and anti-CD166; allophycocyanin (APC)-conjugated anit-CD45; and their respective isotypes from BD Biosciences. After two washes with PBS, samples were resuspended in PBS for FACS analysis.
Western blotting was used to verify HSVtk gene expression after baculoviral or lentiviral transduction. MSCs transduced with the HSVtk baculoviral vector were lysed on day 2 posttransduction. Cell lysates were centrifuged at 10,000 × g for 10 min at 4°C. Samples were mixed with one-quarter volume of NuPAGE LDS sample buffer and one-tenth volume of NuPAGE reducing agent (Invitrogen), heated at 70°C for 10 min, and separated on 4–12% Bis-Tris gels with morpholineethanesulfonic acid (MES) running buffer. Electroblotting was performed with an iBlot dry blotting system (Invitrogen). The membrane was blocked in 5% nonfat milk in Tris buffered saline with Tween 20 (TBST) for 1 hr, and incubated overnight with goat polyclonal anti-HSVtk primary antibody (diluted 1:800; Santa Cruz Biotechnology, Santa Cruz, CA), and for 1 hr with mouse monoclonal β-actin primary antibody (diluted 1:1000; Sigma-Aldrich, St. Louis, MO). After three washes of 5 min each with TBST, the membrane was incubated with horseradish peroxidase (HRP)-conjugated anti-goat IgG or anti-mouse IgG (Abcam, Cambridge, UK) for 1 hr, followed by a second round of washing. Protein bands were visualized on X-ray film after a 10-sec exposure.
In vitro cytotoxicity assay
Baculovirus- or lentivirus-transduced hESC-MSCs were trypsinized and mixed with U87 glioblastoma cells at various ratios. The mixed cells were seeded on 96-well plates (1000 cells per well) and cultivated in MSC medium with ganciclovir. After 5 days, cell viability was examined by CellTiter 96 AQueous one solution cell proliferation assay (Promega).
In vivo animal study
Adult male BALB/c athymic, immuno-incompetent nude mice (weight, 20 g; age, 6–8 weeks) were used. For tumor inoculation, 0.1 × 106 human glioma U87-luc cells were injected into the right side of the striatum of anesthetized animals (designated as day 0).
To test baculovirus-transduced hESC-MSCs, tumor-inoculated animals were distributed into three groups (n = 8 per group) on day 8 after tumor inoculation and received an intratumoral injection of sample solution: PBS, PBS suspension of 2.5 × 105 MSCs transduced with a baculoviral vector carrying the eGFP gene, or PBS suspension of 2.5 × 105 MSCs transduced with a baculoviral vector carrying the HSVtk gene. Five microliters of sample solution was injected into each tumor xenograft, followed by daily intraperitoneal injection of ganciclovir (50 mg/kg body weight) for 2 weeks starting 1 day after MSC injection.
To test lentivirus-transduced hESC-MSCs, tumor-inoculated animals were similarly distributed into three groups (n = 10 per group) on day 5 after tumor inoculation. Sample solution consisting of PBS, PBS suspension of 1 × 106 MSCs transduced with a lentiviral vector carrying the eGFP gene, or PBS suspension of 1 × 106 MSCs transduced with a lentiviral vector carrying the HSVtk gene was injected into the left striatum at a site contralateral to the tumor inoculation site. Ten microliters of sample solutions was injected into the brain, followed by daily intraperitoneal injection of ganciclovir (50 mg/kg body weight) for 2 weeks starting 7 days after MSC injection.
To monitor bioluminescence signals of U87-luc cells, isoflurane gas-anesthetized animals were injected intraperitoneally with 200 μl of
Animal survival was monitored until all the animals were dead. All handling and care of animals was carried out according to the Guidelines on the Care and Use of Animals for Scientific Purposes (National Advisory Committee for Laboratory Animal Research, Singapore).
Statistical analysis
All data are presented as means ± SD. The statistical significance of differences was determined by unpaired Student t test, or by one-factor or two-factor analysis of variance (ANOVA) with replication followed by the Holm–Sidak method. The statistical analysis of survival data was performed by log-rank test followed by the Holm–Sidak method for pair-wise multiple comparison tests. A P value less than 0.05 was considered to be statistically significant.
Results
We used human MSCs generated from two hES cell lines, HuES9 and H1. Generation and characterization of HuES9-derived MSCs (HuES9-MSCs) were published previously (Lian et al., 2007), and H1-derived MSC-like cells (H1-MSCs) were generated under feeder-free conditions according to the method reported by Hwang and colleagues (2008). H1 embryoid bodies were derived and seeded onto gelatin-coated plates, after which fibroblastic outgrowth from the embryoid bodies was isolated after 10 days and expanded. These cells displayed MSC-like morphology, exhibited a surface marker profile characteristic of MSCs, expressing CD44, CD73, CD90, CD105, and CD166 but not CD34 and CD45 (Fig. 1). To determine whether the H1-MSCs were pluripotent and held differentiation potential, the cells were cultured in selection media to undergo adipogenesis, osteogenesis, or chondrogenesis. Adipogenic differentiation was induced in medium containing insulin, dexamethasone, phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine, and indomethacin; osteogenic differentiation was induced in medium containing dexamethasone, ascorbate, and β-glycerophosphate; and chondrogenic differentiation was induced in medium containing transforming growth factor (TGF)-β. Although H1-MSCs did not spontaneously differentiate during cell expansion, their differentiation into various cell types was observed in the selection media and confirmed by RT-PCR analysis of mesodermal lineage markers and specific staining (Fig. 2).

Characterization of H1 hESC-derived MSC-like cells.
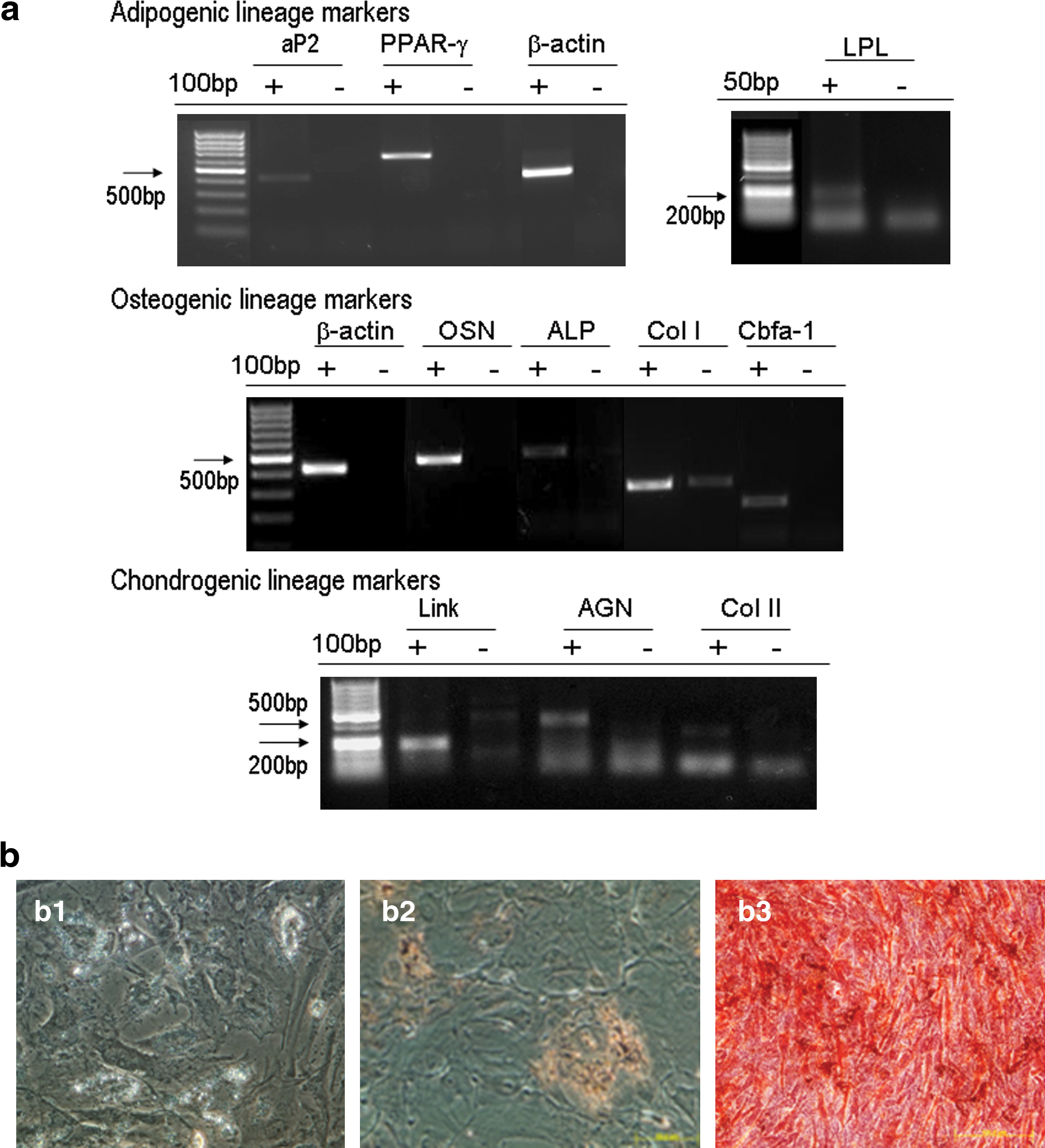
Differentiation of H1 hESC-derived MSC-like cells.
To examine the feasibility of using hESC-MSCs for targeted glioma gene therapy, we investigated whether HuES9-MSCs and H1-MSCs are similar to adult bone marrow MSCs in displaying tropism for tumors. Our results from in vitro Transwell cell migration assays using Boyden chambers showed that, compared with migration toward plain Opti-MEM medium (the blank), there was significantly increased migration of these MSC-like cells toward U87 glioma cells, increasing for HuES9-MSCs from 12% migrated cells toward the blank to 25% toward U87 cells and for H1-MSCs from 7% toward the blank to 12% toward U87 cells (Fig. 3a and b). The U87 cell-directed migration of H1-MSCs was comparable to that directed by Opti-MEM with 15% fetal bovine serum, a positive control for cell migration (Fig. 3a). These results from in vitro assays clearly suggest the tropism of hESC-MSCs toward some chemoattractants secreted by the U87 glioma cells.

Migration of hESC-MSCs toward glioma cells.
To investigate whether this tumor tropism could be observed in an in vivo setting, HuES9-MSCs were labeled with the red fluorescent dye CM-DiI and injected into the left hemisphere of mouse brains inoculated with green fluorescent dye CM-DiO-labeled U87 tumors in the right hemisphere. One week after injection, the mice were killed and their brains were collected for histological sectioning. Red fluorescent dye-labeled HuES9-MSCs were either observed to be en route toward the right brain hemisphere (Fig. 3c), or found located within the tumor (Fig. 3d). The migration tract in the brain appeared specific as the HuES9-MSCs in all three tested mice were observed to stream toward the right hemisphere.
Our previous study demonstrated that insect baculovirus-transduced bone marrow MSCs can be used as targeting vehicles for gene delivery into tumors (Bak et al., 2010). Baculoviral vectors typically mediate transient transduction with transgene expression lasting for days to weeks. To select a proper baculoviral vector that can sufficiently prolong transgene expression in hESC-MSCs for cancer gene therapy, we tested six baculoviral vectors containing different expression cassettes for the eGFP reporter gene in HuES9-MSCs. These vectors were developed previously in the laboratory and contained either the elongation factor (EF)-1α promoter, the EF-1α promoter with a cytomegalovirus (CMV) enhancer element, or the CMV promoter with or without the woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) (Fig. 4a). We observed that the vector containing the CMV promoter in combination with the WPRE gave the highest efficiency, with more than 90% eGFP-positive cells (Fig. 4b). Transgene expression from this baculoviral vector could be maintained at more than 80% in the first week after transduction, followed by a decline to less than 10% at the end of the second week, hence providing a transgene expression window of approximately 2 weeks (Fig. 4c).
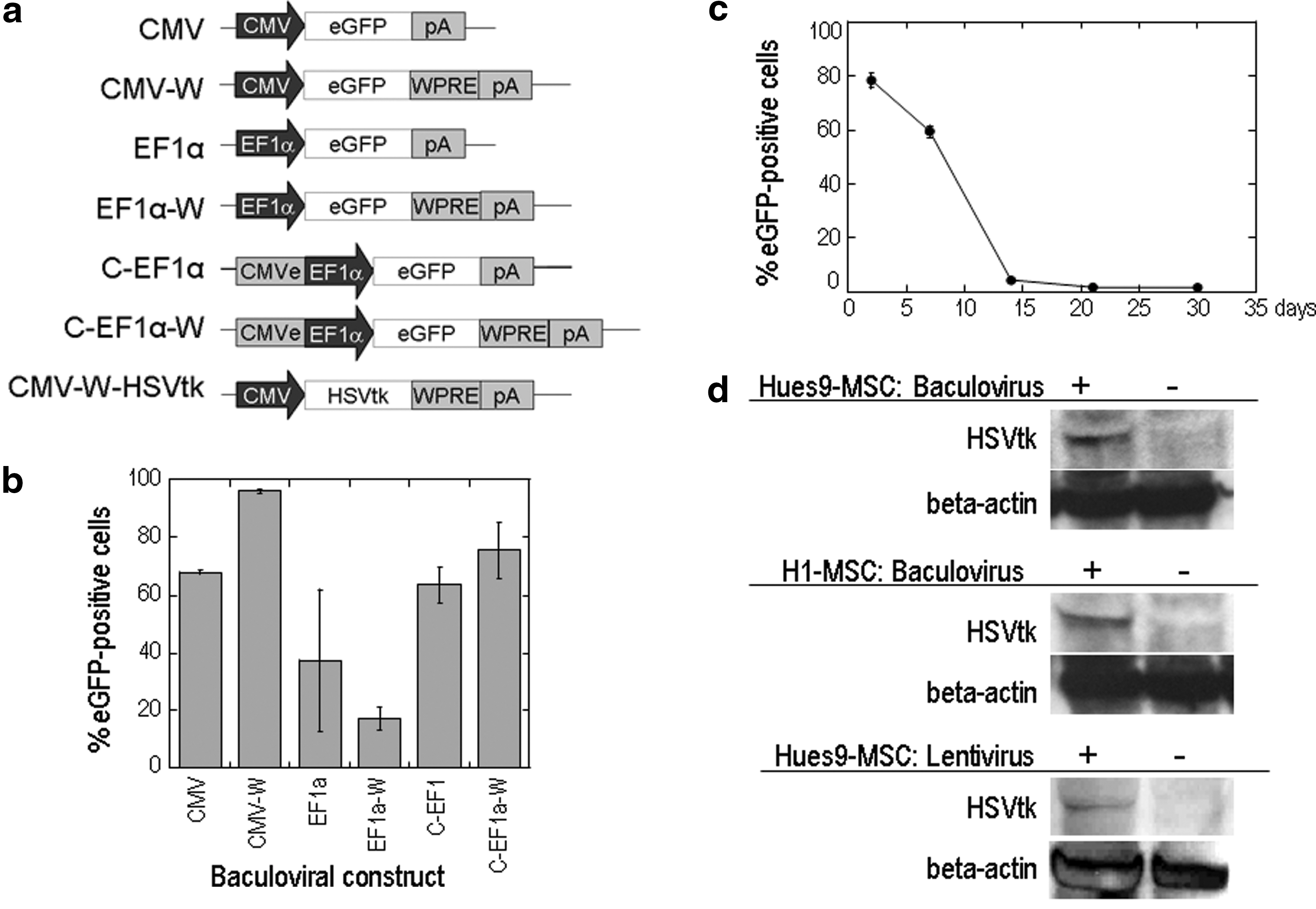
Viral vectors for transduction of hESC-MSCs.
In view of the previously described findings, we used a baculoviral vector with the CMV promoter and WPRE to drive the expression of our suicide gene of interest, the HSVtk gene, in hESC-MSCs (Fig. 4d). A lentiviral vector with the CMV promoter and WPRE was also constructed and tested for stable HSVtk expression in these cells (Fig. 4d). We checked whether viral transduction had any adverse effect on stem cell identity, using antibody staining and FACS analysis, and observed no significant differences in surface marker profile between the transduced and mock-transduced cells, which expressed CD44, CD73, CD90, CD105, and CD166 and did not express CD34 and CD45 (Supplementary Fig. S1; supplementary data are available online at
The tumor tropism property of hESC-MSCs, along with their capacity to be efficiently loaded with the HSVtk transgene, prompted us to test whether these loaded cells could efficiently kill cancer cells when coupled with the HSVtk–ganciclovir system. This system is notable for exhibiting a strong bystander effect by transferring the toxic phosphorylated form of ganciclovir from HSVtk-expressing cells to nearby unmodified tumor cells, thus resulting in the inhibition of DNA replication in these cells and their eventual death (Mesnil and Yamasaki, 2000; Asklund et al., 2003). To test the bystander effect in vitro, we investigated the cytotoxicity effect mediated by mixing U87 glioma cells with HSVtk-expressing hESC-MSCs at various ratios. Whereas 10 μM ganciclovir caused a negligible killing effect on U87 cells in the absence of HSVtk-expressing hues9-MSCs or H1-MSCs (100:0 ratio), this concentration of ganciclovir induced obvious cell death in U87 cells in the presence of HSVtk-expressing cells, regardless of whether the HSVtk gene was introduced by a baculoviral or lentiviral vector. The tumor cell-killing effect increased when the cell-mixing ratio between U87 and HSVtk-expressing cells was altered from 90:10 to 50:50 (Fig. 5a, c, and e). Cell death in the 0:100 groups provided direct evidence that HSVtk-expressing MSCs were susceptible to the cytotoxic effects of ganciclovir. When baculoviral vectors were used to introduce the HSVtk gene into hESC-MSCs, we noted a tumor cell-killing effect even when HSVtk-expressing cells transduced 8 days previously were mixed with U87 cells (Fig. 5a–d), providing evidence for the presence of HSVtk in baculovirus-transduced hESC-MSCs for at least 8 days. To test the dose-dependent cytotoxic effects of ganciclovir, U87 cells were mixed with HSVtk-expressing cells at a cell-mixing ratio of 50:50 and treated with ganciclovir at concentrations ranging from 0 to 100 μM. The drug induced a pronounced cytotoxic effect at a concentration as low as 1 μM, and gave rise to an increased killing effect at higher concentrations, particularly when HSVtk-expressing cells that have been transduced with baculovirus 8 days previously or with lentivirus were used (Fig. 5b, d, and f). There was no obvious cytotoxicity to untransduced hESC-MSCs at all ganciclovir concentrations employed (Fig. 5b, d, and f).

In vitro tumor cell-killing effects of suicide gene-expressing hESC-MSCs. Bystander-killing effects were assessed in a coculture system by mixing U87 glioblastoma cells with HuES9-MSCs or H1-MSCs. A baculoviral vector expressing HSVtk (BVTK) was used for transduction of HuES9-MSCs or H1-MSCs in
We next tested the tumor-killing efficacy of these HSVtk-expressing cells in in vivo settings. U87 cells stably expressing luciferase (U87-luc) were injected into the right striatum of nude mice to establish tumor xenografts. In our first in vivo experiment, baculovirus-transduced, HSVtk-expressing HuES9-MSCs (BVTK-MSCs) were injected directly into the tumors. HuES9-MSCs transduced with a baculoviral vector expressing eGFP (BVeGFP-MSCs) and PBS were, respectively, injected into the tumors as controls. Ganciclovir was administered to the mice by daily intraperitoneal injection. Our previous study (Zhao and Wang, 2010) demonstrated that ganciclovir alone, at the tested dose and in the tested mouse species, has no effects on glioma growth. When the IVIS imaging system was employed for bioluminescence imaging of U87-luc cells in living animals, we observed a slow tumor growth rate in the BVTK-MSC group (Fig. 6a). Bioluminescence photos taken on days 9 and 25 after tumor implantation revealed tumor shrinkage in three of eight animals in this group and a slight increase in tumor bioluminescence for the other five animals, as opposed to the great increase in tumor bioluminescence in the two control groups (Fig. 6a). Quantitative analysis of the bioluminescence signals indicated that mice injected with BVTK-MSCs had 10-fold lower tumor luciferase expression than mice in the two control groups on day 25 after tumor implantation (Fig. 6b). This slowed tumor growth prolonged animal survival by approximately 10 days (Fig. 6c), increasing the maximal survival time of 36 days in the PBS group and 38 days in the BVeGFP-MSC group to 48 days in the BVTK-MSC group (P = 0.0000925 and 0.00222, respectively).

In vivo glioma therapy using hESC-MSCs transduced with baculoviral vectors and injected directly into an established human glioma. Mice were inoculated via their right striatum with U87-luc glioma cells. HuES9-MSCs transduced with a baculoviral vector expressing HSVtk (BVTK-MSCs) were injected into established tumor xenografts. Mice injected intratumorally with HuES9-MSCs transduced with a baculoviral vector expressing eGFP (BVeGFP-MSCs) or with PBS served as controls. Ganciclovir was administered to the mice via intraperitoneal injection right after intratumor injection of MSCs or PBS.
We then investigated in vivo targeted delivery after injection of HSVtk-expressing hESC-MSCs into the left striatum, contralateral to the tumor inoculation site, on day 5 after tumor inoculation. Ganciclovir injection started 7 days after MSC injection, providing the cells ample time to migrate from the contralateral side to the tumor side. We used lentivirus-transduced HuES9-MSCs (LVTK-MSCs) in this experiment as they provide a broadened HSVtk expression window. Histological examination of mouse brains collected 30 days after tumor inoculation revealed significant tumor tissue necrosis in the group of mice that received LVTK-MSC treatment and extensive tumor growth in the control groups of mice that were injected with either eGFP-expressing MSCs or PBS (Fig. 7a). Whereas most of the mice in the control groups died on about day 30, the maximal survival time of the LVTK-MSC group was significantly prolonged to 40 days (P = 0.00174 vs. the PBS control and 0.00088 vs. the LVeGFP-MSC control; Fig. 7b).
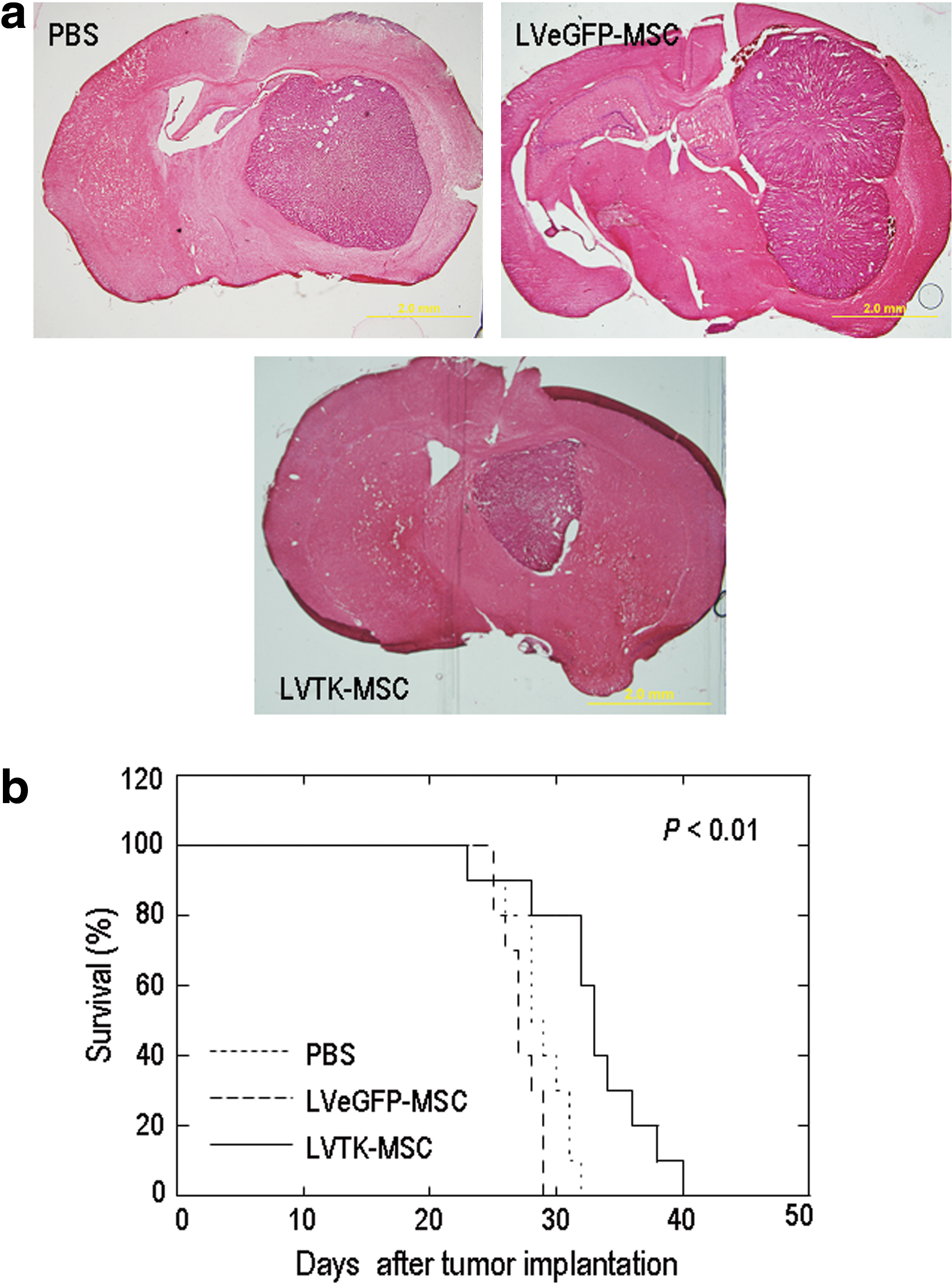
In vivo glioma therapy using hESC-MSCs transduced with lentiviral vectors and injected into the cerebral hemisphere opposite an established human glioma. Mice were inoculated with U87-luc glioma cells via their right striatum. HuES9-MSCs transduced with a lentiviral vector expressing HSVtk (LVTK-MSCs) were injected into the cerebral hemisphere opposite established tumor xenografts. Mice injected with a lentiviral vector expressing eGFP (LVeGFP-MSCs) or with PBS in the same way served as controls. Ganciclovir was administered to the mice via intraperitoneal injection right after brain injection of MSCs or PBS.
Discussion
In this study, we successfully demonstrated the use of hESC-MSCs as therapeutic gene delivery vectors in cancer treatment. hESC-MSCs can be easily differentiated and isolated from embryonic stem cells, and also have a greater life span in vitro as well as a faster proliferation rate than adult MSCs. HuES9 embryonic stem cell-derived MSCs, for instance, can be passaged up to at least 35 population doublings with a fast population doubling time of 72 hr, enabling large-scale derivation of cells within a short period of time (Lian et al., 2007). Interestingly, consistent with reports describing adult MSC migration, the hESC-MSCs we tested in this study also possess the ability to migrate toward tumors both in vitro and in vivo. One week after injection, hESC-MSCs injected into the left brain hemisphere of mice were found streaming toward, or located within, tumor inoculated in the right brain hemisphere. Adult MSCs are well known to home into sites of injury to facilitate wound healing and tissue regeneration. It has been hypothesized that the tumor-tropic property of adult MSCs could be attributed to the tumors secreting factors that mimic those from damaged tissues (Hall et al., 2007). Platelet-derived growth factor (PDGF)-BB, epidermal growth factor (EGF), stromal cell-derived factor (SDF)-1α, and vascular endothelial growth factor (VEGF)-α have been identified as some possible chemoattractants that could have triggered adult MSC tropism for tumors (Nakamizo et al., 2005; Schichor et al., 2006; Menon et al., 2007). The upregulation of matrix metalloproteinase (MMP)-2, tissue inhibitor of metalloproteinase-2, and the Wnt target genes such as membrane type 1 MMP and cyclin D1 is also responsible for enhancing adult MSC invasiveness (Neth et al., 2006; Ries et al., 2007). We postulate that some of these factors might be responsible for hESC-MSC tumor tropism. Further evaluation will need to be done to confirm the exact migratory mechanism.
Our in vitro killing data suggest that both HuES9 and H1 embryonic stem cell-derived MSCs (HuES9-MSCs and H1-MSCs, respectively) exhibit a good bystander-killing effect on U87 glioma cells when they were used 3 days after transduction with baculovirus containing the HSVtk expression cassette. This is similar to the results we obtained in our previous paper, in which bone marrow-derived MSCs were transduced with the same baculoviral vector and tested for their bystander effect on U87 glioma cells (Bak et al., 2010). However, whereas the bystander effect mediated by baculovirus-transduced bone marrow (BM)-derived MSCs was enhanced when the cells were used 8 days after transduction, the bystander effect for baculovirus-transduced HuES9-MSCs and H1-MSCs diminished. We hypothesized that the faster proliferation rate of HuES9-MSCs and H1-MSCs could have led to a rapid dilution of the baculoviral genome, HSVtk transcripts, and/or HSVtk protein in the cells, causing the drop in killing effect. In view of the small window of time for effective bystander effect, we injected baculovirus-transduced HuES9-MSCs into tumors directly to assess their in vivo tumor-killing efficacy. The results of this experiment are comparable to those achieved by other groups using adenovirus to confer IFN-β or IL-2 expression in MSCs for intraglioma gene delivery (Nakamura et al., 2004; Nakamizo et al., 2005), suggesting that baculovirus provides efficiency similar to adenovirus as a transient gene delivery vector.
Compared with baculovirus-transduced cells, lentivirus-transduced HuES9-MSCs appeared to give a weaker tumor-killing effect in our in vitro studies. We hypothesized that this is due to the combined effect of the much lower MOI (less than 1) used during lentiviral transduction, which caused lower level HSVtk protein expression in the lentivirus-transduced cells, and the short period of 5 days over which the in vitro killing effect was assessed. As lentivirus-transduced HuES9-MSCs give long-term HSVtk expression, we decided to inject them at a site contralateral to the U87 tumor inoculation site to test their in vivo tumor-targeting capacity. The results from this experiment clearly indicate that the lentivirus-transduced, HSVtk-expressing HuES9-MSCs were capable of migrating to a remote tumor site and exerting a tumor-killing effect there when coupled with the HSVtk–GCV system.
Despite the long-term expression of HSVtk, our lentivirus-transduced hESC-MSCs were unable to completely terminate tumor growth or eliminate tumor volume in this study. Drawing inference from our migration experiment, in which not all hESC-MSCs were found within the tumor 7 days after injection into a contralateral site, we propose that the amount of HSVtk-expressing MSCs that eventually reached the tumor could be too little to give a killing effect that can overcome the rate of tumor growth. Nakamizo and colleagues (2005) estimated that there was about 25% MSC engraftment into gliomas inoculated into mouse brains after injection of IFN-β-expressing BM-MSCs into the ipsilateral carotid artery. In a contralateral injection model, it is hence reasonable to infer that the long distance over which the MSCs had to migrate would likely lead to an even lower percentage of MSC engraftment into the glioma. One way to improve the killing prowess of this method would be to enhance hESC-MSC migration toward tumors. Clinical low-dose irradiation of murine tumors has been reported to enhance the engraftment of circulating murine MSCs (Klopp et al., 2007), and is one avenue that can be explored and considered. Another way could be to shorten the migratory route of MSCs to the tumor masses. Bexell and colleagues (2009) reported that a single intratumoral injection of MSCs is enough to track down 72 ± 14% of glioma extensions, and 32 ± 6% of distant microsatellites, suggesting that intratumoral injection is an effective mode of stem cell delivery for tumor infiltration.
The natural ability of lentivirus to stably integrate transgenes into the genomes of dividing and nondividing cells makes it a preferred vector for studies requiring long-term transgene expression. It has been used to successfully introduce antitumor agents in other stem cell-coupled studies, such as tumor necrosis factor (TNF)-α against prostate cancer (Zhang et al., 2011), pigment epithelium-derived factor against hepatocellular carcinoma (Gao et al., 2010), and a secreted version of TNF-α-related apoptosis-inducing ligand against glioblastoma (Menon et al., 2009). The use of lentivirus is, however, not without concerns. Derived from the HIV-1 vector, its use poses the risk of subsequent viral replication and viral infection in host cells. Safety features incorporated into the design of the replication-defective lentiviral vector have sought to address these issues. They include a multiplasmid packaging system to minimize the chance of generating replication-competent lentiviruses through spontaneous homologous recombination between the plasmids, the removal of accessory genes crucial to lentiviral pathogenicity, and the deletion of promoter and enhancer elements in the U3 region of the lentivirus transfer vector, which addresses the issues of homologous recombination, insertional oncogenesis, and mobilization of the integrated virus on host infection with wild-type virus (Pauwels et al., 2009). Yet, despite the safety features, the preferential integration of the lentiviral vector into parts of the host genome containing transcriptionally active genes could still potentially give rise to loss or enhancement of host gene function, causing adverse effects when critical genes such as growth-controlling genes, tumor suppressor genes, or oncogenic genes are affected (Schroder et al., 2002; Hacein-Bey-Abina et al., 2003; Liu et al., 2006).
Insect baculovirus bypasses the risk of virus replication and potential viral infection in host cells, risks borne by conventionally used animal viruses such as adenovirus, retrovirus, and adeno-associated virus. A large cloning capacity, ease of virus production, and low cytotoxicity to transduced cells are other advantages associated with the emergence of baculovirus in the gene delivery field (Hu, 2008). The transient gene expression mediated by nonintegrating baculovirus, however, limits its efficacy in studies that necessitate long-term transgene expression. This limitation is not unique to baculovirus, but is also faced by adenovirus, another transient gene delivery vector. Oncolytic adenoviruses have emerged as a possible alternative to prolong transgene expression from a single round of adenoviral infection. Preexisting host immune response against adenoviral vectors and cytotoxicity inflicted by secondary adenoviral infection on the carrier, however, restrict the efficacy of this approach (Sonabend et al., 2008; Tyler et al., 2009; Ahmed et al., 2010). Unlike adenovirus, baculovirus is not targeted by preexisting immunity in humans (Strauss et al., 2007). Baculovirus-transduced MSCs has also been shown to not elicit any immune response in immunocompetent recipients (Chuang et al., 2009). To overcome the limitation of transient gene delivery for long-term studies, ongoing research with baculovirus has seen the development of a hybrid baculoviral vector system that can prolong baculovirus-mediated transgene expression beyond 63 days (Lo et al., 2009). We postulate that incorporation of this baculoviral vector design into future work will help extend the period of HSVtk expression from hESC-MSC delivery vectors for an improved tumor-killing effect.
More research will also need to be done to define the properties of hESC-MSCs. One particular issue concerns culture purity, as the presence of rogue undifferentiated embryonic stem cells in a culture could potentially cause tumorigenesis in downstream applications. Immunogenicity is another issue. Although hESC-MSCs have been shown to exhibit the same immunosuppressive effects as adult MSCs (Trivedi and Hematti, 2008; Yen et al., 2009), the eventual fate of hESC-MSCs after tumor targeting and tumor treatment remains unclear. There may be cells that escape the toxic effects of the HSVtk–ganciclovir system by staying quiescent or differentiating in the brain. The latter prospect may bring about the generation of terminally differentiated cells that are not immunosuppressive, and that may be targeted by the host immune system. Also, among those using MSCs for glioma therapy, controversy has risen over the effects of adult MSCs on tumor growth. In one study, native rat BM-MSCs were reported to suppress the growth of rat glioma (Nakamura et al., 2004). In another, human adipose-derived MSCs were reported to promote the viability of human glioma cells in vivo (Yu et al., 2008). To further confuse matters, Bexell and colleagues (2009) reported that adult MSCs, in fact, have no effects on tumor growth. Thorough investigation into the effects of hESC-MSCs on glioma can shed more light on the actual effect of MSCs on these tumors, and more importantly, validate the suitability of these cells as delivery vectors to tumors.
In conclusion, we have shown that MSCs derived by different methods from two human embryonic stem cell lines could be recruited to tumors, and could bring about a killing effect when coupled with the HSVtk–ganciclovir prodrug cancer gene therapy system. The ease with which large amounts of these cells could be derived and genetically modified makes them an attractive option for stem cell-based gene therapy. The potential of this approach is not limited to hESC-MSCs but can likely be extended to MSCs derived from induced pluripotent stem cell lines, an exciting venture that does not just provide a source for large-scale derivation of MSCs, but also bypasses the ethical and allogeneic issues posed by hESC-MSCs.
Footnotes
Acknowledgments
This work was supported by the Institute of Bioengineering and Nanotechnology, the Biomedical Research Council, and the Agency for Science, Technology, and Research (A*STAR) in Singapore and by grants from the Ministry of Education of Singapore (T206B3110) and the National Medical Research Council in Singapore (NMRC/1203/2009).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.