
Abstract
Dystrophin plays an important role in muscle contraction, linking the intracellular cytoskeleton to the extracellular matrix. Mutations of the dystrophin gene leading to a complete loss of the protein cause Duchenne muscular dystrophy (DMD), frequently associated with severe cardiomyopathy. Early clinical trials in DMD using gene transfer to skeletal muscle are underway, but gene transfer to dystrophic cardiac muscle has not yet been tested in humans. The aim of this study was to develop an optimized protocol for cardiac gene therapy in the mouse model of dystrophin deficiency (mdx), using a cardiac promoter for expression of a microdystrophin (μDys) transgene packaged into an adeno-associated virus serotype 9 vector (AAV9). In this study adult mdx mice were intravenously injected with 1×1012 genomic particles of AAV9 vectors carrying a cDNA encoding μDys under the control of either a ubiquitously active cytomegalovirus (CMV) promoter or a cardiac-specific CMV-enhanced myosin light chain (MLC0.26) promoter. After 10 months, both AAV9 vectors led to sustained μDys expression in cardiac muscle, but the MLC promoter conferred about 4-fold higher protein levels. AAV9-CMV-MLC0.26-μDys resulted in significant protection of cardiac morphology and function as assessed by histopathology, echocardiography, and left ventricular catheterization. In conclusion, we established an AAV9-mediated gene transfer approach for efficient and specific long-term μDys expression in the hearts of mdx mice, resulting in a sustained therapeutic effect. Thus, this approach might be a basis for further translation into a treatment strategy for DMD-associated cardiomyopathy.
Introduction
Some earlier studies in mdx mice investigated whether microdystrophin genes could be transferred to cardiac muscle of normal and dystrophic mice, using AAV vectors as a potential treatment of dystrophin-deficient cardiomyopathy (Townsend et al., 2007; Bostick et al., 2008). In general, cardiomyopathy in mdx mice is relatively mild and becomes clinically relevant only in older animals (Quinlan et al., 2004; Spurney et al., 2008; Willmann et al., 2009). Interestingly, cardiac muscle of dystrophic mice is more readily transduced than skeletal muscle after systemic AAV administration, especially when the gene transfer is carried out at an early age. Certain serotypes (e.g., AAV9) appear to be effective, and widespread microdystrophin (μDys) expression led to the amelioration of electrocardiographic abnormalities in young adult mdx mice on vector injection into newborns (Bostick et al., 2008). Although these reports were encouraging, we wanted to develop an optimized gene therapy approach using delivery of a novel μDys (Jorgensen et al., 2009) with transcriptionally targeted AAV9 vectors in an experimental scenario that is closer to the clinical reality. This included the use of adult mdx mice at the time of gene transfer and assessment of cardiac endpoints in 1-year-old mice.
Materials and Methods
Generation of microdystrophin constructs and AAV vectors
Plasmids pUFCMVenh/MLC0.26-μDys and pUFCMV-μDys were constructed by replacing luciferase in pUF-CMV-Luc and pUFCMVenh/MLC0.26-Luc, a derivate of pUFCMV-MLC1.5-Luc (Müller et al., 2006), with an XbaI-cDNA encompassing the coding sequence of μDys, amplified from m2-pRcCMV2 (Jorgensen et al., 2009) and subcloned in pGEM via XbaI sites immediately flanking the start and stop codon. The μDys construct m2 is characterized by an engineered, large, in-frame rod deletion, as spectrin-like repeats 1–25 were removed, but retains hinges 1 and 4, as well as the full coding sequence of the N and C termini. It gives rise to a 125-kDa protein that localizes to the plasma membrane, recruits the sarcoglycan complex, and stabilizes the sarcolemma in skeletal muscle cells in vitro and in vivo (Jorgensen et al., 2009). AAV vector production and purification were performed as previously described (Hauswirth et al., 2000; Veldwijk et al., 2002). AAV9 pseudotyped vectors were generated by triple transfection of p5E18-VD2-9 (Gao et al., 2004), providing the AAV9 cap sequence, and pDGdelVP (Dubielzig et al., 1999), containing the AAV2 rep gene as well as adenoviral helper sequences, together with either pUFCMVenh/MLC0.26-Luc, pUFCMVenh/MLC0.26-μDys, or pUFCMV-μDys, resulting in AAV9-MLC0.26-Luc, AAV9-MLC0.26-μDys, and AAV9-CMV-μDys.
Animal procedures
mdx and C57BL/10 wild-type mice were obtained as a gift from R. Fink (Department of Physiology, University of Heidelberg, Heidelberg, Germany). Mice were housed in Makrolon cages type II under controlled conditions, with a 12-hr light:dark cycle and a temperature between 20 and 24°C in the animal facility of the University of Heidelberg. All procedures involving the use and care of animals were performed according to the Guide for the Care and Use of Laboratory Animals published by the National Research Council (NIH Publication No. 85-23, revised 1996) and the German animal protection code. Approval was also granted by the local ethics review board (G-87/08). For in vivo gene transfer, 6-week-old male mdx mice were administered 1012 viral genomes of AAV9 via tail vein injection, starting with n=10 per group. Four weeks postinjection, one animal from the AAV9-MLC0.26-μDys group and one animal from the AAV9-CMV-μDys group were killed by cervical dislocation as controls for μDys expression without further phenotyping. The remaining animals were monitored in our long-term study for 10 months. For additional analysis of μDys expression in heart, diaphragm, and skeletal muscle 4 weeks after vector infection, additional animals were injected with 1012 viral genomes of AAV9-MLC0.26-μDys (n=2) and AAV9-CMV-μDys (n=4). At the end of the study mice were killed by cervical dislocation.
Voluntary wheel running
Voluntary wheel running was performed by all groups of mice in the long-term study (AAV9-MLC0.26-Luc, n=10; AAV9-MLC0.26-μDys, n=9; and AAV9-CMV-μDys, n=9), starting at an age of 5 weeks. A hamster-size metal cage wheel with a diameter of 11 cm (Trixi running wheel; Fahrholz, Hamburg, Germany) was placed into a Makrolon cage type II. To measure running distance and running time from outside the cage, a digital magnetic counter (Sigma BC 500; Sigma Sport, Neustadt, Germany) was used. Data were collected every 4 weeks over 10 months and running performance is represented as the decline in running distance, which was calculated as the difference in 8-week median running distance between the start and end of voluntary wheel running for each animal. Notably, because of incomplete recordings of running distance, long-term measurements were performed in n=8 per group.
Western blot
Organ samples were homogenized in radioimmunoprecipitation assay (RIPA) buffer (1 M Tris [pH 7.5], 0.5 M EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 10% sodium dodecyl sulfate, 2 mM dithiothreitol, 0.2% 25× protease inhibitor cocktail [Roche, Mannheim, Germany]), using a TissueLyser (MM301; Qiagen, Hilden, Germany) at a frequency of 30 Hz for 2 min. Lysates were cleared from cellular debris by centrifugation and a total amount of 50 μg of protein was loaded onto a 3–8% gradient gel (Tris–glycine gel; Invitrogen, Karlsruhe, Germany). Protein was blotted on a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA), which was then cut horizontally and incubated for 3 hr with either a polyclonal anti-dystrophin antibody (AB15277; Abcam, Cambridge, UK) or a polyclonal anti-β-tubulin antibody (DLN-15304; Dianova, Hamburg, Germany), diluted 1:2500/1:2000 in 1× Roti-Block (Carl Roth, Karlsruhe, Germany). A fluorophore-labeled goat anti-rabbit antibody (IRDye 800; LI-COR, Lincoln, NE) and a goat anti-mouse antibody (IRDye 680; LI-COR), diluted 1:10,000 in 1× Roti-Block, were used to visualize protein bands by Odyssey infrared imaging (LI-COR). Relative quantification has been performed with ImageJ, using β-tubulin as a reference.
Immunohistochemistry and histology
Frozen 10-μm transverse sections were cut from the midpart of hearts, embedded in Tissue Tek (Leica, Wetzlar, Germany), and stained with an M.O.M. immunodetection kit (Vector Laboratories, Burlingame, CA) in accordance with the manufacturer's instructions. To detect μDys, a monoclonal antibody against the dystrophin C terminus (NCL-DYS2; Leica) was applied at a 1:20 dilution for 3 hr to the sections, followed by incubation with biotinylated mouse anti-mouse secondary biotinylated antibody, provided as a component of the M.O.M. immunodetection kit (Vector Laboratories). For quantitative analysis, sections were costained with Alexa Fluor 546-conjugated phalloidin (Life Technologies, Karlsruhe, Germany). On being embedded in VECTASHIELD HardSet mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories), sections were analyzed by fluorescence microscopy, using a Nikon Eclipse 90i upright automated microscope with a D1QM camera (Nikon, Düsseldorf, Germany).
For analysis of cardiac fibrosis, 10-μm transverse cryosections were stained with Masson trichrome and mounted in Roti-Histokitt (Carl Roth). Sections were evaluated by transmission microscopy with a Nikon Eclipse 90i upright automated microscope with a DS-Ri1 color camera (Nikon).
Quantitative real-time PCR analysis
To measure mRNA expression levels of transforming growth factor (TGF)-β and brain natriuretic peptide (BNP), total RNA was extracted from cardiac tissue lysates of treated mdx, untreated mdx, and C57BL/10 control mice with an RNeasy mini kit (Qiagen). SuperScript III reverse transcriptase (Invitrogen) was used to randomly reverse transcribe 1 μg of RNA into cDNA according to the manufacturer's specifications. Quantitative real-time PCR was performed on an ABI PRISM 7000 (Applied Biosystems, Foster City, CA), using the QuantiTect primer assay (Qiagen) in combination with SYBR GreenER qPCR SuperMix (Invitrogen).
To quantify viral genomes in heart and quadriceps femoris muscle of mdx mice treated with AAV9-MLC0.26-μDys (n=9) or AAV9-CMV-μDys (n=9), whole DNA was extracted from tissue samples, using a DNeasy blood and tissue kit (Qiagen) according to the manufacturer's specifications. An amount of DNA equivalent to 100 ng was used to perform quantification of viral genome copies as previously described (Hauswirth et al., 2000; Veldwijk et al., 2002).
Transthoracic echocardiography
The thorax area of mice was depilated 1 day before echocardiography. Echocardiography was performed at the end of the treatment period (10 months postinjection) on a Sonos 5500 (Philips, Eindhoven, The Netherlands) with an S12 transducer (12 MHz) by an investigator blinded to the experimental groups. Three consecutive beats were used to obtain left ventricular end-diastolic diameters (LVEDD) and left ventricular end-systolic diameters (LVESD). Furthermore, thicknesses of left ventricular posterior wall and septum were measured. Cardiac function is represented as fractional shortening (FS), which was calculated as FS %=[(LVEDD – LVESD)/LVEDD] ×100.
Pressure–volume loops
Measurements were made in closed-chest, spontaneously breathing mice, essentially as described previously (Bauer et al., 2009). Mice were anesthetized by single intraperitoneal injections of medetomidine (0.5 μg/g body weight), fentanyl (0.05 μg/g body weight), and midazolam (5 μg/g body weight) and monitored by ocular reflexes. Body temperature was maintained at 37°C with a homoeothermic blanket (Harvard Apparatus, Edenbridge, UK).
A 1.2 Fr catheter (model FT111B; Scisense, London, ON, Canada) was inserted into the left ventricle (LV) of each mouse through the carotid artery to simultaneously measure pressure and volumes. Left ventricular volumes were extrapolated from admittance magnitude, and admittance phase in real time, using the ADVantage PV system (Scisense). Pressure and volume data were recorded with a Scisense 404 16-bit four channel recorder with LabScribe2 software (Scisense). Indices of systolic function included end-systolic pressures, cardiac output, stroke work, and maximal rate of pressure development (dP/dt max). It was not possible to perform meaningful inferior vena caval occlusions to obtain the end-systolic pressure–volume relationship (ESPVR), a parameter to determine load-independent myocardial contractility, as in our experimental setting 1-year-old control mdx mice were extremely vulnerable to stressful procedures. Diastolic function was assessed by end-diastolic pressure, τ (Weiss method, regression of log of pressure vs. time) and the maximal rate of pressure decay (dP/dt min). Left ventricular size was determined by end-diastolic and end-systolic volumes.
Statistical analysis
All data, if not declared otherwise, are presented as means±standard error. To test for statistical significance between groups, one-way analysis of variance with Fisher least significant difference test was applied. Differences in data were considered significant at p<0.05.
Results
Running distance and mortality of treated mdx mice
It has been suggested that dystrophy and cardiomyopathy in dystrophin-deficient mice are aggravated by prolonged exercise (Townsend et al., 2008). To ensure that changes in cardiac structure and function are due to μDys gene transfer and not to reduced hemodynamic stress (e.g., inactivity) we systematically compared the running distance of all groups of mice from the beginning to the end of the study (n=8). Although we found a significant difference (p<0.05) in the decline in running distance between the AAV9-MLC0.26-μDys and AAV9-CMV-μDys groups, there was no difference between control mice injected with AAV9-MLC-Luc. Furthermore, all mdx mice showed a reduction in running distance regardless of treatment (Fig. 1A), indicating that cardiac μDys expression does not significantly influence the running performance of mdx mice. Notably, we lost 40% of luciferase-treated control mdx mice under stress (depilation), whereas all mdx mice treated with μDys survived (Fig. 1B, arrow: depilation). Because the death of animals occurred under direct observation, tissue samples could be collected immediately.
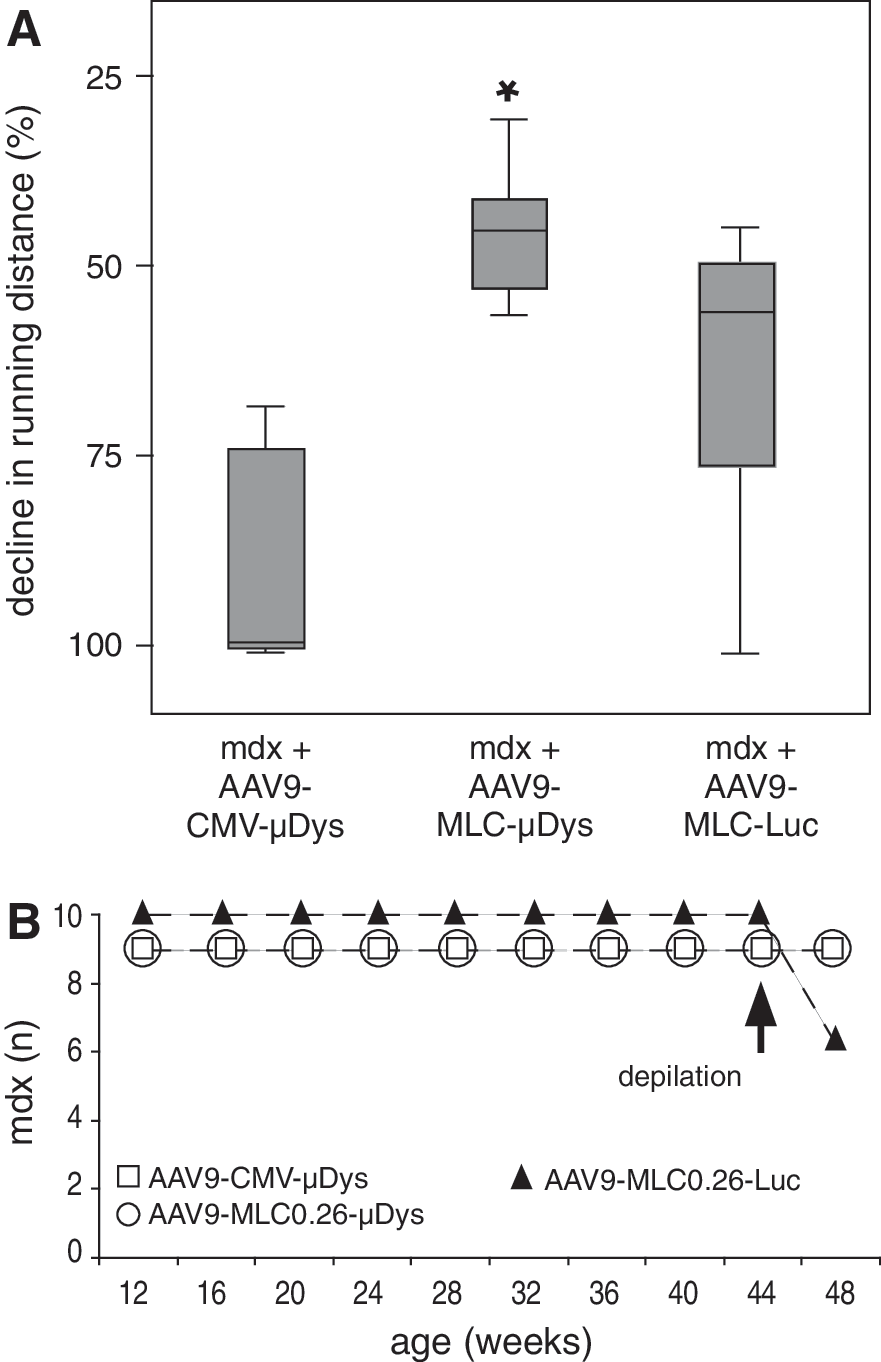
Long-term expression of microdystrophin
To evaluate expression of μDys, organ samples from mdx mice treated with AAV9-MLC0.26-Luc, AAV9-MLC0.26-μDys, or AAV9-CMV-μDys were taken 4 weeks and 10 months after injection. Transverse cardiac cryosections were cut and stained with an anti-dystrophin antibody recognizing μDys. There was widespread μDys expression detectable by immunofluorescence with both AAV9-CMV-μDys and AAV9-MLC0.26-μDys 4 weeks after treatment (Supplementary Fig. S1A; supplementary data are available online at
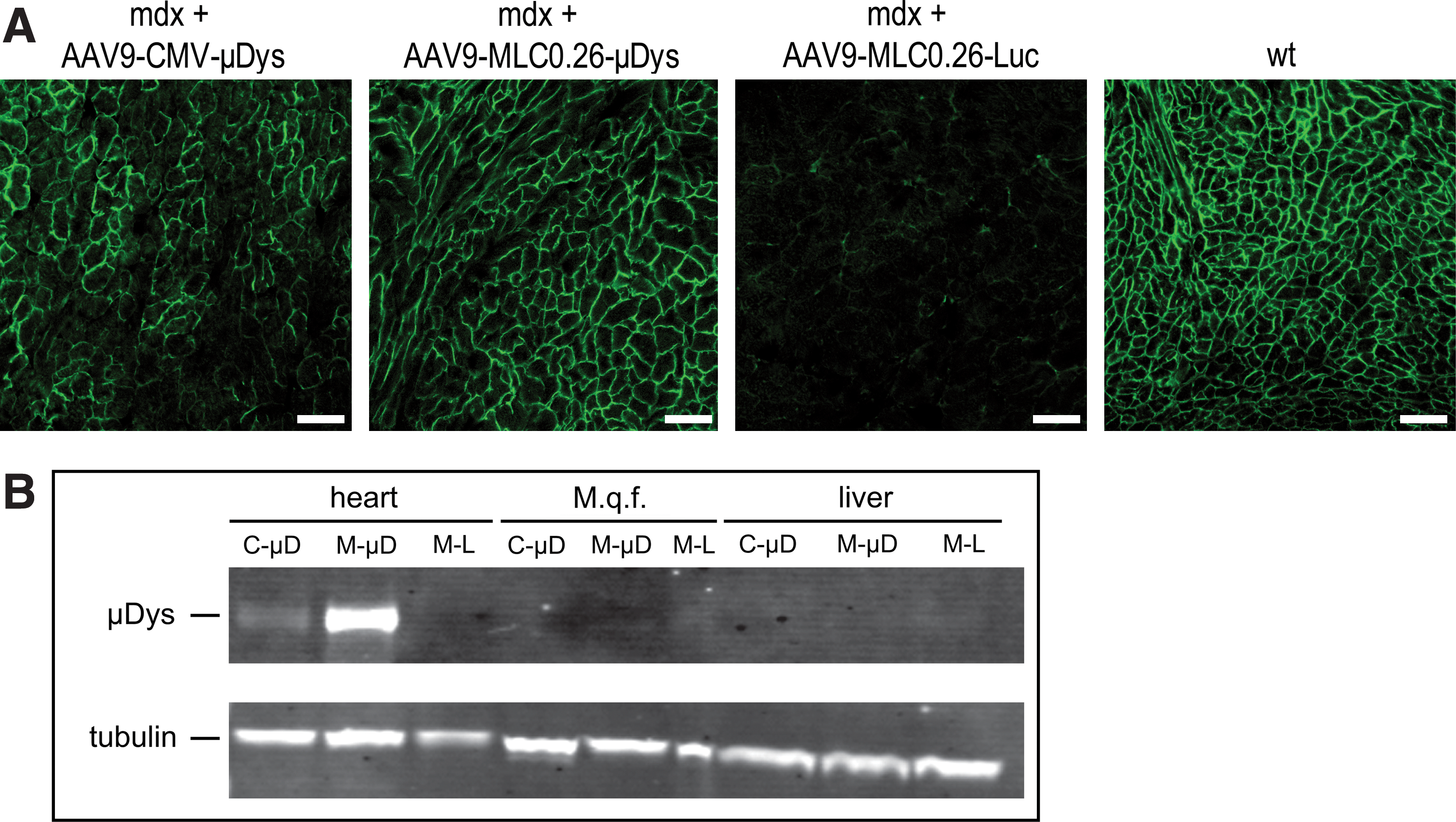
Long-term expression of μDys.
In contrast to cardiac tissue, quantitative analysis of viral genome copies in quadriceps femoris muscle revealed no significant difference in number of genome copies per microgram of genomic DNA between the groups of mdx mice treated long term (10 months) (Supplementary Fig. S1B). In addition, μDys protein was not detected in quadriceps femoris muscle and liver of mdx mice treated with any of the constructs after 10 months or 4 weeks (Fig. 2B and Supplementary Fig. S1A).
Because extracardiac μDys expression at an earlier time point might have influenced cardiac function, we analyzed additional mice 4 weeks after gene transfer. Whereas all mice revealed high cardiac gene expression in Western blot analysis, only one of four mdx mice injected with AAV9-CMV-μDys showed a faint signal in diaphragm (data not shown). Thus, the lack of extracardiac gene expression is not an effect of long-term gene transfer and diaphragmal μDys expression cannot explain the improvement in cardiac function in our approach.
Cardiac microdystrophin expression prevents myocardial fibrosis but not cardiac hypertrophy
To investigate a potential protective effect of μDys treatment on cardiac fibrosis, transverse cardiac sections were cut 10 months after injection of mice treated with AAV9-MLC0.26-Luc (n=10), AAV9-MLC0.26-μDys (n=9), or AAV9-CMV-μDys (n=9) and stained with Masson trichrome. Whereas hearts of mdx mice injected with AAV9-MLC0.26-Luc revealed diffuse fibrotic lesions (Fig. 3A, arrow), no signs of myocardial fibrosis were detected in hearts of μDys-treated mdx mice, comparable to wild-type controls (Fig. 3A). These histological findings were further confirmed by quantitative real-time PCR analysis of cardiac mRNA expression of TGF-β subtypes 1 and 2. TGF-β subtypes 1 and 2 are profibrotic cytokines, which are considered to play a key role in the development of cardiac fibrosis. The expression of TGF-β1 and TGF-β2 mRNAs is significantly increased in luciferase-treated mdx mice compared with wild-type mice (Fig. 3B and C). μDystrophin-treated mdx mice showed decreased levels of TGF-β1 (0.85±0.38-fold) and TGF-β2 (0.72±0.38-fold) compared with mice injected with the luciferase construct, although the difference was not statistically significant for the CMV-μDys construct (Fig. 3B and C).

Prevention of cardiac fibrosis in hearts of mdx mice.
Treatment of mdx mice did not change measures of myocardial hypertrophy assessed by echocardiographic determination of left ventricular wall thickness (Fig. 4A and B).

Ventricular wall thickness. No significant difference in the thickness of
Attenuation of heart failure after microdystrophin gene transfer
Echocardiographic evaluation of cardiac function was performed at the end of the study for mdx mice treated with AAV9-MLC0.26-Luc, AAV9-MLC0.26-μDys, and AAV9-CMV-μDys. Twelve-month-old mdx mice of the luciferase control group (n=6) revealed a significant reduction in fraction of shortening when compared with wild-type mice (n=6) (66.6±6.9 vs. 77.9±2.2%; p<0.05) (Fig. 5A). The fractional shortening was significantly higher in μDys-treated mdx mice, both with the cardiac-specific MLC0.26 promoter (77.7±2.3%, n=9) and the ubiquitous CMV promoter (77.6±1.6%, n=9) as compared with luciferase-treated mice.
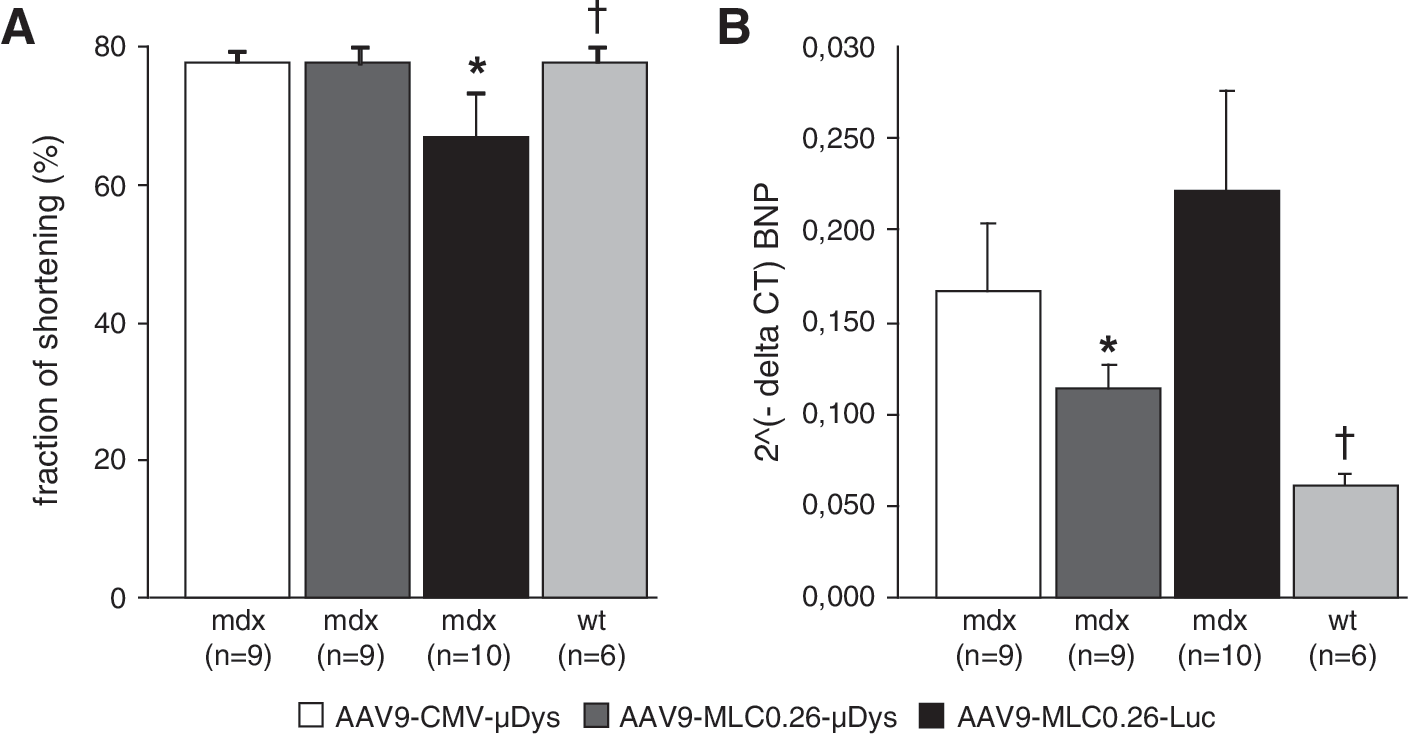
Preservation of cardiac function.
In addition, assessment of in vivo cardiac function was performed by measuring pressure–volume loops (Table 1). We found a reduction of end-systolic pressure and dP/dt max in the hearts of luciferase-injected mdx mice compared with wild-type mice, an indicator of reduced systolic function. mdx mice treated with AAV9-MLC0.26-μDys revealed a significant increase in these parameters compared with control luciferase-injected mdx mice, indicating a significant preservation of systolic function. Furthermore, we observed a tendency toward increased cardiac output and stroke work in mdx mice after transfer of AAV9-MLC0.26-μDys compared with luciferase-injected mdx mice. Increased dP/dt min in the hearts of mdx mice treated with MLC0.26-controlled μDys as compared with luciferase controls points toward improved diastolic function. There were no significant differences in left ventricular volumes between the groups.
Values are presented as means±(standard error).
p<0.05, wt versus mdx-Luciferase.
p<0.05, mdx-μDys versus mdx-Luciferase.
To address the question concerning whether gene transfer of μDys is able to attenuate molecular markers of heart failure in mdx mice, expression levels of cardiac BNP mRNA were analyzed by quantitative real-time PCR. Although there was a trend toward decreased BNP expression in AAV9-CMV-μDys-treated mdx mice compared with luciferase controls (Fig. 5B), AAV9-mediated transfer of the MLC0.26-controlled μDys resulted in a significant reduction of BNP, confirming attenuation of heart failure also at the molecular level.
Discussion
Progressive pathological changes within the myocardium lead to the development of cardiomyopathy in most patients with DMD and frequently determines their survival, as pulmonary dysfunction is increasingly treated by noninvasive ventilation (Nigro et al., 1990; Vianello et al., 1994). Current treatment modalities delay disease progression and may include corticosteroids, ACE inhibitors, and β-blockers (Sejerson and Bushby, 2009). There is a clear need for better, novel therapies, and exon skipping using antisense oligonucleotides may be one of the most advanced in humans (van Deutekom et al., 2007; Kinali et al., 2009). However, exon-skipping therapies need to be tailored toward the individual dystrophin mutation, restricting the number of patients eligible for this therapy. Studies in mice have raised some concern that not all oligonucleotide chemistries may be equally effective in targeting the heart muscle. Dystrophin gene transfer using viral vectors may overcome some of these limitations. AAV is receiving much attention as a vector for in vivo gene transfer in humans because of its favorable safety profile. However, AAV vectors have a limited packaging capacity, which has led to the construction of various small dystrophin molecules, so-called microdystrophin (μDys) (Wang et al., 2000; Townsend et al., 2007; Bostick et al., 2008; Foster et al., 2008; Jorgensen et al., 2009; Koo et al., 2011). Whereas clinical gene transfer experiments using μDys constructs are underway for skeletal muscle in boys with DMD (Mendell et al., 2010), gene therapy for heart failure in dystrophin deficiency has barely been evolved.
Systemic AAV-mediated transfer of a microdystrophin cDNA resulted in amelioration of clinical signs of cardiomyopathy in mdx mice (Townsend et al., 2007; Bostick et al., 2008). Whereas one study used vascular endothelial growth factor (VEGF) to facilitate transvascular gene transfer of AAV6 vectors in adult mdx mice (Townsend et al., 2007), the study by Bostick and colleagues investigated systemic administration of AAV9 vectors into neonatal mdx mice, taking advantage of the high efficiency of AAV9 vectors for cardiac gene transfer in mice (Inagaki et al., 2006; Pacak et al., 2006; Zincarelli et al., 2008). Because a systemic gene transfer approach with coadministration of VEGF might be limited by the side effects of VEGF, and because DMD is usually not diagnosed at the newborn stage, we believe that our approach of transvascular AAV9 vector delivery in adult mice might be closer to the clinical setting.
Reduction or loss of transgene expression after intravenous injection of AAV vectors is frequently observed with unspecific viral promoters, limiting their application for gene transfer in genetic disorders that require long-term and sustained gene expression. Possible reasons include promoter shutdown by methylation (Brooks et al., 2004) or other mechanisms such as an immune response against the transferred gene product elicited by expression in antigen-presenting cells (Cordier et al., 2001; Dressman et al., 2002; Fougerousse et al., 2007). Therefore, in this study we compared the ability of an enhanced heart-specific promoter (MLC0.26) with that of the ubiquitously active CMV promoter to express a truncated dystrophin cDNA (μDys). This is in contrast to previous studies, in which exclusively viral promoters were used to drive μDys expression in the cardiac muscle of mdx mice after systemic AAV application. We detected widespread μDys expression in cardiac muscle 10 months after a single, systemic AAV delivery with both promoters. Direct comparison by immunoblotting revealed 4.8-fold higher μDys expression with the MLC0.26 promoter as compared with the CMV promoter. A similar increase in efficiency of cardiac transgene expression mediated by a heterologous promoter was observed previously with the CMV-enhanced 1.5-kb MLC promoter driving a luciferase reporter gene in adult NMRI mice (Müller et al., 2006). These findings underline the advantage of using nonviral, tissue-specific promoters for long-term and sustained μDys expression.
We did not detect significant levels of μDys protein in skeletal muscle and liver with either promoter 10 months after AAV9-mediated delivery. Because the CMV-MLC promoter already demonstrated reduced gene expression in skeletal muscle, when compared with cardiac tissue (Müller et al., 2006), and gene transfer of AAV9-CMV-μDys revealed a low signal in diaphragm only in one of four mice after 4 weeks, extracardiac expression cannot explain the protective effect on cardiac function in our approach, as previously shown by Crisp and colleagues for diaphragmal gene transfer (Crisp et al., 2011).
Because a strong skeletal muscle promoter was able to drive sufficient levels of μDys in skeletal muscle after intramuscular injection of an AAV9 vector (Koo et al., 2011), lacking expression in extracardiac muscle after intravenous AAV9 vector injections might rather be explained by lower promoter activity in skeletal muscle than in cardiac muscle or by low transduction of skeletal muscle in adult mice after systemic injection of AAV9 vectors as observed earlier (Inagaki et al., 2006; Pacak et al., 2006; Zincarelli et al., 2008). The limitation of a systemic AAV9-based approach with a CMV promoter for μDys expression in skeletal muscle was also confirmed by a study that reported only an unexpectedly low level of skeletal muscle transduction after intravenous injections in juvenile mice with three times higher vector doses than in our study (Shin et al., 2011).
The μDys construct used in this study has been tested only in skeletal muscle, not in cardiac muscle (Jorgensen et al., 2009). Although it contains a large in-frame deletion of the rod domain, like other reported μDys constructs, it retains the entire coding sequence of the C terminus (C+). The most frequently used μDys constructs carry a deletion of the C terminus (ΔC) with binding sites to a number of proteins including syntrophins. To our knowledge there has not been a thorough, side-to-side comparison of C+ and ΔC, but otherwise identical μDys in skeletal or cardiac muscle. However, we may suggest, based on the data of this study, that μDys (C+) provides long-term protection in cardiac muscle of dystrophin-deficient mice if sufficient expression levels are sustained. Because the μDys in the present study differs from that originally tested in skeletal muscle (Jorgensen et al., 2009) by lack of an untranslated 5′ region, which was excised from the original m2 construct in order to fit within the AAV packaging limit, we cannot rule out a loss of potential stabilizing mechanisms in skeletal muscle.
Higher levels of cardiac μDys expression levels mediated by the MLC0.26 promoter were associated with an increased protective effect for the heart muscle in aged mdx mice. The development of cardiac fibrosis was attenuated or prevented, and contractile function was preserved. This cannot be attributed to lower hemodynamic stress (inactivity in treated animals), as we monitored physical activity throughout the study. The structural and functional improvement in μDys-transduced mdx mice are remarkable and extend previous observations (Bostick et al., 2008; Shin et al., 2011) because young adult mdx mice were used rather than newborn or juvenile mice and mice were analyzed 10 months after AAV delivery, which is far longer than in previous studies.
In contrast to previous publications, which did not report an activation of the TGF-β signaling pathway in the hearts of mdx mice at an age of 6–9 months (van Erp et al., 2010), our study revealed increased mRNA expression of the profibrotic protein TGF-β in mdx hearts compared with wild-type hearts at the age of 12 months. This may be explained by the higher age of our animals. Fibrosis, which is the pathological end stage of cardiomyopathy in dystrophin deficiency, was clearly reduced in AAV9-MLC0.26-μDys-treated hearts as compared with controls assessed both by histological assessment and TGF-β studies.
Moreover, echocardiography and pressure–volume loops show improved cardiac function after μDys treatment in mdx mice at the age of 12 months. In accordance with these findings, a decrease in expression of BNP, an established molecular marker for heart failure, was noted especially when levels of μDys expression were high (MLC0.26 promoter construct). However, it needs to be noted that functional measurements of μDys-expressing mdx hearts, albeit significantly improved, did not reach wild-type levels. This may be due to several factors such as accumulated damage before gene transfer, incomplete gene transfer as not all cardiac cells express μDys, and incomplete protection of cardiac cells by μDys as compared with full-length dystrophin (Hor et al., 2009).
Our echocardiographic examinations show that mdx mice develop substantial myocardial hypertrophy, which remains unaffected by our treatment strategies. No significant differences in thickness of left ventricular posterior wall or septum between treated and untreated mdx mice could be observed in echocardiography. However, because accurate molecular alterations in cardiomyopathy of mdx mice are still not fully understood, causal mechanisms still remain speculative (Crisp et al., 2011).
In conclusion, expression of μDys with the MLC0.26 promoter leads to long-term and robust μDys levels within myocardium after AAV9-mediated gene transfer, resulting in preservation of cardiac function and prevention of cardiac fibrosis. Although DMD is a disorder affecting all muscles and a generalized treatment may be preferred, cardiac muscle may have different requirements and offer other opportunities than skeletal muscle, which justifies specific treatment. Further studies in large-animal models with delivery systems suitable for gene transfer into human hearts, such as coronary venous retroinfusion (Raake et al., 2004), are necessary to translate this approach into a clinical perspective for patients suffering from this devastating disease.
Footnotes
Acknowledgments
The authors thank Barbara Leuchs and the German Cancer Research Center (DKFZ) vector core production unit for their support in generating AAV vector stocks; and Ulrike Gärtner, University of Heidelberg, for expert assistance in animal experiments. The authors further thank Prof. James M. Wilson (University of Pennsylvania) for providing p5E18-VD2-9, Prof. Dr. Rainer Fink (Institute of Physiology, University of Heidelberg) for providing mdx mice, and the Nikon Imaging Center Heidelberg for support with microscopy. This work was supported by grants from the Deutsche Forschungsgemeinschaft (MU 1654/3-2 to O.J.M.) and Benni & Co-Muscle Dystrophy Parent Organization (to O.J.M., H.L., and R.B.). The muscle group at Newcastle University is supported by the Medical Research Council UK through the Centre for Neuromuscular Diseases (G0601943).
Author Disclosure Statement
The authors declare no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.