
Abstract
Duchenne muscular dystrophy is a severe X-linked inherited muscle wasting disorder caused by mutations in the dystrophin gene. Adeno-associated virus (AAV) vectors have been extensively used to deliver genes efficiently for dystrophin expression in skeletal muscles. To overcome limited packaging capacity of AAV vectors (<5 kb), truncated recombinant microdystrophin genes with deletions of most of rod and carboxyl-terminal (CT) domains of dystrophin have been developed. We have previously shown the efficiency of mRNA sequence–optimized microdystrophin (ΔR4-23/ΔCT, called MD1) with deletion of spectrin-like repeat domain 4 to 23 and CT domain in ameliorating the pathology of dystrophic mdx mice. However, the CT domain of dystrophin is thought to recruit part of the dystrophin-associated protein complex, which acts as a mediator of signaling between extracellular matrix and cytoskeleton in muscle fibers. In this study, we extended the ΔR4-23/ΔCT microdystrophin by incorporating helix 1 of the coiled-coil motif in the CT domain of dystrophin (MD2), which contains the α1-syntrophin and α-dystrobrevin binding sites. Intramuscular injection of AAV2/9 expressing CT domain–extended microdystrophin showed efficient dystrophin expression in tibialis anterior muscles of mdx mice. The presence of the CT domain of dystrophin in MD2 increased the recruitment of α1-syntrophin and α-dystrobrevin at the sarcolemma and significantly improved the muscle resistance to lengthening contraction–induced muscle damage in the mdx mice compared with MD1. These results suggest that the incorporation of helix 1 of the coiled-coil motif in the CT domain of dystrophin to the microdystrophins will substantially improve their efficiency in restoring muscle function in patients with Duchenne muscular dystrophy.
Introduction
Adeno-associated virus (AAV) vectors can efficiently transfer genes into skeletal muscle (Fabb et al., 2002; Gregorevic et al., 2008). To overcome the DNA packaging limitation of AAV (<5 kb), several microdystrophin genes have been developed (Athanasopoulos et al., 2004; Yoshimura et al., 2004; Foster et al., 2008; Gregorevic et al., 2008). In particular, ΔR4-23/ΔCT microdystrophin with deletions of the rod domains 4–23 and the CT domain of the dystrophin is one of the best characterized microdystrophin genes, displaying efficient dystrophin expression along with significant improvement of muscle function in skeletal muscles of the mdx mouse (Yue et al., 2006; Foster et al., 2008; Gregorevic et al., 2008), a widely used mouse model for muscular dystrophy (Turk et al., 2005). However, the importance of the CT domain of dystrophin in DPC restoration and muscle protection from lengthening contraction–induced muscle damage (LCIMD) remains unclear. In this study, we demonstrated that the delivery of AAV2/9-microdystrophin incorporating helix 1 of the coiled-coil motif in the CT domain of dystrophin increased the recruitment of α1-syntrophin and α-dystrobrevin at the sarcolemma of skeletal muscle fibers of mdx mice and efficiently protected mdx muscles from LCIMD.
Materials and Methods
Construction of AAV-ITR2–based murine-specific mRNA sequence–optimized microdystrophin plasmids
Murine-specific mRNA sequence–optimized microdystrophin (MD1) cDNAs have been previously described (Foster et al., 2008). Briefly, the MD1 cDNA incorporating deletions of spectrin-like repeat domain 4–23 and CT domain (exons 71–78) contains the last three amino acids of exon 79 of dystrophin followed by three stop codons and includes the SV40 poly-adenylation site. The CT domain–containing microdystrophin (MD2) cDNAs were generated by incorporating helix 1 of the coiled-coil motif in the CT domain of dystrophin, which interacts with α1-syntrophin and α-dystrobrevin based on an MD1 construct (Supplementary Fig. S1; Supplementary Data are available online at
AAV vector production and titration
HEK293T cells were cultured in roller bottles in Dulbecco modified Eagle medium, supplemented with 10% (v/v) fetal bovine serum and incubated at 37°C, 5% CO2. Reagents for cell culture were purchased from Invitrogen. Recombinant pseudotyped AAV2/9 vector stocks were generated by using calcium phosphate coprecipitation and triple transfection with the AAV2ITR-MD1 or -MD2, pAdΔF6, and pAAV5E18-VD2/9 plasmids (the latter two plasmids were kind gifts from Jim Wilson, University of Pennsylvania, Philadelphia), at a molar ratio of 1:1:1 in HEK293T cells. After 72 hr incubation, cells were lysed and particles were purified by using iodixanol (Sigma-Aldrich) step-gradient ultracentrifugation. The number of vector genomes (vg) was determined using dot blot hybridization. The titers of AAV2/9-MD1 and MD2 were 3.7 × 1012 and 3.1 × 1012 vg/ml, respectively.
In vivo gene delivery
For electrotransfer of plasmid DNA, skin covering mouse tibialis anterior (TA) muscles was shaved and the mice were injected with 10 U of bovine hyaluronidase (25 μl at 0.4 U/μl; Sigma) using a 28-gauge needle. After 2 hr, the mice were anesthetized with 2%–4% isoflurane and the TA muscle was injected intramuscularly with 25 μg of plasmid DNA in 25 μl of sterile saline solution. Control muscle was injected with 25 μl of saline solution only. Electrode gel (MediSupplies) was placed on the electrode plates to increase the contact area with the skin of mouse. Electrical field was then applied to the injected TA muscle using external electrodes at 175 V/cm in 20-msec square wave pulses at 1 Hz using a BTX ECM 830 electroporator (BTX, Tweezertrodes, Kramel Biotech). AAV administration was performed in neonatal and adult mice. Six-day-old mice were anesthetized by indirect contact with ice and injected intramuscularly with AAV expressing mouse microdystrophin in 5 μl of saline solution. Two-month-old adult mdx mice were anaesthetized with 2%–4% isoflurane and injected intramuscularly with AAV expressing mouse microdystrophin in 30 μl of saline solution. Wild-type C57BL/10 mice were purchased from Harlan. C57BL/10ScSn-Dmdmdx (mdx) mice were bred in house and maintained in minimal disease facilities (Royal Holloway, University of London) with food and water ad libitum. In vivo experimentation was conducted under statutory Home Office recommendation, regulatory, ethical and licensing procedures, and under the Animals (Scientific Procedures) Act 1986.
Measurement of the in situ lengthening contraction
Protection from LCIMD of TA muscles was evaluated by measuring the in situ isometric muscle contraction in response to nerve stimulation. Mice were anesthetized using pentobarbital (60 mg/kg intraperitoneally). The knee and foot were fixed with pins and clamps and the distal tendon of the muscle was attached to a lever arm of a servomotor system (305B, Dual-Mode Lever, Aurora Scientific) using a silk ligature. The sciatic nerve was proximally crushed and distally stimulated by a bipolar silver electrode using supramaximal square wave pulses of 0.1 msec duration. Absolute maximal isometric tetanic force was measured during isometric contractions in response to electrical stimulation (frequency of 75–150 Hz, train of stimulation of 500 msec). The sciatic nerve was stimulated for 700 msec (frequency of 150 Hz). All isometric contractions were made at an initial length L0 (length at which maximal tension was first obtained during tetanic contraction). Maximal isometric force was measured after each eccentric contraction and expressed as a percentage of the initial maximal isometric force. A maximal isometric contraction of the TA muscle was initiated during the first 500 msec. Then, muscle lengthening (1.1 mm, 10% L0) at a velocity of 5.5 mm/sec (about 0.5 L0/sec) was imposed during the last 200 msec. Six isometric lengthening contractions of the TA muscles were performed, each separated by a 60-sec rest period.
Histological analysis
TA muscles were excised from tendon to tendon, weighed, and rapidly frozen in liquid nitrogen–cooled isopentane. To assess muscle pathology, 10-μm cryosections of muscles were prepared and fixed in cold acetone at −20°C. Slides were stained with hematoxylin and eosin. The percentage of centrally nucleated fibers was calculated with SigmaScan Pro image analysis software (Systat Software) in four randomly captured fields ( × 20 magnification) from the largest section of individual muscles analyzed. For immunohistochemistry, muscle sections were stained using Mouse on Mouse (M.O.M) kit (Vector Labs) as previously described (Fabb et al., 2002). Antibodies for dystrophin (MANEX1011C, mouse monoclonal, 1:100, Glenn Morris; P6, rabbit polyclonal, 1:800, Sherratt et al., 1992), α1-syntrophin (rabbit polyclonal, 1:200, Abcam), α-dystrobrevin (mouse monoclonal, 1:200, BD Biosciences), nNOS (R20; rabbit polyclonal, 1:50, Santa Cruz Biotechnology), and laminin (rat monoclonal, 1:1000, Sigma Aldrich) were used. The signal was visualized using Alexafluor 568–conjugated anti-mouse or anti-rabbit IgG (1:200, Invitrogen). For laminin staining, an anti-rat biotinylated antibody followed by Alexafluor 568–conjugated streptavidin was used. For intensity measurement, all immunofluorescence images were captured and digitalized using identical parameters of exposure, saturation, and γ-levels between treated and untreated specimens. The intensity of dystrophin (MANEX1011C and P6), α1-syntrophin, α-dystrobrevin, and nNOS immune-stained fibers were analyzed by Metamorph software as previously reported (Arechavala-Gomeza et al., 2010) using a rat anti-laminin for normalization and expressed as a percentage of the intensity level of C57BL/10 muscles. As an indicator of sarcolemma permeability, endogenous intracellular IgG was evaluated in muscle fibers by direct staining using an Alexafluor 488–conjugated anti-mouse IgG (1:200, Invitrogen).
Western blotting
Muscles were homogenized in 300 μl of homogenization buffer (75 mM Tris HCl [pH 6.8], 10% SDS, 0.1% bromophenol blue, 20% glycerol and 100 mM DTT). Proteins were separated on 3%–8% polyacrylamide Tris-acetate gel (Invitrogen) and transferred onto a nitrocellulose membrane (Hybond ECL membrane; Amersham Biosciences). Nonspecific binding was blocked in 0.1 M PBS, 0.1% Tween-20 containing 5% (w/v) nonfat milk powder at 4°C overnight with gentle agitation. Dystrophin expression was detected using the Manex1011B (mouse monoclonal, 1:100; Glenn Morris) or GAPDH (rabbit polyclonal, 1:1000; Abcam) as an internal control and incubated at room temperature for 1 hr. HRP-conjugated anti-mouse or anti-rabbit IgG (1:1000, Dako) was added for 1 hr, followed by a washing step. The membrane was exposed to the Western blotting ECL chemiluminescent detection system (Amersham Biosciences) and exposed to ECL Hyperfilm (Amersham Biosciences).
Statistical analysis
All data are shown as mean values ± SEM (cohort size stated per experiment). For all statistical analyses, either Student t-tests or one-way ANOVAs were used, as appropriate. GraphPad Prism software package was used for the analysis (version 4, GraphPad Software Inc.). Levels of significance and p values are indicated in Results.
Results
Microdystrophin incorporating helix 1 of the coiled-coil motif in the CT domain of dystrophin leads to efficient dystrophin expression in both neonatal and adult skeletal muscles of mdx mice
Mouse microdystrophin plasmids that either lacked the carboxyl terminal (CT)-domain (MD1) or had helix 1 of the coiled-coil motif in the CT domain of dystrophin (MD2) were transduced by electrotransfer into TA muscles of 5-month-old mdx mice. Eight days after electroporation, muscles were harvested and cryosections were immunostained for dystrophin expression (data not shown). In pAAV-MD1 and MD2 plasmid-injected muscles, 321 ± 106 and 385 ± 92 dystrophin positive fibers were counted, respectively (data not shown). No statistically significant difference was observed in dystrophin expression levels in mdx muscles treated with the two plasmids (n = 4, t-test, p > 0.05, not significant [NS]). Dystrophin proteins of the expected molecular weights of 427, 138, and 154 kDa were expressed in total tissue extracts of wild-type C57BL/10 muscles and MD1- and MD2-expressing muscles, respectively (data not shown). These results indicated that MD1 and MD2 cassettes were functional and generated AAV-microdystrophins of expected molecular weights.
We then produced AAV2/9-MD1 or -MD2, and 5 × 109 vg of AAV2/9-MD1 or -MD2 was injected into TA muscles of 6-day-old neonatal mdx mice. Microdystrophin was expressed and correctly localized to the sarcolemma at 4 months after intramuscular injection (Fig. 1a,b). The number of dystrophin-positive fibers in the widest section of the TA muscles injected with AAV2/9-MD1 or -MD2 was 2113 ± 146 (corresponding to 72% ± 4.6% of the total fibers) and 1063 ± 338 (corresponding to 45% ± 13.7%), respectively (n = 4, t-test, p > 0.05, NS) (Fig. 1c). The TA muscles injected with AAV2/9-MD1 or -MD2 showed significantly reduced weight compared with saline-injected contralateral TA muscles of mdx mice (n = 4, t-test, p < 0.001 and p < 0.01 for MD1 and MD2, respectively), although the MD1- or MD2-expressing TA muscle weight was still significantly different compared with wild-type C57BL/10 muscles (Fig. 1d).
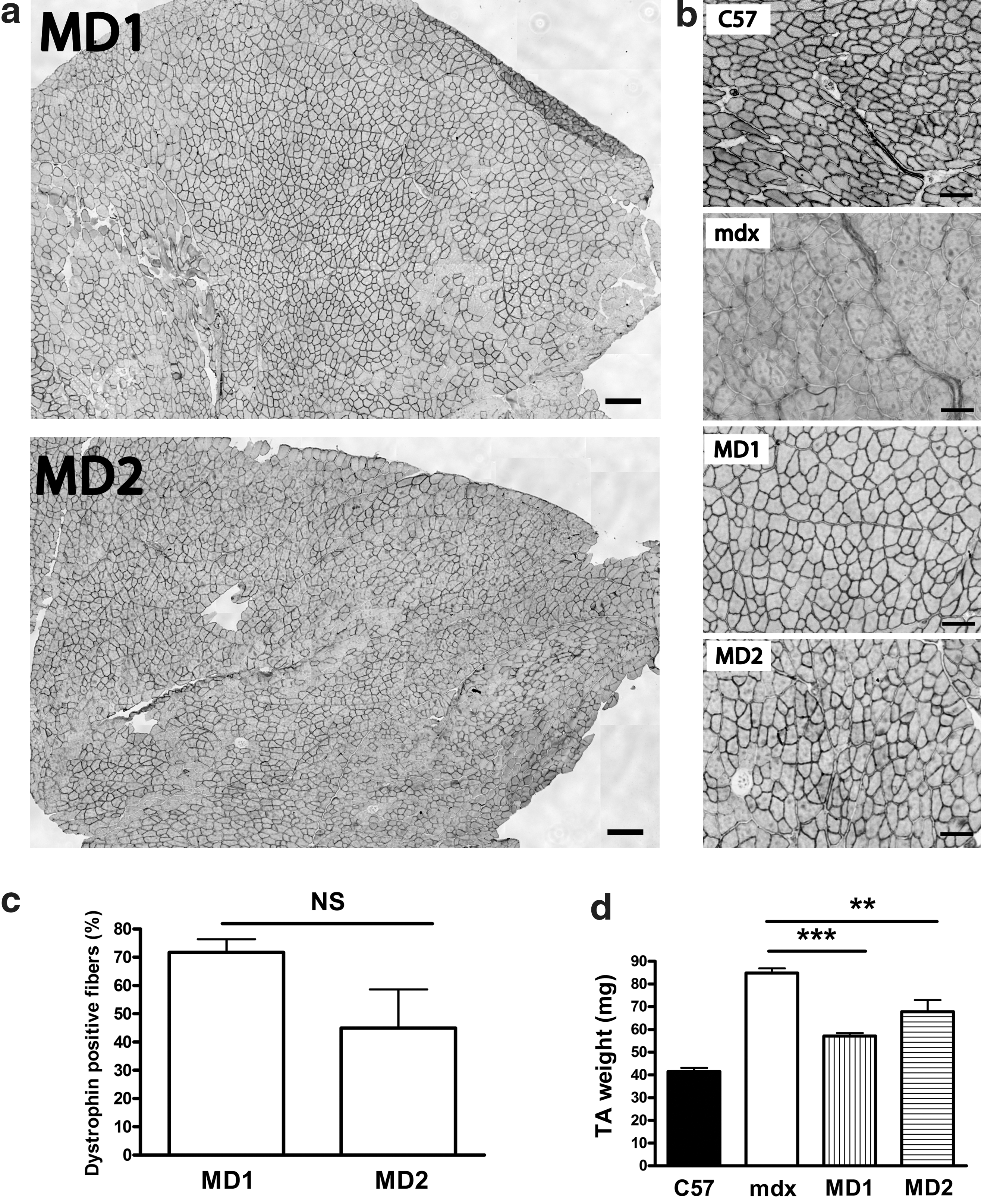
Efficient dystrophin expression in tibialis anterior (TA) muscles of neonatal mdx mice injected with AAV2/9-MD1 or -MD2. TA muscles of 6 day old mdx neonatal mice were injected with 5 × 109 vector genome (vg) of AAV2/9-MD1 or -MD2. TA muscles were stained for dystrophin expression using MANEX1011C antibodies at 4 months after injection. Control muscles from age-matched mdx and C57BL/10 mice were injected with saline only. MD1, mRNA sequence-optimized microdystrophin lacking the CT-domain; MD2, mRNA sequence-optimized microdystrophin containing the coiled coil region of the CT-domain.
These data demonstrate that intramuscular injection of AAV2/9-MD1 or -MD2 into TA muscles of neonatal mdx mice leads to stable and long-term microdystrophin expression up to 4 months after AAV delivery. Microdystrophin expression was then evaluated in 2-month-old adult mdx mice. TA muscles injected with 2 × 1010 vg of AAV2/9-MD1 or -MD2 exhibited microdystrophin expression at 2 months after AAV injection (Fig. 2a), and 563 ± 182 (corresponding to 36.0% ± 5.4%) and 658 ± 103 (41.3% ± 9.9%) of dystrophin-positive fibers were counted in AAV2/9-MD1 and -MD2 injected muscles, respectively (Fig. 2b) (n = 4, t-test, p > 0.05, NS). These results confirm that both MD1 and MD2 are equally and efficiently expressed in TA muscles of adult mdx muscles after injection of AAV2/9-MD1 or -MD2.
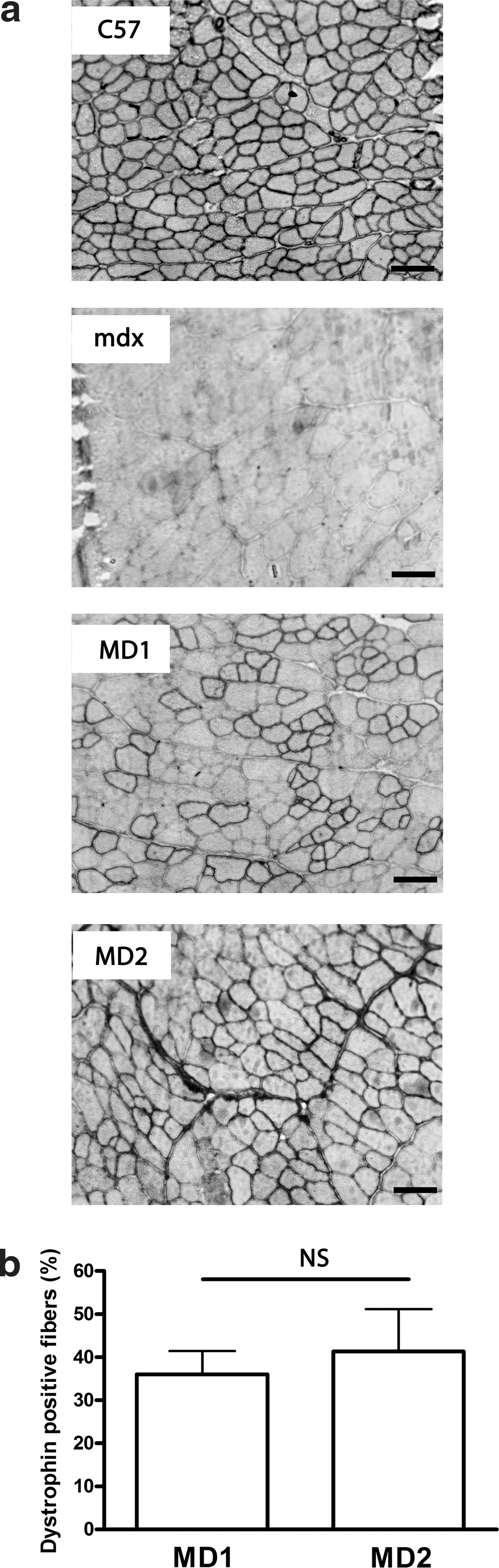
Efficient dystrophin expression in adult mdx TA muscles injected with AAV2/9-MD1 or -MD2. TA muscles of 2-month-old mdx mice were injected with 2 × 1010 vg of AAV2/9-MD1 or -MD2. Control muscles from age-matched mdx and C57BL/10 were injected with saline only. TA muscles were stained for dystrophin expression using a MANEX1011C antibody.
Microdystrophin incorporating helix 1 of the coiled-coil motif in the CT domain of dystrophin increases the dystrophin-associated protein complex recruitment at the sarcolemma
In order to verify the level of DPC recruitment at the sarcolemma, a semiquantitative intensity analysis was performed in TA muscles injected with AAV2/9-MD1 or -MD2. Similar intensity levels of MD1 and MD2 were detected using an antibody raised against repeat region 1 of the rod domain of dystrophin (n = 4, t-test, p > 0.05, NS) (Fig. 3a). However, the levels of intensity of both α1-syntrophin and α-dystrobrevin were significantly higher at the sarcolemma of TA muscles treated with AAV2/9-MD2 compared with muscles treated with AAV2/9-MD1 (Fig. 3b,c). More specifically, the percentage of α1-syntrophin intensity level in MD1- and MD2-positive fibers was 70.7% ± 3.6% and 88.2% ± 4.2%, respectively (n = 4, t-test, p < 0.01 compared with MD1), whereas the intensity level of α-dystrobrevin in MD1- and MD2-positive fibers reached 87.1% ± 2.9% and 97.5% ± 3.2%, respectively (n = 4, t-test, p < 0.05 compared with MD1). We observed a level of α1-syntrophin sarcolemmal association in MD2-positive fibers very close to wild-type C57BL/10 muscles (n = 4, t-test, p = 0.03), whereas the α-dystrobrevin sarcolemmal association was fully restored to the level of wild-type C57BL/10 muscles (n = 4, t-test, p = 0.53). As expected, the expression of MD1 or MD2, which both lack the spectrin-like rod domains 16–17 for interaction with nNOS, failed to recruit nNOS at the sarcolemma (Fig. 3d). Importantly, an antibody recognizing exon 57 to 63 in rod domain of dystrophin, which is equally present in both MD1 and MD2, positively stained only the muscles treated with AAV2/9-MD2 (95.3% ± 2.7%) and was not significantly different from that observed in wild-type C57BL/10 muscles (n = 4, t-test, p = 0.21) (Supplementary Fig. S2).

Intramuscular injection of AAV2/9-MD2 restores the level of α1-syntrophin and α-dystrobrevin at the sarcolemma of treated TA muscles. Quantification of fluorescence intensity was performed in cryosections of TA muscles of 2-month-old mdx mice injected with 2 × 1010 vg of AAV2/9-MD1 or -MD2. TA muscles were immunostained for
Microdystrophin incorporating helix 1 of the coiled-coil motif in the CT domain of dystrophin improves muscle morphology at the sarcolemma
Histological analysis was performed on TA muscles injected with AAV2/9-MD1 or -MD2 after lengthening contraction induced muscle damage. In MD1- or MD2-expressing muscles, central nucleation, one of the indicator of muscle degeneration and regeneration, was decreased (n = 4, t-test, p < 0.001, Fig. 4a). Dystrophin deficiency in muscle is associated with aberrant signal transduction caused by influx of endogenous extracellular proteins such as albumin, immunoglobulin (Ig) G, and IgM due to abnormal membrane permeability in DMD muscle cells (Blake et al., 2002). The permeability of membrane in myofibers of dystrophic muscles is increased after mechanical stress or electric stimulation (Petrof et al., 1993). We did not observe IgG influx in MD1- or MD2-positive myofibers indicating a protective role of de novo dystrophin expression on the membrane permeability in treated muscles (Fig. 4b).
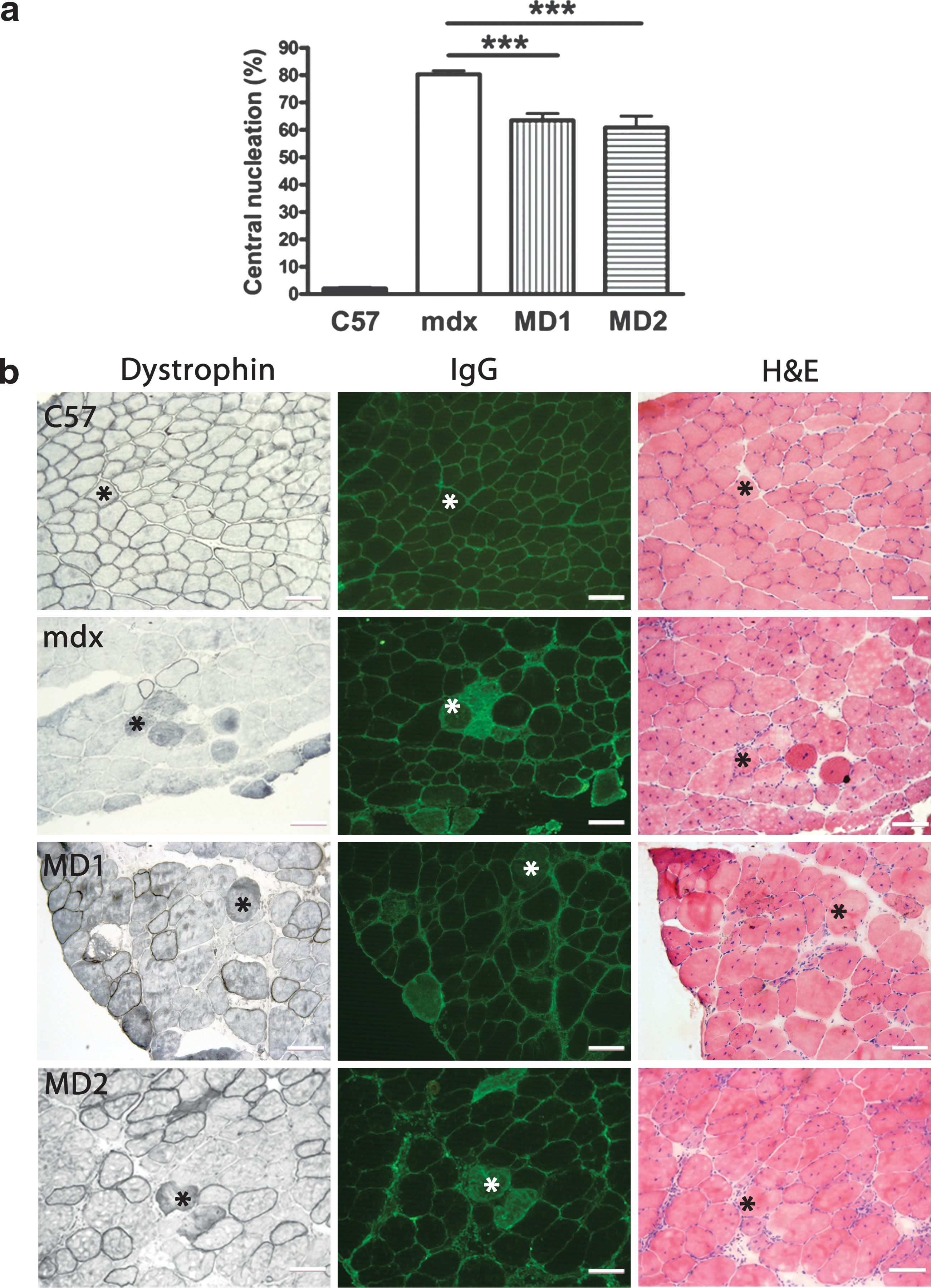
Improvement of morphological properties of mdx TA muscles injected with AAV2/9-MD2. TA muscles of 2-month-old mdx mice were injected with 2 × 1010 vg of AAV2/9-MD1 or -MD2. TA muscles were recovered and histology of muscle was assessed at 2 months after injection.
Delivery of AAV2/9-microdystrophin incorporating helix 1 of the coiled-coil motif in the CT domain of dystrophin improves muscle protection from exercise-induced muscle injury in mdx mice
The ability of microdystrophin to protect muscles from in situ LCIMD was examined by measuring the deficit in isometric maximal force following a series of six lengthening (eccentric) contractions. TA muscles of 2-month-old mdx mice were injected with 2 × 1010 vg of AAV2/9-MD1 or -MD2. The saline-injected contralateral TA muscles from the mdx group were used as a negative control to compare the resistance to eccentric contractions. At 2 months after AAV injection, muscles injected with AAV2/9-MD2 were protected compared with saline-injected contralateral TA muscles of mdx mice (n = 4–6, one-way ANOVAs, p < 0.05 and p < 0.001 following the fourth and sixth eccentric contraction, respectively) and did not show any significant difference in maximal force deficit compared to TA muscles of wild-type C57BL/10 mice (n = 4, one-way ANOVAs, p > 0.05, NS) (Fig. 5a). In contrast, no improvement was observed in muscles injected with AAV2/9-MD1 at the same dosage of viruses (Fig. 5b). Notably the injection of three times higher titer of AAV2/9-MD1 (6 × 1010 vg) was able to protect mdx muscles compared with saline-injected contralateral TA muscles of mdx mice (n = 4–6, one-way ANOVAs, p < 0.05), and treated muscles showed a similar maximal force deficit compared with TA muscles of wild-type C57BL/10 mice following the fourth lengthening contraction (n = 4–6, one-way ANOVAs, p > 0.05, NS) (Fig. 5c). However, following the sixth lengthening contraction, AAV2/9-MD1 failed to protect the muscle and did not show any significant difference in maximal force deficit compared with saline-injected contralateral mdx muscles (Fig. 5c). These data show that MD2 treatment has a significantly greater impact on LCIMD than MD1. The magnitude of resistance to eccentric contraction following the sixth lengthening contraction in the various experimental groups is ordered as follows: C57BL/10 wild-type muscles = MD2 (low dose) > MD1 (high dose) > MD1 (low dose) = mdx negative control.
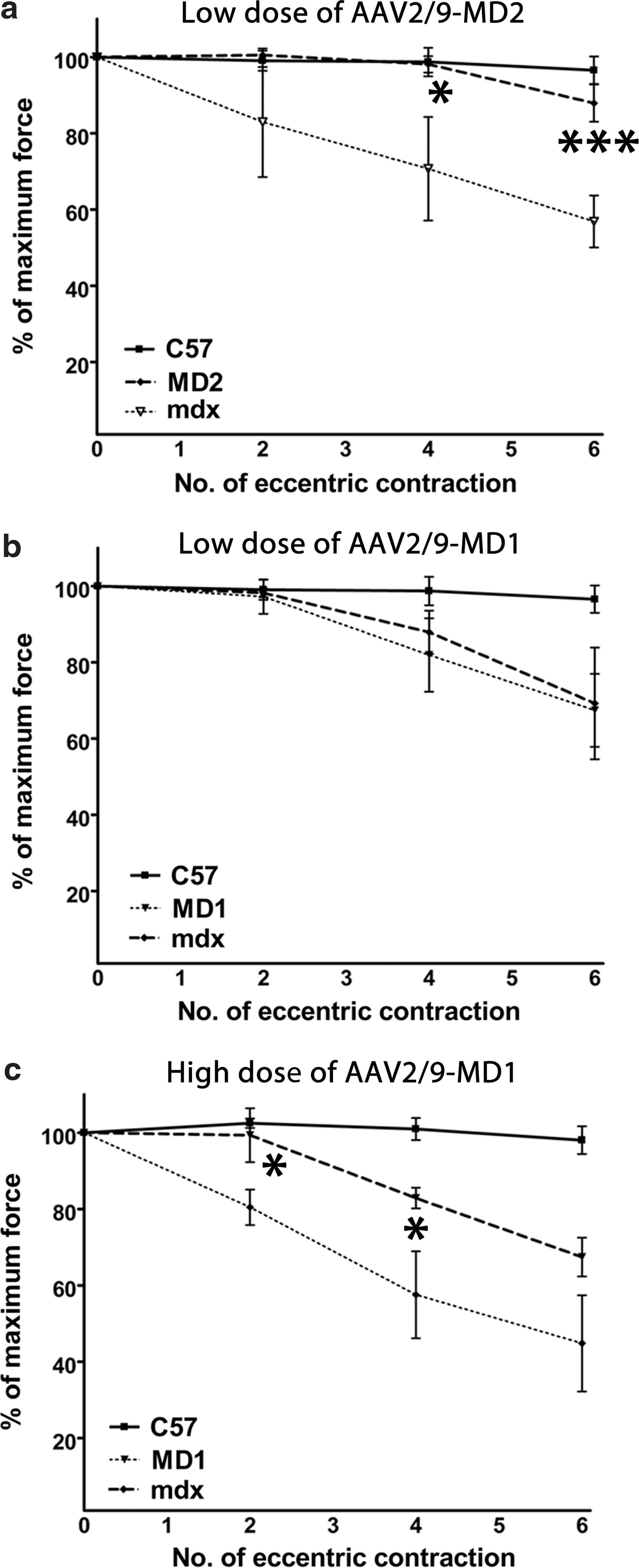
Intramuscular injection of AAV2/9-MD2 leads to muscle protection from lengthening contraction–induced muscle damage (LCIMD) in TA muscles of mdx mice. TA muscles of 2-month-old mdx mice were injected with either 2 × 1010 vg of AAV2/9-MD1, -MD2, or 6 × 1010 vg of AAV2/9-MD1. TA muscles were assessed for isometric maximal force deficit following a series of six eccentric contractions at 2 months after injection.
Discussion
In DMD muscles, the lack of dystrophin induces the disassociation of the DPC in muscles (Blake et al., 2002). The sequence alignment analysis we have performed (based on Blake et al., 1995) demonstrated that helix 1 and 2 of the coiled coil motif in the CT domain of dystrophin are highly conserved among vertebrates (Table 1). This homology suggests that this domain may play a crucial role, presumably by allowing direct interaction of dystrophin with DPC, including α1-syntrophin and α-dystrobrevin (Blake et al., 2002).
We previously tested mRNA sequence–optimized, murine-specific microdystrophin based on deletion of the rod domain 4 to 23 and CT domain of dystrophin (MD1) and demonstrated significant improvement in mdx pathology and function following neonatal delivery (Foster et al., 2008). In this study, a structurally similar version incorporating helix 1 of the coiled-coil motif in the CT domain of dystrophin (MD2) was used to demonstrate that the presence of the CT domain substantially increases DPC restoration and protection from LCIMD. Transduction of some 40% of myofibers by using either MD1 or MD2 was able to decrease the level of central nucleation from 80% to 65% in treated mdx muscles. Moreover, both MD1 and MD2 recruit part of DPC, α1-syntrophin and α-dystrobrevin, at the sarcolemma of muscle fibers. This result supports a previous report that showed that α1-syntrophin and α-dystrobrevin can bind to dystrophin even in the absence of the CT domain of dystrophin (Crawford et al., 2000). However, data obtained by a semiquantitative intensity study showed that the MD2 recruits significantly higher level of α1-syntrophin and α-dystrobrevin at the sarcolemma compared to MD1. In particular, the intensity level of α-dystrobrevin was equivalent to wild-type levels. This finding confirms the substantial role of the helix 1 of the coiled-coil motif in the CT domain of dystrophin on DPC restoration at the sarcolemma. Furthermore, we observed that an antibody recognizing a region between exon 57 to 63 of rod domain of dystrophin, which is equally present in both MD1 and MD2, failed to detect MD1 proteins. This observation suggests that the lack of helix 1 of the coiled-coil motif in the CT domain of dystrophin may partially lead to a misfolded conformational structure, further decreasing the efficiency on DPC recruitment at the sarcolemma.
The PDZ domain of α1-syntrophin can directly interact with the NT domain of nNOS (Thomas et al., 2003). However this interaction does not seem strong enough to recruit nNOS at the sarcolemma of fibers positive for α1-syntrophin and microdystrophin. This result supports the recent demonstration that spectrin-like domains 16 and 17 of dystrophin are required to recruit nNOS protein at the membrane of muscle fibers (Lai et al., 2009). Development of improved microdystrophin by addition of spectrin-like repeat domains 16 and 17 of dystrophin based on MD2 construct could be a next step for further improvement of MD2 to restore dystrophin function through interaction with nNOS. However, due to DNA packaging limitation of conventional AAV vector systems (<5 kb), this strategy may be difficult to attain. Trans-splicing AAV vector approaches using dual vector systems, which have been shown to transfer larger cDNA cassettes (Lai et al., 2005; Yan et al., 2007; Li et al., 2008), could be used to transfer the AAV cassettes including MD2 incorporating rod domains 16 and 17 of dystrophin (approximately 5.4 kb) into mdx muscles.
We previously demonstrated that high levels of MD1 expression lead to significant improvement of muscle resistance to LCIMD at 2 months after systemic injection of AAV2/8 in neonatal mdx mouse (Foster et al., 2008). Here we showed that the addition of the helix 1 of the coiled-coil motif in the CT domain of dystrophin into MD1 induced a significant improvement of muscle resistance to LCIMD following intramuscular injection of a lower dose of AAV2/9-MD2 compared with AAV2/9-MD1 at either an equivalent low dose or a higher dose. Furthermore, these experiments were performed in adult mdx mice, a much more reliable model when extrapolating to any clinical studies in DMD patients. A future step will be an examination of the effects of MD2 on the locomotor physiological behavior following systemic delivery of AAV2/9-MD2 in mdx mice.
These data suggest that the CT domain of dystrophin may improve muscle protection from LCIMD via stable association of DPC at the sarcolemma. DPC is composed of more than 10 different proteins, which form a transmembrane link acting as a mediator of physical and molecular interaction between the extracellular matrix and cytoskeleton (Tinsley et al., 1994; Campbell, 1995). α-dystrobrevin deficiency in mice is associated with muscular dystrophy with secondary loss of nNOS from the sarcolemma (Grady et al., 1999). In addition, α1-syntrophin–deficient mice show abnormal neuromuscular junction and reduction of nNOS level, although lack of syntrophin does not induce myopathy (Kameya et al., 1999). In contrast, it has been reported that α1-syntrophin–deficient mice showed remarkable muscle hypertrophy and reduced force generation during regeneration in mdx mice (Hosaka et al., 2002). These reports further suggest that restoration of level of α1-sytrophin and α-dystrobrevin at the sarcolemma induced by MD2 may have a crucial role in muscle protection from LCIMD in mdx muscles.
In conclusion, this study suggests that the inclusion of helix 1 of the coiled-coil motif in the CT domain of dystrophin to microdystrophin enhances the association with DPC such as α1-syntrophin and α-dystrobrevin, leading to an improvement of the resistance to LCIMD. These data could contribute to the optimization of the AAV-mediated microdystrophin gene delivery for the treatment of DMD.
Footnotes
Acknowledgments
We are grateful to Julie Johnston and James Wilson of the University of Pennsylvania for supplying plasmid vectors to allow AAV2/9 viral production, Ruxandra Draghia-Akli (VGX Pharmaceuticals) for supplying the SPc5-12 promoter, and to Glenn Morris at the Centre for Inherited Neuromuscular Disease for supplying monoclonal antibody MANEX1011B,C to detect dystrophin. This work was supported by grants from Clinigene European Network of Excellence, Duchenne Ireland, and the Muscular Dystrophy Campaign. Luisa Boldrin is funded by the Muscular Dystrophy Campaign, grant number RA3/776, held by Jennifer Morgan.
Author Disclosure Statement
No conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.