
Abstract
The promise of the RNA interference (RNAi) technology is equally dependent on the efficiency and stability of gene silencing. The aim of the present study was the development of foamy virus (FV) vectors for stable RNAi, utilizing two potent RNA polymerase III (Pol III) promoters. Using green fluorescent protein as a target gene, we examined the efficiency of mouse U6 (mU6) and human H1 Pol III promoters in different human cell lines and mouse hematopoietic stem cells (HSCs) ex vivo and in vivo, following bone marrow transplantation. Both our mU6 and H1 FV vectors mediated very efficient gene silencing with as low as one vector copy per cell. However, transduction of human cell lines with FV vectors expressing short hairpin RNA from mU6 led to the gradual elimination of cells in culture, as opposed to H1-harboring cells, underscoring the importance of the expression system or cellular context in the evaluation of the overall RNAi effects. The efficiency and stability of the H1 vectors were further shown by the successful silencing of BCR-ABL in K562 cells. Accordingly, mU6 vectors induced efficient and stable gene silencing in mouse HSCs following bone marrow transplantation. Our work is the first in vivo study on the efficiency and stability of RNAi gene silencing in HSCs with FV vectors, currently a safe alternative for viral gene transfer.
Introduction
Along with the numerous studies that demonstrate the extensive potential of RNAi technology, there has been a significant accumulation of reports on the pitfalls of RNAi-based approaches, sparking the discussion on the validity of gene-silencing experiments. Common limitations include the induction of innate immune responses (Sledz et al., 2003), saturation of the intracellular RNAi processing machinery (Grimm et al., 2006), and off-target silencing effects (Jackson and Linsley, 2004). The extent of such phenomena depends on the particular RNAi sequence itself (Jackson and Linsley, 2004; Scacheri et al., 2004), the expression system deployed (Bridge et al., 2003; An et al., 2006; McBride et al., 2008), and the cell-type specific ability to buffer perturbations to the physiological microRNA (miRNA)/RNAi mechanism (Grimm et al., 2006). Such reports question the therapeutic potential of systems aiming at a sustained gene-silencing effect, highlighting the need for optimal experimental planning and careful controlling to avoid unintended adverse effects.
Foamy viruses (FVs) have been used successfully for the silencing of disease-related and viral genes, encoding shRNAs against the simian immunodeficiency virus rev/env (Park et al., 2005), the S and X genes of the hepatitis B virus (Sun et al., 2007), against the HBsAg from the human H1 promoter (Moore et al., 2005), and against HIV proteins from the human U6 RNA Pol III promoter (Taylor et al., 2008). FVs, belonging to the Spumaviridae subfamily of retroviruses, have distinct features compared with the widely deployed gammaretro- or lentiviruses and are considered a promising vehicle for viral gene transfer. Among their main advantages are the lack of pathogenicity, the persistent long-term expression in transduced cells with low copy numbers, and a favorable integration profile (Rethwilm, 2007).
The present study focuses on the development of an FV-based system for stable RNAi expression using the mouse U6 (mU6) and human H1 RNA Pol III promoters both in vitro and in vivo. Initially, we demonstrated the efficiency of our vectors against a green fluorescent protein (GFP) transgene in human cell lines, both short- and long-term, as well as the efficiency of the H1-FV constructs against an endogenous target, the BCR-ABL oncogene. In addition, we used the mU6-FV vectors to efficiently silence GFP in primary mouse hematopoietic stem cells (HSCs), following bone marrow (BM) transplantation (BMT). Collectively, our experiments allowed us to comparatively evaluate our FV-RNAi vectors with the two different Pol III promoters and further identify an mU6 promoter-related cytotoxic RNAi effect, in human cell lines. This study comprises a detailed report on the use of shRNA expression systems from FVs and reports for the first time on the efficient use of an FV-RNAi system for stable gene silencing in vivo.
Materials and Methods
Cell lines
All adherent cell lines were grown in Dulbecco's modified Eagle's medium, and suspension lines in RPMI, both supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin (all from Invitrogen, San Diego, CA), at 37°C in a 5% CO2, 100% humidified atmosphere. The GFP-expressing lines were generated after transduction with GFP-expressing FV vectors, followed by fluorescence cell sorting of the GFP+ cell pools (BD FACSAria; BD Biosciences, Franklin Lakes, NJ).
shRNA sequences and cloning
We have designed all shRNA molecules according to currently accepted rules for siRNA design (Khvorova et al., 2003; Mittal, 2004; Reynolds et al., 2004). Detailed sequences of all the shRNAs used in the study are given below. The oligonucleotides contained a sense target sequence, the published loop sequence TTCAAGAGA (Brummelkamp et al., 2002), the antisense sequence, a termination sequence of five thymidines, and appropriate sequences for restriction enzymes at both ends, to allow cloning into the human H1 or mU6 promoters. Specifically, the target sequence for GFP was GCAAGCTGACCCTGAAGTTCA, based on a previous publication (Tiscornia et al., 2003), and the target sequence for BCR-ABL was GCAGAGUUCAA-AAGCCCUU, as published (Rangatia and Bonnet, 2006). All mU6-encoded shRNA sequences begin with a G at the U6+1 sequence to assure efficient transcription (Pebernard and Iggo, 2004). All the shRNA sequences were chemically synthesized as forward and reverse complement of 56–63-nucleotide (nt) oligos (Sigma-Genosys, The Woodlands, TX), annealed in vitro, and ligated back to the respective restriction sites of the Pol III promoters. All plasmids were verified by sequencing. The oligos designed were as follows: GFP: sense GCAAGCTGACCCTGAAGTTCA and antisense TGAACTTCAGGGTCAGCTTGC (Tiscornia et al., 2003); NS1: sense GTTCTCCGAACGTGTCACGT and antisense ACGTGACACGTTCGGAGAAC (Qiagen no. 1022076); NS2: sense CTTCATTGTCGGCATGGGT and antisense ACCCATGCCGACAATGAAG (McLaughlin et al., 2007); and BCR-ABL: sense GCAGAGTTCAAAAGCCCTTCA and antisense TGAAGGGCTTTTGAACTCTGC (Rangatia and Bonnet, 2006).
Construction of shRNA-expressing foamy vectors
All pΔΦ are safety-enhanced third-generation FV vectors that do not produce replication-competent viruses (Trobridge et al., 2002a,b). The FV vector plasmid pΔΦmU6F, harboring the mU6 promoter, was a gift from D.W. Russell (University of Washington, Seattle, WA). The H1 promoter (NR_002312, nt 145–486) was amplified by PCR from the commercially available pSUPER vectors (OligoEngine, Seattle, WA) and inserted as an NcoI fragment in the exact same position as the mU6. For H1 amplification, the sense primer was ATCCCATGGAATTCGAACGCTGACGTC and the reverse ATCCCATGGACGCGTGTCGACGGTATCGA. All shRNA oligos were cloned as BglII-MluI fragments at the H1 promoter.
For the double mU6-shRNA–expressing vectors, the mU6 promoter was cloned within the delU3 region of the 3’ long terminal repeat (LTR), as an XbaI fragment. Cloning was performed in an NdeI-PstI deletion intermediate of the standard pΔΦ vector backbone. FV vector plasmids containing marker gene cassettes harbor either the GFP or the deleted low-affinity nerve growth factor receptor (ΔNGFR) genes under regulation of the murine stem-cell virus (Mscv) promoter, denoted as MscvF or MscvN (MF/N), respectively. The pΔΦMscvF vector (Trobridge et al., 2002a,b) was used for subsequent cloning of the MF cassette to all our FV-RNAi backbones as an EcoRI-NotI fragment, within the multiple cloning site. The ΔNGFR gene was cloned from plasmid pBBSNF2’hmp (kind gift from R. Richards, University of Washington), as an AgeI-NotI fragment, with AGTCACCGGTATGGGGGCAGGTG forward and ACTATGCGGCCGCCTAGACCTCTTG reverse primers.
Vector production and titration
Vector stocks were produced by calcium phosphate cotransfection of 293T cells with three distinct packaging plasmids coding the gag, pol, and env accessory genes, as previously described (Vassilopoulos et al., 2001; Trobridge et al., 2002a,b). The vector supernatants were collected 72 hr post transfection, centrifuged at 200 g for 10 min, and filtered through 0.45-mm Durapore filters (Millipore, Bedford, MA). Viral stocks were further concentrated by centrifugation at 20,000 rpm for 4 hr at 20°C and resuspended in appropriate culture medium for transduction. Biological stock titers were determined on HeLa or HT1080 human cells and were calculated by flow cytometry (FCM) as GFP or NGFR transducing units per milliliter of supernatant (TU/ml), according to marker gene expression from the MF or MN marker cassettes. Cell line transductions at low multiplicity of infection (MOI; 1–2) were carried out in six-well plates.
Real-time PCR
We developed a real-time PCR method for the determination of provirus copy numbers, adapted from a previous study (Kiem et al., 2007). Genomic DNA from transduced cells was extracted by standard protocol (Sambrook et al., 1989), and 250 ng was amplified in duplicate 25-μl reactions using an ABI Prism 7000 instrument (Applied Biosystems, Foster City, CA). For FV genome detection, the Fermentas SYBR Green Master Mix reagent (Fermentas Inc., Glen Burnie, MD) was used with primers CTGGAATGTTACTCAAAGAGCTGTTT and TGGAACAGGATGCTGCATTCT, whereas human RNase P gene or rodent glyceraldehyde-3-phosphate dehydrogenase was used for the determination of sample diploid cell number, as previously described (Andrianaki et al., 2010). Our negative control was DNA from untransduced cells, animal BM cells before BMT, and water. Proof-of-principle experiments that comprised the transduction of HeLa cells with an equal amount of viral supernatant from the different vectors showed an almost linear correlation of GFP expression and integrated viral vector copies (R 2 = 0.9174), supporting the validity of our methodology (data not shown).
Flow cytometry and fluorescence microscopy
Transduction efficiency, as determined by the percentage of GFP/ΔNGFR–expressing cells, and mean fluorescence intensities (MFIs) were measured at various time points on a Becton Dickinson FC500 flow cytometer. ΔNGFR expression was measured using an anti-NGFR-phycoerythrin (PE) antibody (BD Pharmingen, Athens, Greece). Exclusion of dead cells was based on propidium iodide or 7-aminoactinomycin D staining (Sigma) and subsequent acquisitions after gating on live cells. For detection of apoptosis, cells were washed two times in phosphate-buffered saline, and 1 × 105 cells were stained per sample, using the Annexin V-PE reagent (BD Pharmingen) as per the manufacturer's instructions.
Bone marrow transplantation studies
All C57Bl/6J and GFP-transgenic (JAX Mice Database: strain 003291-C57BL/6-Tg(CAG-EGFP)1Osb/J; The Jackson Laboratory, Bar Harbor, ME) animals were kept at the Biomedical Research Foundation of the Academy of Athens (BRFAA) Animal Facility. The study was performed in the animal facility of the Center for Experimental Surgery of the BRFAA. The facility is registered as a “breeding” and “experimental” facility by the Veterinary Service of the Prefecture of Athens according to Presidential Decree 160/91 in accordance with the European Directive 86/609/EEC for the protection of animals used for experimental and other scientific purposes (EL25BIO1 and EL25BIO3, respectively). The study was reviewed and approved by the competent Veterinary Service of the Prefecture of Athens (ref. number K715/2010) in accordance with the national existing legislation referred above.
For BMT studies, we performed partial myeloablative conditioning, adapted from a published protocol (Hsieh et al., 2007). Eight- to 12-week-old female recipients were injected intraperitoneally with Busilvex at 20 mg/kg for 4 consecutive days prior to BMT. Sex-mismatched BMTs were performed using lineage-depleted (Lin–) BM cells (StemSep Mouse Hematopoietic Progenitor Cell Enrichment Kit; Stem Cell Technologies, Vancouver, BC, Canada) according to the manufacturer's protocol. Transduction was performed at MOI = 10 for 18–20 hr on RetroNectin-coated plates (Takara Biomedicals, Otsu, Japan) in StemSpan serum-free medium (Stem Cell Technologies) supplemented with 3% fetal calf serum, 100 ng/ml Flt-3 ligand, 100 ng/ml murine stem cell factor, and 50 ng/ml human interleukin-6 (all Peprotec, London, UK). Following transduction, the cells were either plated in methylcellulose colony assays (Methocult H 4230; Stem Cell Technologies) or administered intravenously for BMT studies (0.5–1.5 × 105 per mouse). Donor cell engraftment was determined on animal BM upon termination of the experiments by real-time PCR for the mouse single-copy testis-specific Y pseudogene (TSPY) (Wang et al., 2002).
Statistical analysis
Two-tailed Student's t test was performed to assess the level of significance between different mean values, using GraphPad software. Each assay analyzed examined at least n = 3 independent experiments.
Results
Efficient production and stable target cell transduction from FV-RNAi vectors
To test the efficiency and stability of RNAi with FV vectors, we cloned RNA Pol III promoter cassettes (human H1 or mU6) (Fig. 1A) in the standard FV ΔΦ backbone, where the majority of the viral cis-acting sequences and a significant portion of the U3 region have been deleted (Trobridge et al., 2002a,b). Our vector constructs were engineered to coexpress the GFP or the deleted low-affinity NGFR marker genes from the Mscv LTR promoter (designated as MF or MN, respectively). We also constructed a double-copy mU6 vector, through the insertion of the shRNA-expressing cassette within the U3 region of the 3’LTR, thus generating two shRNAs per integrated provirus (pΔΦ2mU6.MF/N) (Tiscornia et al., 2003). The titers of the crude FV vector stocks reached 105 TU/ml, similar to the standard Mscv-GFP vector (MF), indicating that the presence of the mU6 or H1 Pol III promoters, with or without expression of the nonspecific hairpin (shNS), did not affect vector production and packaging (Fig. 1B). This also indicates the lack of vector genome self-targeting by the expressed shRNAs, which could occur due to the existence of the hairpin-coding sequence within the vector. Furthermore, cloning of the H1 or mU6 promoter in forward (H1f.MF, mU6f.MF) or reverse orientation (H1r.MF, mU6r.MF) relative to FV vector transcription did not affect vector titers (data not shown). Compared with the standard MF vector, a titer drop of 28% and 19% was observed for the double-copy vectors 2mU6.MF and 2mU6shNS.MF, which was not statistically significant (p = 0.07 and p = 0.8, respectively). As a proof of principle for the silencing potential of our FV vector system, we cloned into a GFP-expressing vector an anti-GFP hairpin (Tiscornia et al., 2003) (mU6shGFP.MF). As opposed to the negative control shNS, the expression of shGFP resulted in significantly reduced titers (6 × 104 TU/ml) compared with those of the standard MF and shNS.MF vectors (1.78 × 105 and 2.02 × 105 TU/ml, respectively), because the expressed shRNAs target the GFP-containing vector genome in the packaging cells (Fig. 1C). This titer drop was also indirect evidence for the efficiency and specificity of the shRNA against GFP. To compare the transduction efficiency and stability of our FV-RNAi vectors in the absence of hairpin expression, we transduced HeLa cells and measured the GFP expression over time in culture. Comparable gene transfer efficiency (20–55% GFP+ cells) and expression were observed for all constructs, whereas the percentage of GFP-expressing cells was stable over time (Fig. 1D).
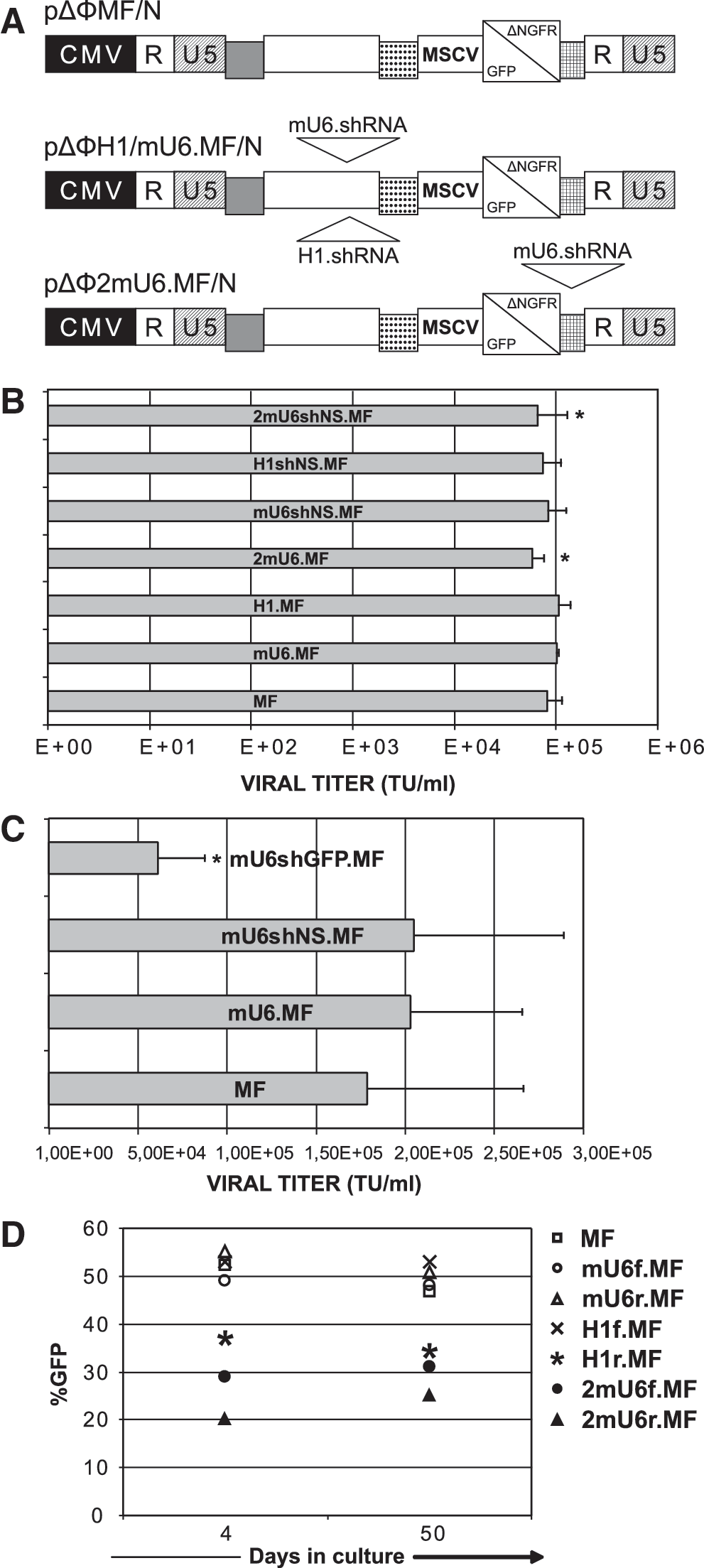
Construction and efficient transduction of target cells with FV-RNAi vectors.
Efficient short-term silencing by H1 and mU6 FV constructs in human cell lines
To study the gene-silencing potential of our FV vectors, we cloned the shRNA against GFP in FV vectors harboring the mU6 or H1 promoter cassettes without any reporter genes. We then transduced human cell lines (293T, HT1080, and HeLa) stably expressing GFP from an integrated FV vector, with low MOI (0.5–2) and monitored changes in the percentage of GFP+ cells and in the MFI levels by FCM. At day 4 post transduction and in all three cell lines tested, we observed a reduction in the GFP+ cells (Fig. 2A) and a pronounced effect in GFP down-regulation, as evidenced by MFI reduction (Fig. 2B). Overall, our results showed that the FV-RNAi effect was more evident as MFI down-regulation and less as a decrease in the fraction of the GFP+ cells. Relative to the control, untransduced cells, we observed GFP MFI levels that ranged from 82% to 37% of the original, despite the low MOI (Fig. 2B). Regarding promoter efficiency, both mU6 and H1 performed comparably. However, there was a statistically significant difference in MFI reduction (p < 0.01) in HT1080 cells (Fig. 2B), where the H1 promoter was more efficient than the mU6 (55% versus 33%, respectively), an effect that was offset when the latter was in the double-copy configuration (2mU6). Comparing vector performance in the different cell lines tested reveals that the 293T cells are the least amenable to RNAi manipulation, indicating the existence of cell-specific elements that also affect the silencing potential.
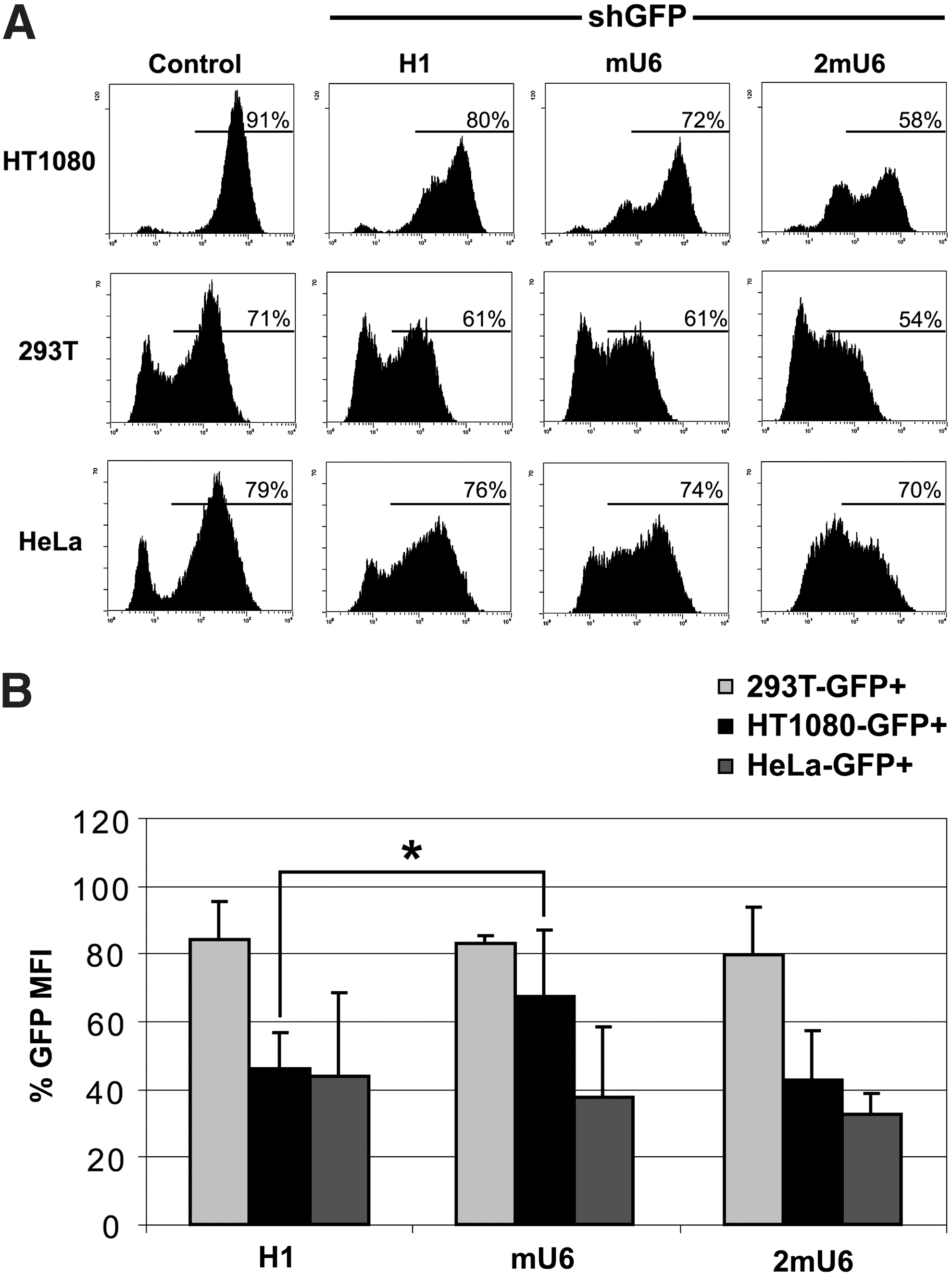
Comparison of H1 and mU6 promoter efficiency in human cell lines.
H1 mediates stable long-term gene silencing in human cell lines
To monitor the stability of the silencing effect mediated by the FV-RNAi vectors, we transduced GFP-expressing HeLa and HT1080 cells with H1 vectors harboring the shGFP and the MscvNGFR cassette (MN). This enabled long-term analysis of transduced cells using an antibody to track NGFR+ cells. As shown in Fig. 3A, at day 4 the NGFR+/GFP– cells were 7.1% and 11.2% for the HT1080 and HeLa cells, respectively; at day 25 the NGFR+/GFP– population was further increased to 17.7% and 17.4%. The latter values correspond to the 54.3% and 67% of transduced NGFR+ HT1080 and HeLa cells, respectively. This clearly showed that the H1 promoter can induce a gene-silencing effect that was stable over time.
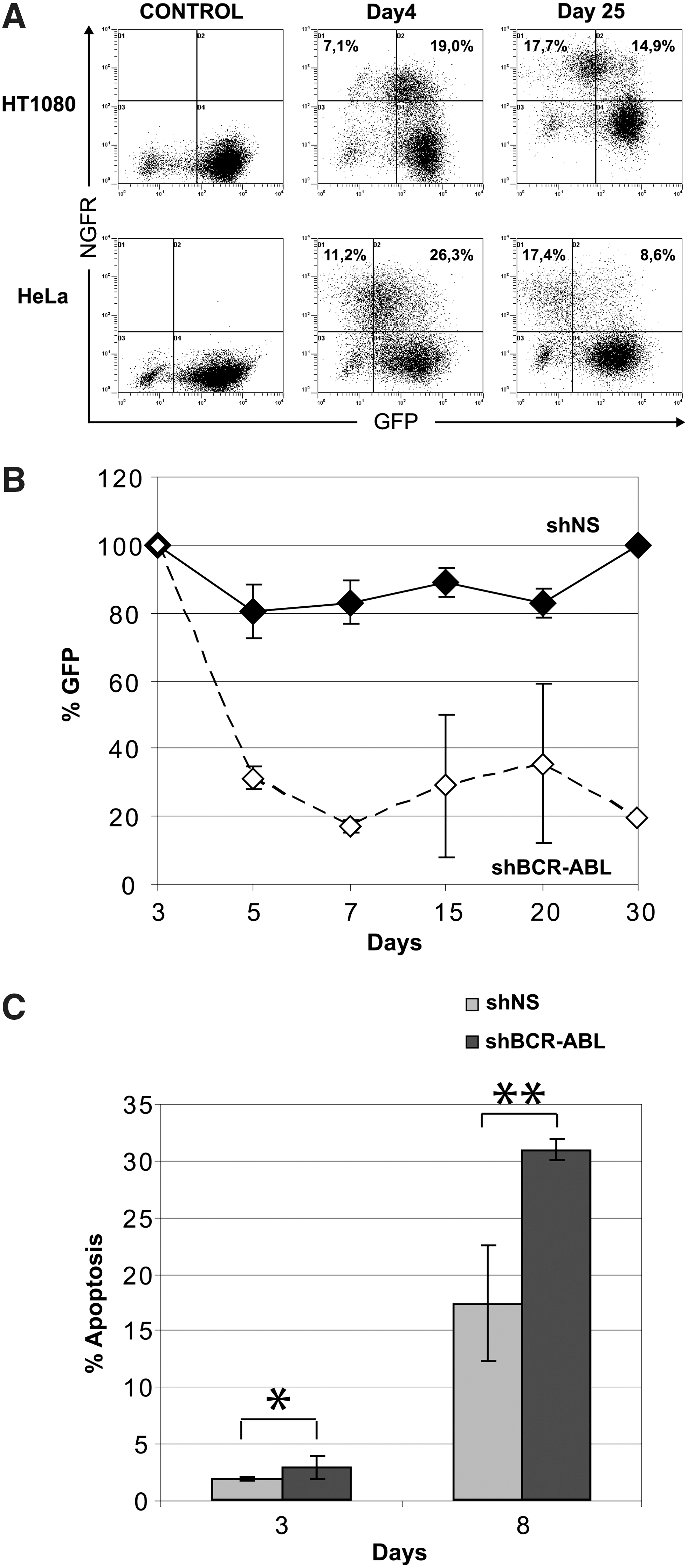
H1 promoter mediates stable and prolonged gene down-regulation in human cell lines.
As a further proof of principle, we moved on to check the efficiency of our H1 vector against an endogenous target, the BCR-ABL mRNA in the human erythroleukemic cell line K562. These cells express the BCR-ABL fusion transcript at levels exceeding even those of primary chronic myelogenous leukemia (CML) cells, and they constitute one of the best models for studying the in vitro silencing of BCR-ABL (Scherr et al., 2003). Following transduction with our H1.MF vector, the cells were monitored for GFP expression. We observed that shRNA expression against BCR-ABL led to an acute and significant loss of transduced, GFP-expressing cells within 1 week of cell culture, as opposed to nonspecific shNS-transduced cells, which retained a stable number of FV-transduced, GFP-expressing cells (Fig. 3B), indicating the absence of any vector-silencing phenomena in this cell line. As shown by Annexin V staining (Fig. 3C), loss of shBCR.ABL-expressing K562 cells was accompanied by increased apoptosis, verifying that down-regulation of BCR-ABL renders these cells more prone to apoptosis (Wilda et al., 2002). These data provided further proof for the efficiency and long-term stability of the H1-FV–mediated silencing effect against a highly expressed transcript, in a human cell line.
Toxicity of mU6-mediated RNAi in human cell lines
We have previously shown (Fig. 2A and B) that FV-mediated silencing of GFP by either the H1 or mU6 promoter constructs was efficient short-term. Although cells expressing shGFP from the H1-FV vector retained the GFP silencing effect even 30 days post transduction, the effect was lost in HT1080 and 293T cells harboring the mU6-shGFP or the 2mU6-shGFP FV (data not shown). To exclude the possibility that this was an RNAi off-target effect, we studied two distinct nonspecific shRNA sequences (denoted shNS1 and shNS2), which we cloned in the H1.MF and mU6.MF vectors, and monitored GFP expression over time, in transduced HT1080 and 293T cells (Fig. 4A). After 1 month in culture, the percentage of H1-harboring cells was stable in both cell lines, whereas a significant reduction in the mU6-harboring cells was observed even by day 8 post transduction, regardless of the transcribed nonspecific shRNA sequence. These data showed that this phenomenon was mU6 promoter–related; in order to investigate whether loss of transgene expression was due to a gradual selection against mU6-transduced cells or due to vector silencing, we measured vector copy numbers in the transduced cells by Taqman real-time PCR. Calculated vector copies were corrected for GFP expression at each time point and plotted against the percentage of GFP (Fig. 4B). We observed that the reduction in GFP expression in the mU6-transduced cells was accompanied by a gradual loss of vector copies, as opposed to H1-harboring cells, which maintained a stable number of integrated proviruses. This confirmed that the observed effect was due to the elimination of mU6-harboring cells from culture and not due to vector silencing. This cell loss was mediated by apoptotic cell death, as shown in Fig. 4C. The induction of apoptosis was more pronounced in HT1080 cells, highlighting a cell-type specific element in RNAi-related effects. No interferon (IFN)-response gene induction was observed in these cultures, as measured by ELISA (data not shown). Collectively, our results indicate that shRNA expression from the mU6 vectors in human cell lines was accompanied by the gradual selection against transduced cells, in sharp contrast to the human H1 promoter-harboring cells.
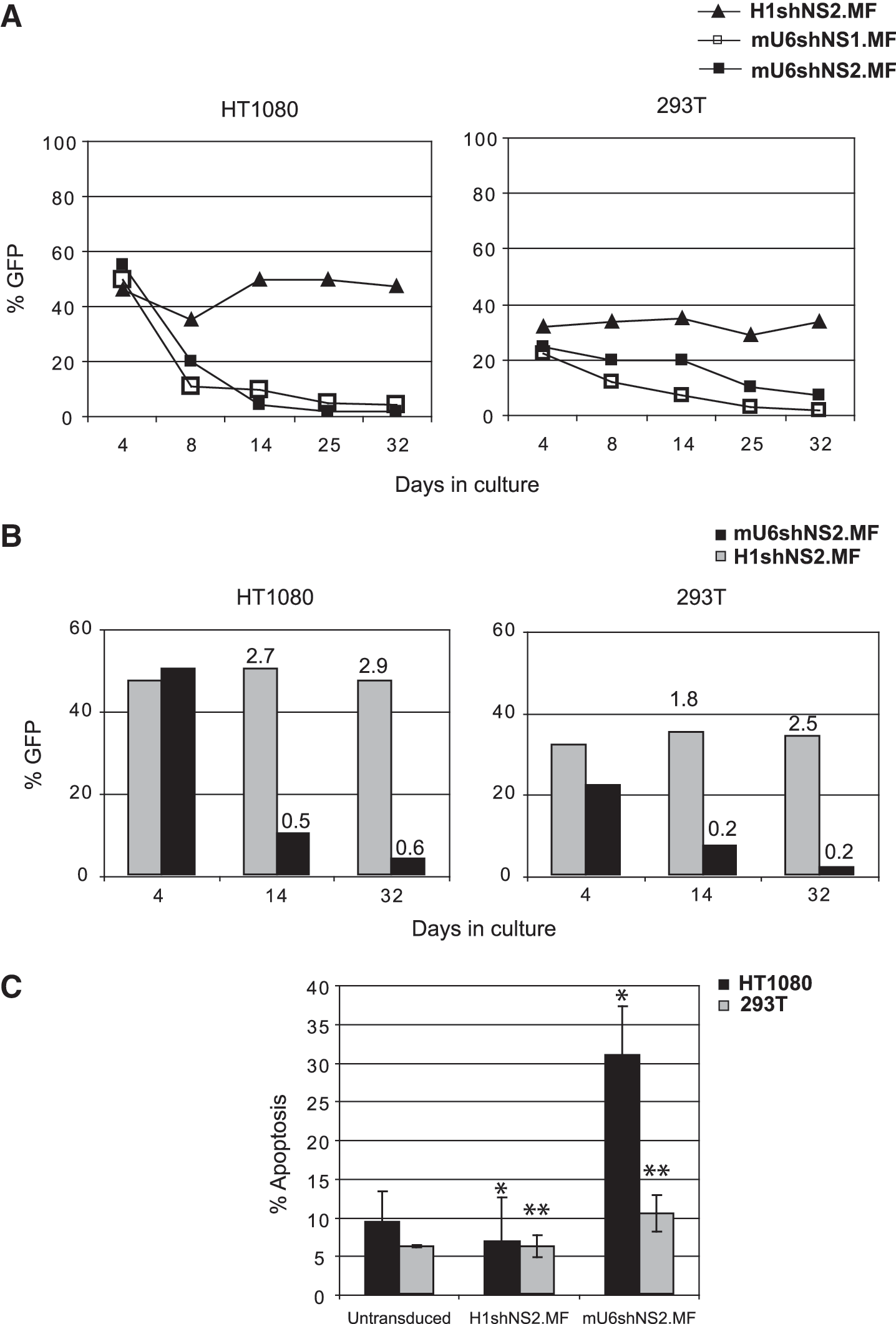
mU6-shRNA expression causes toxicity in human cell lines.
Stable long-term gene silencing by the mU6 vectors in mouse primary Lin– cells ex vivo and in vivo following BMT
In parallel with the studies with the human cell lines, we validated the efficiency of the mU6-FV vectors in murine primary hematopoietic cells. To test whether FV-mediated RNAi could affect HSC function, we transduced mouse C57Bl/6J BM Lin– cells with the mU6shNS1.MF vector (MOI = 10–14) and analyzed donor-cell chimerism and transgene expression up to 4 months post BMT. We used a partially myeloablative protocol for conditioning and obtained an average of 50.6% donor cell chimerism (range 30–76%; Table 1); at sacrifice, GFP+ cells in the BM were on average 38% (Fig. 5A). The sustained marker gene expression was obtained with a provirus copy number that ranged from 0.67 to 4.2 (average 2.5; Table 1). Furthermore, we did not observe any signs of hematological toxicities upon experiment termination. These results illustrate that the mU6-FV vector expressing nonspecific hairpin was stable in vivo and nontoxic to the mouse HSCs.
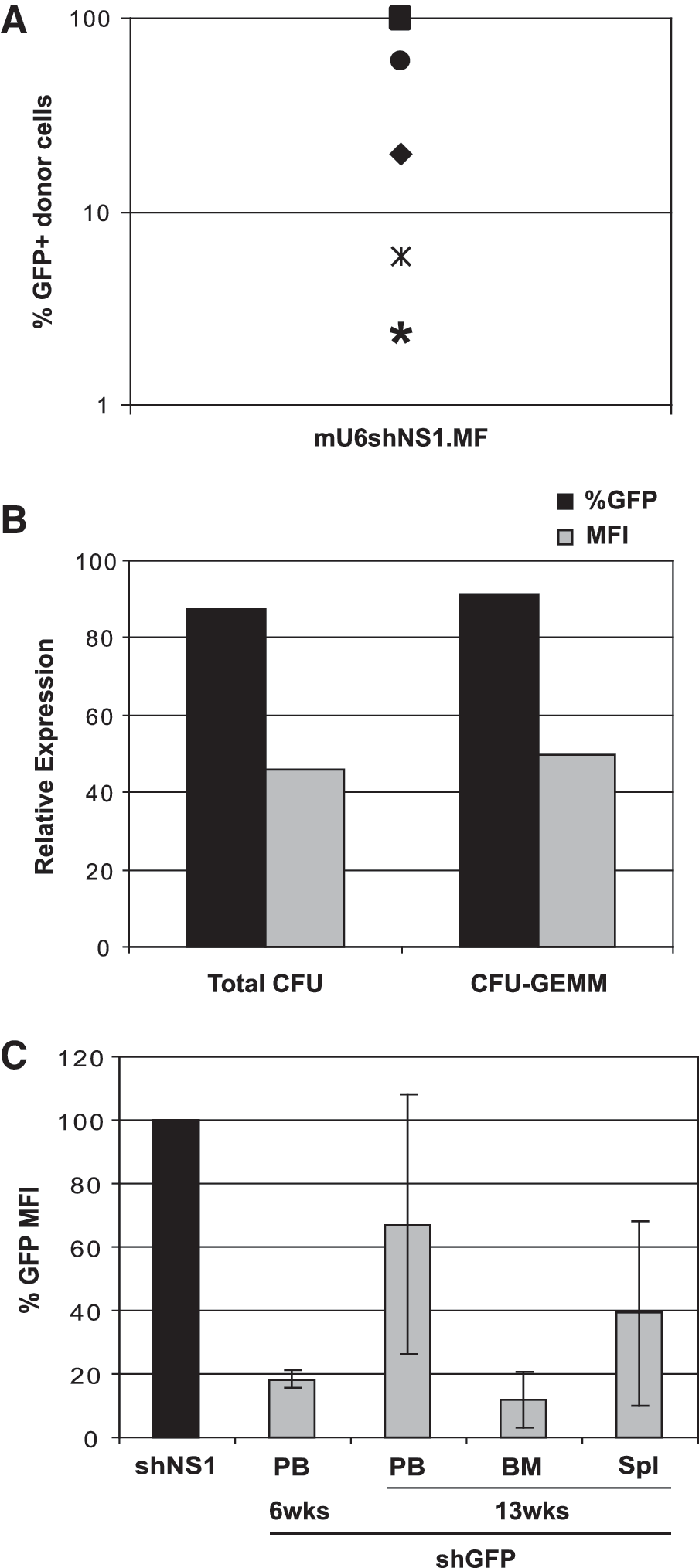
Stable mU6-encoded RNAi effect in mouse primary Lin– cells in vitro and in vivo after BMT.
BM, bone marrow; MOI, multiplicity of infection; PB, peripheral blood; provirus copies, number of integrated proviruses calculated by real-time PCR; % engrafted, % of male donor cells calculated by real-time PCR; % Tx, fraction of engrafted cells that express marker GFP.
To test the efficiency of the FV vectors in primary HSCs, we transduced BM Lin– cells from a GFP transgenic mouse line (Okabe et al., 1997) with the mU6shGFP.MN vector (MOI = 1). These transgenic animals express GFP under the chicken β-actin promoter and cytomegalovirus enhancer, with variable expression levels in distinct hematopoietic cell types, reaching 40%, on average, in the BM. Following transduction, the cells were cultured in methylcellulose and the levels of GFP expression in the colonies were estimated by FCM. GFP MFI reduction reached 55% and 50%, for total colony-forming units (CFUs) and primitive granulocyte/erythrocyte/monocyte/megakaryocyte (GEMM) colonies, respectively, compared with the cultures of nontransduced BM cells (Fig. 5B). In addition, the plating efficiency of the transduced cells was not affected (data not shown).
To investigate whether the GFP silencing could be sustained long-term in vivo, we performed BMT using Lin– cells from the same GFP transgenic donors transduced with our mU6.MN constructs expressing shNS1 (nonspecific) or shGFP and NGFR reporter (MOI = 10–15). Target GFP down-regulation was evaluated by FCM upon gating on the NGFR-expressing cells in the BM and the peripheral blood. As shown in Fig. 5C, we observed efficient reduction in the GFP MFI in all hematopoietic tissues examined, for up to 13 weeks post BMT. Especially in the BM, this reduction reached 82% and 94% relative to the shNS1 vector, with an average vector copy number of 2.1 (range 1.6–2.7). The donor cell engraftment was 40% (±5%) for all mice examined. Collectively, these findings showed that the mU6 vector can sustain stable and prolonged target down-regulation in primary mouse hematopoietic cells, both ex vivo and in vivo, following transplantation of FV-shRNA–expressing cells.
Discussion
The present study describes in detail the development of an FV vector system for the efficient and stable expression of shRNA molecules for in vitro and in vivo gene silencing. In addition, it provides a comparative analysis of the efficiency and limitations of the mU6 and the human H1 RNA Pol III promoters in human cell lines. Our data support earlier observations showing that vectors derived from FVs hold a promising position among stable RNAi systems (Moore et al., 2005; Sun et al., 2007; Taylor et al., 2008). We also confirm that FV-RNAi constructs can be produced at high titers, similar to standard transgene-expressing FV vectors. Our results indicate that neither the cloning of RNA Pol III promoters nor the expression of shRNAs interfered with vector stock production. Furthermore, the production of the nonspecific (shNS) expressing constructs at titers similar to the standard FV vectors showed the lack of self-targeting of the FV-RNAi vector genome by the expressed shRNAs, similar to what has been reported for lentiviral vectors (ter Brake and Berkhout, 2007). In addition, all our mU6 and H1 promoter-harboring constructs gave a high and sustained expression of marker genes in prolonged cell culture, thus ruling out any vector-silencing phenomena.
Our short-term results showed that, at 4 days post transduction and at similar MOIs (≤2), both the mU6 and the H1 promoters were equally efficient in 293T and HeLa cells. In contrast, in HT1080 cells, the mU6 promoter was required in double-copy configuration to achieve similar levels of silencing to the H1. Although previous studies with the human U6 in murine and human cells have reported on its higher efficiency compared with H1 (Boden et al., 2003; An et al., 2006; Mäkinen et al., 2006), our results show that the mU6 promoter is also active in human cell lines and the activity of mU6 and H1 is influenced by the cellular context.
Our long-term stability studies with the H1 promoter revealed that the efficient reduction in target-GFP expression was augmented in culture, up to 1 month post transduction, even with MOI ≤1. The efficiency and stability of the H1-FV vector were further evaluated against an endogenous target, the BCR-ABL oncogene in the human erythroleukemic cell line K562. Our results showed that H1-shRNA expression against BCR-ABL resulted in an acute loss of transduced K562 cells within 1 week of culture, which was accompanied by a reduction in apoptosis resistance. Our findings are in accordance with previous studies showing that silencing of BCR-ABL induces apoptotic cell death (Wilda et al., 2002; Rangatia and Bonnet, 2006). As reported in these studies, the extent of cytotoxicity depends on the level of BCR-ABL silencing; complete silencing triggers acute apoptosis, whereas lower but sustained down-regulation can generate a subpopulation of long-term survivors with cell-cycle defects. This dual effect was also observed in our study, with a 20–30% fraction of transduced, GFP-expressing cells surviving in culture, after the initial rapid population decline.
Long-term monitoring of the mU6-transduced cells revealed a rapid loss of the RNAi effect in culture, in contrast to what we observed with the H1 vectors in the human cell lines. In addition, when we used two distinct nonspecific shRNAs with mU6 and H1 vectors, we still observed a consistent loss of mU6-transduced cells in culture, indicating that this was an shRNA sequence-independent phenomenon. The kinetics of this cell loss varied among cell lines (HT1080 > HeLa > 293T), indicating a cell-type specific sensitivity to RNAi (Reynolds et al., 2006). Cultured mU6-transduced cells carried a significantly lower number of integrated vector copies compared with H1, reflecting a gradual elimination of the mU6-transduced population rather than vector silencing. In addition, an overall induction of apoptotic death was observed in the different mU6-shRNA cell cultures, which was proportional to the kinetics of transduced cell loss. Collectively, our results show that the use of the mU6 promoter in human cell lines triggers a sequence-independent cytotoxic effect.
Besides our studies using human cell lines, we also examined the potential of the mU6 vectors to provide RNAi in the murine hematopoietic system. To assay mU6-mediated shRNA expression, we used a vector (mU6shNS.MF) expressing a nonspecific hairpin in mouse BMT assays; we observed efficient engraftment and marker gene expression that exceeded 40% in some mice and was sustained for up to 4 months. To investigate further the gene-silencing efficiency of the mU6-FV vector, we transduced Lin– hematopoietic progenitors from transgenic GFP mice with our FV vector expressing shRNA against GFP (mU6shGFP.MN). Following 10 days of methylcellulose culture, we observed efficient and stable target gene silencing without any effect on colony plating efficiency or morphology. When transduced Lin– cells were transplanted in vivo, efficient GFP silencing (by 33–88%, on average) was achieved with as low as two vector copies per engrafted cell in all hematopoietic tissues examined. Our in vivo experiments are in complete accordance with previous reports on gene therapy experiments with the use of FV vectors in animal models, which are performed with low MOIs (5–10), achieving an average number of ∼2.5 integrated vector copies (Vassilopoulos et al., 2001; Kiem et al., 2007; Trobridge et al., 2009). These results are the first to provide solid evidence for the successful in vivo deployment of the FV-derived systems for RNAi, in the context of BMT approaches, further enhancing their potential for stem-cell–based gene therapy applications.
In light of both previous and present studies, it becomes evident that exogenous si/shRNA molecules are implicated in a much broader range of unintended effects than the sequence-specific silencing of their target genes. General RNAi-mediated toxicity is variable, including the shRNA-dependent induction of innate cellular immunity (Bridge et al., 2003; Sledz et al., 2003), sequence-related off-target effects (Jackson and Linsley, 2004; Scacheri et al., 2004), as well as the saturation of the RNAi and miRNA pathways (Grimm et al., 2006) and a general perturbation of the endogenous miRNA status (Khan et al., 2009). One of the mechanisms implicated in RNAi-mediated apoptosis in mammalian cells is the induction of type I IFN responses, an effect shown to be shRNA sequence–dependent (Bridge et al., 2003; Sledz et al., 2003). However, we and others (Fish and Kruithof, 2004; An et al., 2006) did not identify such a phenomenon (data not shown). A possible explanation could be that our shRNAs were smaller than the minimum of 30 nt known to trigger the response (Kim et al., 2005). Alternatively, the use of established cells lines that are frequently deficient in immune effectors (Stojdl et al., 2000) may hamper the detection of IFN responses, contributing to the lack of consensus in the various reports on RNAi immune activation.
It becomes evident that the interpretation of the observed RNAi effects is complicated and varies significantly with the cell and expression systems studied. Previous studies with RNA Pol III systems using the H1 (Moore et al., 2005; Park et al., 2005; An et al., 2007) or the human (Zhao et al., 2004; Taylor et al., 2008) and mouse (Robbins et al., 2006) U6 promoters provide direct and indirect evidence of the toxic side effects accompanying RNAi responses. Although the transcriptional activity of the promoter of choice dictates the shRNA expression levels (Boden et al., 2003; Mäkinen et al., 2006), in our study the mU6-mediated cytotoxicity does not seem to be related to shRNA expression, because the mU6 and H1 promoters showed comparable silencing efficiency. We cannot, however, rule out the possibility that the buildup of anti-sense shRNAs (McBride et al., 2008), unprocessed hairpins (Grimm et al., 2006), or potential variability in the shRNA length (Olejniczak et al., 2010) may be responsible for the observed cytotoxicity. All of the above highlight the incomplete current understanding of RNAi-mediated toxicity, the importance of which is just starting to become appreciated (Olejniczak et al., 2010). The vast majority of RNAi-toxicity mechanisms beg for careful analysis, toxicity monitoring, and effect interpretation, in order for such promising technology to be expanded. Collectively, our results provide solid evidence that current RNAi systems from FV vectors can efficiently deliver the dynamic and promise of the technology. Furthermore, we show that FV vectors with the mU6 promoter can provide sustained down-regulation of target genes in mouse BMT assays. However, the possibility of RNAi immune activation or toxicity mediated by the FV-shRNA vectors should be specifically addressed in future in vivo experiments.
Footnotes
Acknowledgments
We thank Dr. D.W. Russell (Division of Hematology, University of Washington, Seattle, WA) for providing the original mU6-containing ΔΦ vector. This work was supported by the Greek General Secretariat of Research and Technology grant no. 03ED602 (PENED).
Author Disclosure Statement
No competing financial interests exist.