
Abstract
Ad[I/PPT-E1A] is an oncolytic adenovirus that specifically kills prostate cells via restricted replication by a prostate-specific regulatory element. Off-target replication of oncolytic adenoviruses would have serious clinical consequences. As a proposed ex vivo test, we describe the assessment of the specificity of Ad[I/PPT-E1A] viral cytotoxicity and replication in human nonprostate primary cells. Four primary nonprostate cell types were selected to mimic the effects of potential in vivo exposure to Ad[I/PPT-E1A] virus: bronchial epithelial cells, urothelial cells, vascular endothelial cells, and hepatocytes. Primary cells were analyzed for Ad[I/PPT-E1A] viral cytotoxicity in MTS assays, and viral replication was determined by hexon titer immunostaining assays to quantify viral hexon protein. The results revealed that at an extreme multiplicity of infection of 500, unlikely to be achieved in vivo, Ad[I/PPT-E1A] virus showed no significant cytotoxic effects in the nonprostate primary cell types apart from the hepatocytes. Transmission electron microscopy studies revealed high levels of Ad[I/PPT-E1A] sequestered in the cytoplasm of these cells. Adenoviral green fluorescent protein reporter studies showed no evidence for nuclear localization, suggesting that the cytotoxic effects of Ad[I/PPT-E1A] in human primary hepatocytes are related to viral sequestration. Also, hepatocytes had increased amounts of coxsackie adenovirus receptor surface protein. Active viral replication was only observed in the permissive primary prostate cells and LNCaP prostate cell line, and was not evident in any of the other nonprostate cells types tested, confirming the specificity of Ad[I/PPT-E1A]. Thus, using a relevant panel of primary human cells provides a convenient and alternative preclinical assay for examining the specificity of conditionally replicating oncolytic adenoviruses in vivo.
Introduction
Within the European Union GIANT consortium (
As an alternative preclinical approach to animal testing, we have used human nonprostate primary cells in in vitro cell culture to test whether Ad[I/PPT-E1A] replication is limited only to the prostate target cells by assessing the specificity and sensitivity of Ad[I/PPT-E1A] replication and cytolytic killing in vitro. The primary cell types were chosen for study, because the parental organs are either close to the site of adenoviral administration or should be exposed to the highest percentage of cardiac output (blood flow) and would likely have increased contact with any circulating Ad[I/PPT-E1A] virus in vivo in the phase I GIANT human clinical trial. They included human vascular endothelial cells, bronchial epithelial cells, urothelial cells, and hepatocytes. In all cases, no replication of the retargeted Ad[I/PPT-E1A] virus was seen, despite retention of the ability to attach to and penetrate the various cell types. We conclude that in vitro preclinical testing procedures using a relevant panel of human primary cells is a feasible approach for the preclinical analysis of the efficacy and specificity of oncolytic adenoviruses, and in this respect will have significant additional value next to animal models.
Materials and Methods
Maintenance of primary cells and cell lines
LNCaP cells (malignant human prostate adenocarcinoma cell line derived from a lymph-node metastasis; European Collection of Animal Cell Cultures, Porton Down, UK) were cultured in RPMI (Invitrogen, UK) supplemented with 10% heat-inactivated fetal calf serum (FCS; PAA, UK) and 2 mmol/L L-glutamine (Invitrogen, UK). HEK293 cells (human embryonic kidney cell line; ATCC, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM; GIBCO, UK) supplemented with 10% heat-inactivated FCS (PAA) and 2 mmol/L L-glutamine (Invitrogen). The following human primary cells were maintained in their associated media: primary human bronchial epithelial cells (HBEC; TCS/Cellworks, UK) were cultured in bronchial epithelial cell growth medium with supplements (TCS/CellWorks, UK); primary human umbilical vein endothelial cells (HUVEC; Lonza, USA) were cultured in Endothelial Cell Basal Medium with supplements (Lonza, USA); primary human urothelial cells (HUC; kindly donated by Professor J. Southgate, Jack Birch Unit for Molecular Carcinogenesis, University of York, UK) were cultured in Primaria flasks (BD Biosciences, UK) in keratinocyte serum-free medium (KSFM; Invitrogen, UK) supplemented with 2 mmol/L L-glutamine, epidermal growth factor (EGF), and bovine pituitary extract (GIBCO, UK); primary human hepatocytes (kindly provided by Prof. A. Dhawan and Dr. R. Mitry, Hepatocyte Biology and Transplantation Group, King's College Hospital, UK) were cultured in Eagle's minimum essential medium, supplemented with 10% heat-inactivated FCS, 2 mmol/L L-glutamine; primary human prostate cells were isolated with consent, from thick-needle cores of patient tissue removed by radical prostatectomy (York District Hospital, York, UK; Castle Hill Hospital, Hull, UK). Prostate cancer was confirmed by a uropathologist, by histology of adjacent tissue. Undifferentiated prostate cells were maintained in complete KSFM supplemented with EGF (Invitrogen), bovine pituitary extract (Invitrogen), 2 ng/ml leukemia inhibitory factor (Sigma, UK), 2 ng/ml stem cell factor (Sigma, UK), 1 ng/ml Gm, and 100 ng/ml cholera toxin (Sigma, UK). Irradiated STO cells (mouse embryonic fibroblasts) were routinely added as feeders. Differentiated primary prostate cells were cultured in 1:1 DMEM:Ham's F-12 medium (vol/vol; DMEM Culture Medium, Gibco, UK; Ham's F-12 Culture Medium, Lonza, UK), 2 mmol/L L-glutamine, 10% heat-inactivated FCS, and 10 nM dihydrotestosterone (Innovative Research of America, USA). All cell culture was performed without the addition of antibiotics.
Adenoviral samples
Ad[I/PPT-E1A] was manufactured at the GMP Facilities at the Center for Cell and Gene Therapy at Baylor College of Medicine (Houston, TX), specifically for the European Union GIANT project. The virus was supplied at a concentration of 1×1011 VP/ml in 20 mM Tris, pH 8.0, 25 mM NaCl, 2.5% glycerol. As a positive viral control, wild-type adenovirus (AdWT, serotype 5) was used (sourced from ATCC) (batch no. 001504, purity >99% in buffer: 20 mM Tris, pH 8.0, 25 mM NaCl, 2.5% glycerol). Viral titer was 5.8×1011 VP/ml. As a negative viral control, a replication-incompetent adenovirus (AdMock) was used (Carlsson et al., 2003). This virus has an E1-deleted and E3-deleted replication-deficient adenoviral vector of serotype 5. It contains no extra transgene and was produced by recombination of pShuttle and pAdEasy1 (He et al., 1998). AdMock was sourced from Professor Magnus Essand (University of Uppsala, Sweden) (batch no. 20090505, purity 99%, in buffer: 10 mM Tris-HCl, pH 7.9, 1 mM MgCl2, 4% sucrose). The viral titer was 4.1×1012 VP/ml. Two replication-incompetent reporter adenoviruses, AdCMV-gfp and AdI/PPT-gfp, were also sourced from Professor Essand.
MTS assays
LNCaP and primary cells were seeded in triplicate at 80% confluency in 48-well plates, 500 μl/well cell culture medium. The following day, cells were infected at room temperature (RT) for 1–2 hr with gentle agitation with 0, 50, 500, and/or 5,000 multiplicity of infection (MOI) (VP/cell) of either Ad[I/PPT-E1A] or the AdWT. Virus was prepared in the appropriate cell culture medium for each primary cell type assayed. After viral incubation, the virus was removed. The cells were washed with medium and incubated for up to 14 days, at 37°C, 5% CO2. Medium was changed on days 3, 6, 7, 10, and 11. On days 0, 3, 6, 10, and 14, 60 μl/well MTS reagent (CellTiter 96 AQueous One Solution Cell Proliferation Assay; Promega, UK) was added and incubated for 2 hr at 37°C, 5% CO2, followed by ultraviolet irradiation for 15 min. The absorbance at 485 nm and 620 nm was determined for each sample on a POLARstar OPTIMA plate reader (BMG Labtech, UK). Average final absorbance values for the triplicate wells were calculated and standard error propagation determined by using Microscoft Excel (Redmond, WA). The percent cell viability for each time point was determined by normalization against MOI 0, plotted with SigmaPlot (Hounslow, London, UK) and presented as bar graphs.
Adenoviral replication assays using hexon titration
Primary cells and cell lines were seeded at 80% confluency in 12-well plates. Twenty-four hours later, cells were infected in triplicate with Ad[I/PPT-E1A] or AdWT at MOI 0 and 500 for 1–2 hr at RT, with gentle agitation. Virus was removed by washing the cells with culture medium, and subsequently the cells were incubated at 37°C, 5% CO2 for 14 days. Fresh medium was added every 3 days to maintain cell viability. Culture media from the cells were collected on days 3, 6, 10, and 14 post infection and stored at or below −75°C. On days 7 and 11, medium was changed and discarded. Cells were harvested on day 14 where possible, and total cell lysates were prepared by repeated freeze/thaw. Hexon titer assays were performed in triplicate on 2.5×105 HEK293 cells (ATCC) using serially diluted viral-infected medium samples, following the Adeno-X Rapid Titre Kit procedure (Clontech, UK). By using this kit, adenoviral plaques were detected by immunostaining using an anti-hexon antibody (Clontech, UK), and viral plaques were counted under light microscopy. Mean viral titers for each time point and corresponding standard deviations were calculated with Microsoft Excel. Bar graphs were plotted in SigmaPlot with standard errors shown for each sample.
Ad[I/PPT] promoter expression in primary human hepatocytes using green fluorescent protein (gfp) reporter adenovirus
Primary prostate and hepatocyte cells were infected with either AdCMV-gfp or AdI/PPT-gfp virus at MOI 500 for 2 hr. Virus was removed by washing the cells with culture medium, and the cells were incubated at 37°C, 5% CO2 for 48 hr. After this time, gfp fluorescent images were taken at ×60 magnification, under oil emersion, using a Nikon Eclipse TE300 fluorescent microscope.
Quantitative PCR (qPCR) analysis of adenoviral genome replication
Primary hepatocytes were infected in triplicate with AdMock and AdWT at MOI 500 and with Ad[I/PPT-E1A] at MOI 5, 50, and 500 for 2 hr at RT. Virus was removed by washing the cells with culture medium, and the cells were incubated at 37°C, 5% CO2. Cells were harvested at 2, 24, and 48 hr, and genomic DNA was extracted using a DNeasy Blood and Tissue kit (Qiagen). qPCR was performed on an ABI Step-One-Plus Real Time PCR machine. Amplification of an 84-bp fragment of the adenovirus fiber gene was carried out using the primers 5’ TGGCTGTTAAAGGCAGTTTGG 3’ and 5’ GCACTCCATTTTCGTCAAATCTT 3’ with detection of amplified sequences by a Taqman probe (5’ TCCAATATCTGGAACAGTTCAAGTGCTCATCT 3’), which was labeled at the 5’ end with the FAM fluorophore and at the 3’ end with the TAMRA quencher. Primers and probe were purchased from Sigma Genosys, UK. Reactions were carried out in a volume of 20 μl in ABI Taqman Fast Master mix (Applied Biosystems, UK), containing primers and probe at concentrations of 1.0 μM and 0.1 μM, respectively. Thermocycling parameters were 2 min at 50°C, 10 min at 95°C followed by 40 cycles of 95°C (30 sec) and 60°C (2 min). Analysis of the data was carried out using the software provided, and test samples were compared to standards of known viral DNA content. Standard curves were prepared by spiking serial dilutions of virus into hepatocyte genomic DNA. Data are presented as bar graphs plotted in SigmaPlot with standard errors shown for each sample.
Localization of adenovirus in primary cells with immunogold labeling of the hexon capsid protein using transmission electron microscopy (TEM)
Primary cells were seeded in appropriate cell culture media, onto Thermanox plastic coverslips (NUNC, UK) coated in a thin layer of Matrigel (Invitrogen). The cells were infected with MOI 500 of either AdWT, Ad[I/PPT-E1A], or AdMock for 1 hr. The virus was removed, and the cells were cultured for a further 36 hr. Cells were then fixed in 2% glutaraldehyde for 20 min. After washing in 100 mM sodium phosphate buffer, adherent cells were osmicated and dehydrated in a series of graded alcohols. A final solution of 50% TAAB resin (TAAB Laboratories Ltd., Australia) and 50% absolute ethanol was applied with warming to evaporate off all traces of alcohol. Resin-filled capsules were inverted over the coverslips and left to polymerize overnight at 60°C. Ultrathin (70 nm) sections were cut and collected on nickel grids. Adenovirus-infected cells were observed on an FEI Tecnai G2 transmission electron microscope, operated at 120 kV. Images were captured on an SIS MegaView II camera.
To localize the hexon capsid protein by immunogold labeling of virus particles, primary cells were infected with Ad[I/PPT-E1A] at MOI 500 for 1 hr at RT. The virus was removed, and the cells were cultured for a further 60 hr. Cells were washed in PBS, removed from the well surface using a cell scraper, and collected in a 1.5-ml Eppendorf tube. Cells were then fixed and processed for cryosectioning following the method of Tokuyasu (1986). Immunogold labeling of infected adenoviruses within the primary cells using a rabbit polyclonal anti-hexon antibody to adenovirus serotype 5 (catalog no. ab24240; Abcam, UK) was performed following the procedure of Mitry et al. (2000). Adenovirus within primary cells was observed on an FEI Tecnai G2 transmission electron microscope, operated at 120 kV. Images were captured on an SIS MegaView II camera.
Immunocytochemical detection of coxsackie adenovirus receptor (CAR) protein in nonprostate primary cells
CAR (green) was detected in nonprostate primary cells and prostate cell lines by immunofluorescent staining using a primary anti-CAR mouse monoclonal antibody (clone RmcB; Millipore Ltd.) (at 1:50) and a goat anti-mouse Alexa 488 secondary antibody (at 1:500). For cell morphology, actin (red) was detected using an anti-actin rabbit monoclonal antibody (clone EP184E; Millipore Ltd.) (at 1:300) and a goat anti-rabbit Alexa 568 secondary antibody (at 1:500). Cells were mounted in VECTORSHIELD containing DAPI (Vector Laboratories, Inc.) to visualize the nuclei (blue). Fluorescent images were taken at ×60 magnification, under oil, using a Nikon Eclipse TE300 fluorescent microscope.
Results
Cytotoxicity of Ad[I/PPT-E1A] infection of human primary cells
Using a colorimetric MTS assay to determine cytotoxicity, cell viability was measured to determine whether the prostate-specific Ad[I/PPT-E1A] could kill nonprostate primary cells. The cytotoxicity data for the infection of primary cells with a positive control adenovirus, AdWT, compared with the prostate-specific adenovirus, Ad[I/PPT-E1A], are shown in Fig. 1. AdWT virus was cytotoxic to all primary cells in a dose- and time-dependent manner, and at all concentrations tested all cells were dead by the final day (day 14) of the assay. In two positive control cell types (prostate cells), Ad[I/PPT-E1A] showed significant cytotoxic effects at MOI 500 and 5,000 in LNCaP cells and in basal primary prostate cells by day 14 (Fig. 1A and B). This confirmed that the MTS assay was able to detect cell death caused by Ad[I/PPT-E1A].
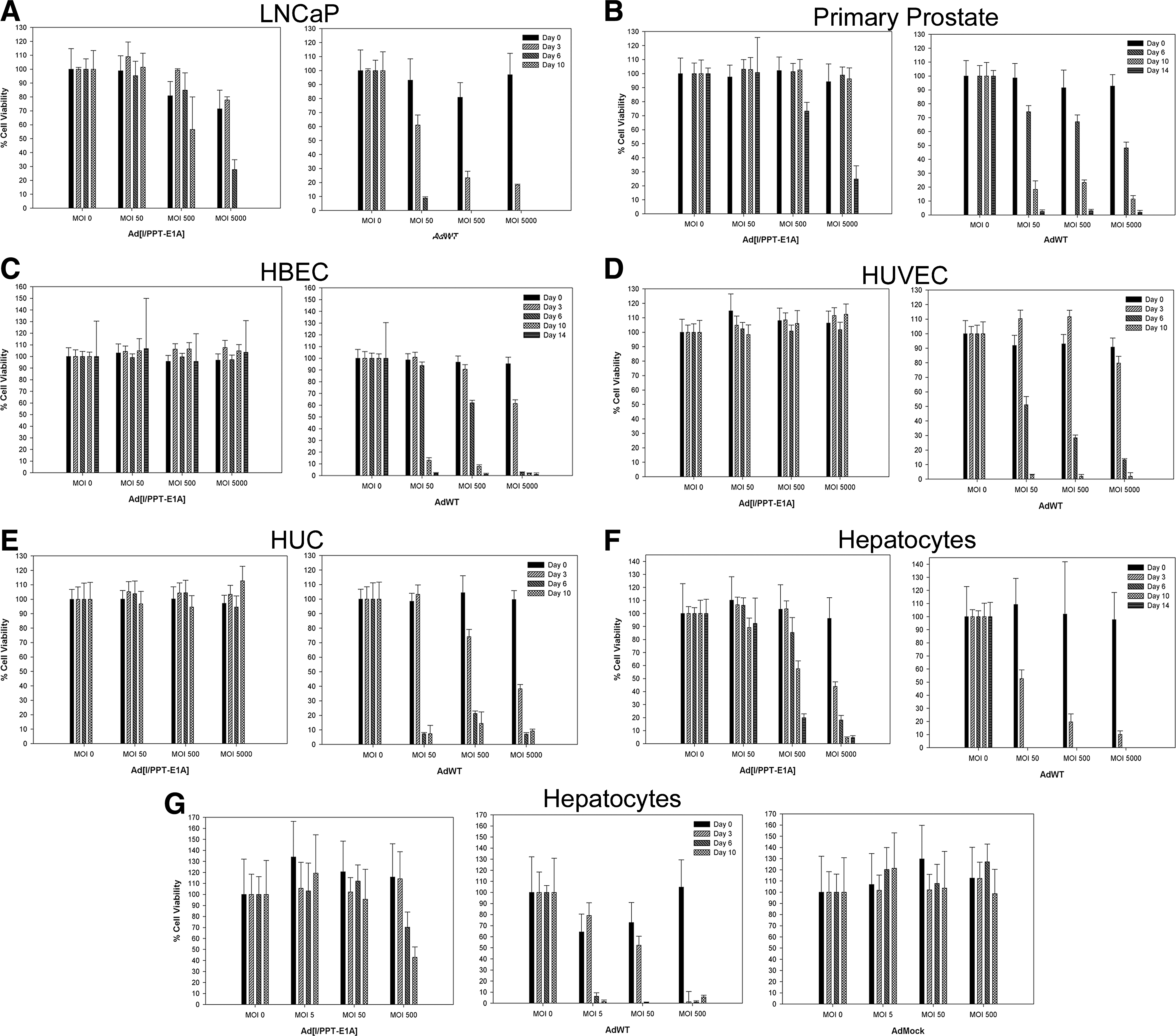
Cytotoxicity bar graphs for LNCaP cells, human primary basal prostate cells, human primary bronchial epithelial cells (HBEC), human primary umbilical vein endothelial cells (HUVEC), human primary urothelial cells (HUC), and human primary hepatocytes infected with Ad[I/PPT-E1A] or AdWT at MOI 0, 50, 500, and 5,000, and human primary hepatocytes treated with Ad[I/PPT-E1A], AdWT, or AdMock at MOI 0, 5, 50, and 500. MTS assays were performed on cells at days 0, 3, 6, 10, and 14 where possible. Viability is expressed as a percentage of untreated cells (MOI 0), and the means±SD are shown for triplicate wells.
There was no significant cytotoxicity associated with Ad[I/PPT-E1A] infections in bronchial, endothelial, or urothelial cells at any virus concentration (Fig. 1C–E and Table 1). In primary human hepatocytes, there was no significant cytotoxicity associated with Ad[I/PPT-E1A] infections at lower virus concentration (MOI 50). However, at higher viral amounts (MOI 500 and 5,000), a large number of dead hepatocytes were observed at day 6 with Ad[I/PPT-E1A] virus (Fig. 1F and G and Table 1). One possible mechanism of action for the cytotoxicity of Ad[I/PPT-E1A] in the fragile hepatocytes is an excessive virus uptake and overloading of the cell with virus particles. A replication-incompetent adenovirus (AdMock) was used to transduce primary hepatocytes to examine the potential of viral load for initiating the observed cytotoxic effects (Fig. 1G). These results showed no cytotoxicity associated with the replication-incompetent adenovirus, indicating that the cytotoxicity observed in hepatocytes infected with Ad[I/PPT-E1A] was most likely not due to nonspecific adenoviral particle overloading. However, digital images of hepatocytes in culture throughout the duration of the assay showed that the uninfected cells were dying by day 6, indicating their fragile nature when maintained in prolonged in vitro cell culture conditions (Fig. 2).
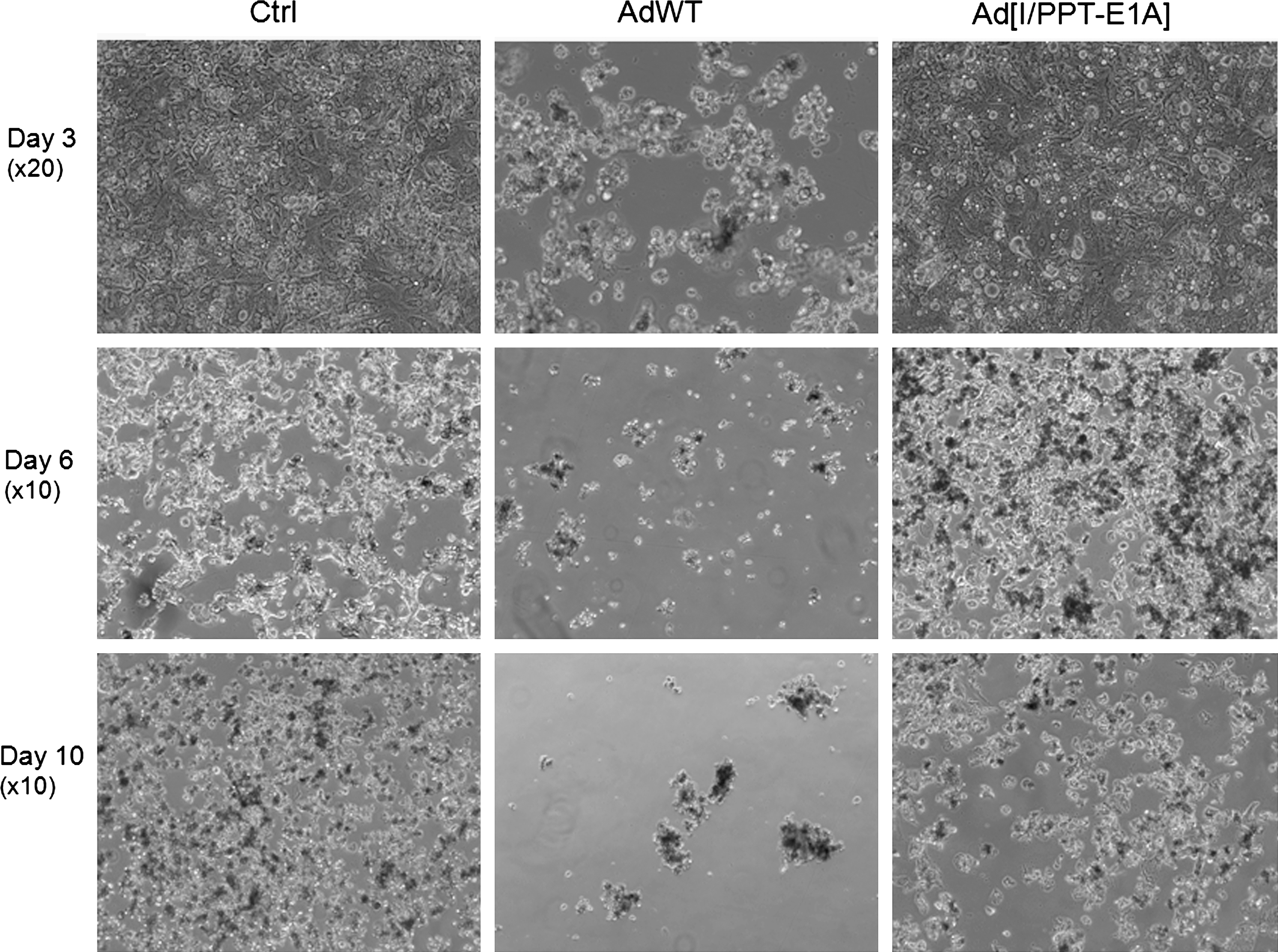
Digital images of cultures of uninfected primary hepatocytes (no virus) and hepatocytes infected with AdWT or Ad[I/PPT-E1A] at MOI 500. Images were taken on days 3, 6, and 10 post infection. ×10 and ×20 refer to the microscope magnification of the images.
Viability at day 6 for human primary urothelial cells, at day 10 for human primary umbilical vein endothelial cells and human primary hepatocytes, and at day 14 for human primary bronchial epithelial cells after treatment with Ad[I/PPT-E1A] or AdWT at MOI 500 is expressed as a percentage of untreated cells (MOI 0). The means±SD are shown for triplicate wells.
The cytotoxicity seen in hepatocytes was further investigated by adenovirus gfp reporter assays, genome-replication studies, adenoviral replication assays, and TEM studies to determine whether the Ad[I/PPT-E1A] virus was actively replicating, or whether this observation was due to another effect (see below).
Ad[I/PPT-E1A] replication in human primary cells
The replication capacity of Ad[I/PPT-E1A] in vascular endothelial cells, bronchial epithelial cells, urothelial cells, and hepatocytes was assessed by determining the presence of active viral particles secreted into the cell culture media at various times after viral infection, and in cell lysates at the final time point of the assay. Primary prostate cells along with a prostate-derived cell line, LNCaP, were used as positive cell-type controls for Ad[I/PPT-E1A] replication. The collected supernatants and cell lysates were titered by infection of HEK293 cells, and the infectious plaques resulting from active Ad[I/PPT-E1A] virus were detected by immunostaining for adenovirus hexon protein (Fig. 3). As a reference for nonspecific viral replication, wild-type adenovirus (AdWT) was included in each assay. The infectivity of the cell-culture supernatants collected at the different time points was compared with the level of infectivity of the input virus (AdWT or Ad[I/PPT-E1A]), namely, the “infection titer.” To define the infection titers, HEK293 cells (2.5×105) were directly infected with the same concentrations (in VP/ml) of AdWT or Ad[I/PPT-E1A] as were used to infect the prostate cancer cell line and human primary cells. These concentrations differed between the various cell types (see Table 2). In cases where the data showed viral titers above the infection titer, we concluded that the virus was actively replicating in these cells (Fig. 3A–C). Viral titers below the infection titer suggested that the virus had been sequestered internally, and active viral replication was not occurring. Throughout the duration of the assay, cells undergoing apoptosis released their sequestered virus into the medium, which was observed in titers at day 6, 10, and 14 in bronchial epithelial cells, urothelial cells, and hepatocytes (Fig. 3D, F, and G).
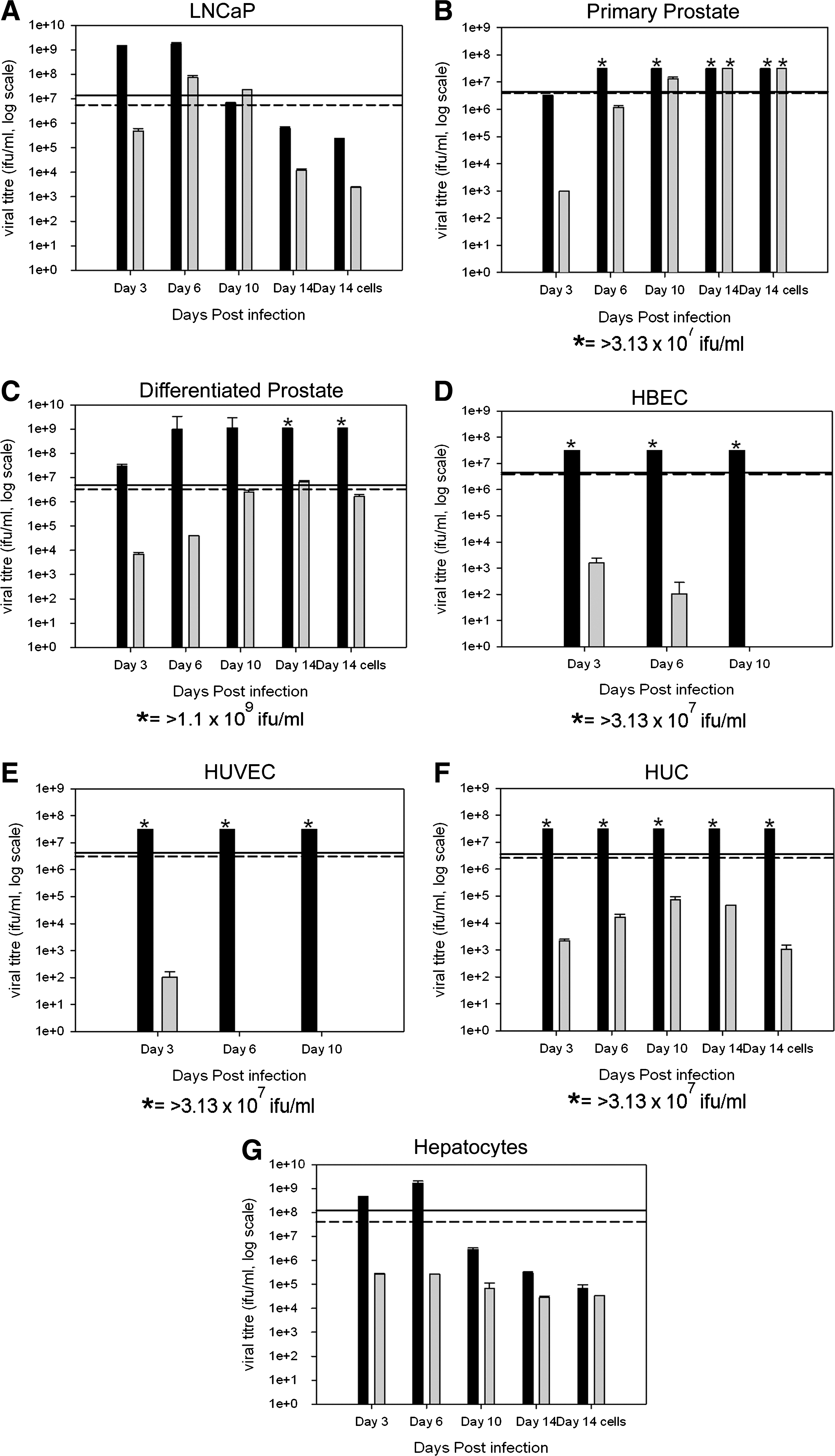
Viral replication bar graphs for LNCaP cells, human primary basal prostate cells, human primary differentiated prostate cells, human primary bronchial epithelial cells (HBEC), human primary umbilical vein endothelial cells (HUVEC), human primary urothelial cells (HUC), and human primary hepatocytes infected with Ad[I/PPT-E1A] and AdWT at MOI 500. Infected culture media collected on days 3, 6, 10, and 14 (where possible) were titered in hexon titer assays, and the means±SD are shown for triplicate wells. The lines indicate the input load of infectious virus: solid line, AdWT; dashed line, Ad[I/PPT-E1A]. Black bars, AdWT virus; gray bars, Ad[I/PPT-E1A] virus. *Viral titer is greater than maximum titer measured in the hexon titration assay, i.e., >3.13×107 ifu/ml or >1.1×109 ifu/ml.
Human primary cell and LNCaP cell input viral titer counts (in VP/ml) and directly assayed active viral infection titers (in ifu/ml) were measured by hexon titer assays for AdWT and Ad[I/PPT-E1A]. The mean values are shown for triplicate wells.
To establish the sensitivity and time parameters for the assay, infections of permissive LNCaP cells with AdWT and Ad[I/PPT-E1A] were carried out (Fig. 3A). The data revealed that both AdWT and Ad[I/PPT-E1A] actively replicated in LNCaP cells. The replication level titers were 10-fold less for Ad[I/PPT-E1A], and there was also a slight delay compared with the AdWT virus (Fig. 3A). Similar replication assays were carried out with cultures from primary prostate tissue, both undifferentiated (basal cells) and cultures induced to differentiate to a luminal phenotype, both at MOI 500 (VP/cell). This MOI value was chosen to eliminate even inefficient attachment and replication as a possibility in vivo, although such viral titers would be impossible to achieve except in close proximity to the needle-injection site. We observed active replication of Ad[I/PPT-E1A] in basal prostate epithelial cells, increasing to >3.13×107 infectious units (ifu)/ml by day 14 (ifu >795% of the viral load) compared with a peak titer of 6.72×106 ifu/ml at day 14 in differentiated prostate epithelia (ifu 172% of the viral load) (Fig. 3B and C). Furthermore, the rate of replication of Ad[I/PPT-E1A] in differentiated prostate epithelial cells was much slower than that in both LNCaP cells and basal prostate epithelial cells, suggesting that viral replication in these cells is attenuated.
In the cases of bronchial, endothelial, and urothelial cells, there was no detectable replication of Ad[I/PPT-E1A] when compared with the input infection titers (Fig. 3D–F). However, there was evidence of sequestered viral release throughout the different time points of the assay, as the cells naturally died in culture. In contrast, efficient replication of AdWT was observed in these cell types.
We examined four independent donor primary hepatocyte samples, all of which showed high levels of AdWT replication. Although significant amounts of Ad[I/PPT-E1A] were retained by the cells, no net viral increase was observed, suggesting that Ad[I/PPT-E1A] does not replicate in hepatocytes (Fig. 3G). The data indicated that hepatocytes can ingest both AdWT and Ad[I/PPT-E1A]. Table 3 summarizes viral yields per cell for each cell type studied, as well as the maximum percentage viral load. When compared with active replication in primary prostate samples and LNCaP cells, the replication data for Ad[I/PPT-E1A] indicate a greatly reduced number of virus particles per cell in the nonprostate primary cells, including hepatocytes, after infection with the high MOI 500, indicating the presence of residual, nonreplicating virus.
Replication status of AdWT and Ad[I/PPT-E1A] in the primary cell types and LNCaP cell line is shown with the peak viral titers, the number of infectious viral particles per cell, and the active virus represented as a percentage of the input virus load.
The number of infectious units (ifu) per cell calculated by dividing the peak viral titer by the number of cells available for infection at day 0.
I/PPT transcriptional activity in primary hepatocytes and prostate cells
To establish the possibility of active Ad[I/PPT-E1A] replication in primary hepatocytes, we examined I/PPT promoter activity in both primary hepatocytes and primary prostate cells using serotype 5 adenoviruses that express gfp. The primary cells were infected for 48 hr with MOI 500 AdI/PPT-gfp or AdCMV-gfp. Figure 4 shows gfp expression from AdI/PPT-gfp virus in prostate cells, but not in hepatocytes, whereas the control AdCMV-gfp virus expressed gfp in both cell types after this time.
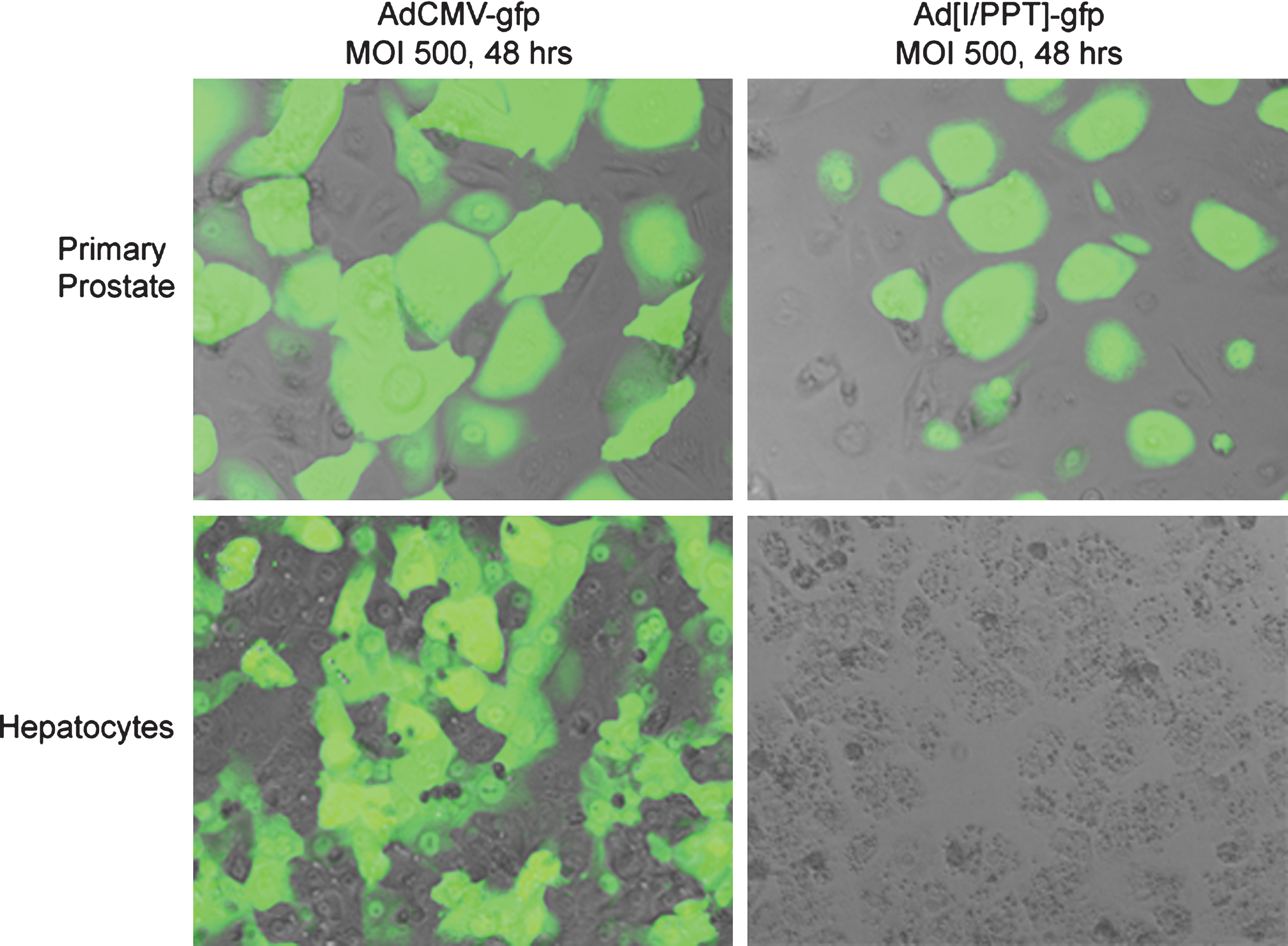
Fluorescent images showing I/PPT-specific promoter activity in primary human prostate cells, but not in human hepatocytes, 48 hr after infection with MOI 500 AdCMV-gfp virus or Ad[I/PPT]-gfp virus. Images were taken at ×60 magnification under oil.
Ad[I/PPT-E1A] genome replication in hepatocytes
qPCR studies were performed in primary hepatocytes to monitor any increase in adenoviral genome content between 2 and 48 hr post infection (Fig. 5). Hepatocyte cultures were infected in triplicate with AdMock or AdWT at MOI 500 and with Ad[I/PPT-E1A] at MOI 5, 50, and 500. Genomic DNA was extracted from the cells at 2, 24, and 48 hr, and adenoviral genome quantities were analyzed by qPCR targeting an 84-bp fragment of the adenoviral fiber gene. At 48 hr, at MOI values under 500, which are not associated with a cytotoxic effect in hepatocytes, Ad[I/PPT-E1A] genome-replication levels were equivalent to those of the replication-incompetent AdMock, i.e., 1.21×10–3 ng compared with 1.36×10–3 ng for MOI 5, respectively. This level was 3,600 times lower than levels with AdWT and comparable to the hepatocyte hexon titer replication data. However, at the much higher MOI 500, although the Ad[I/PPT-E1A] genomic DNA quantity had increased, it was still significantly lower (45 times) than that of the AdWT control. The data suggest that at extreme MOI values at or above 500, Ad[I/PPT-E1A] genomic DNA replication occurs, but as the replication assays do not show active viral particle production even at MOI 500, it is unlikely that these genomes are packaged into infective viral particles that could actively establish secondary infections. TEM studies were performed to examine this finding further.
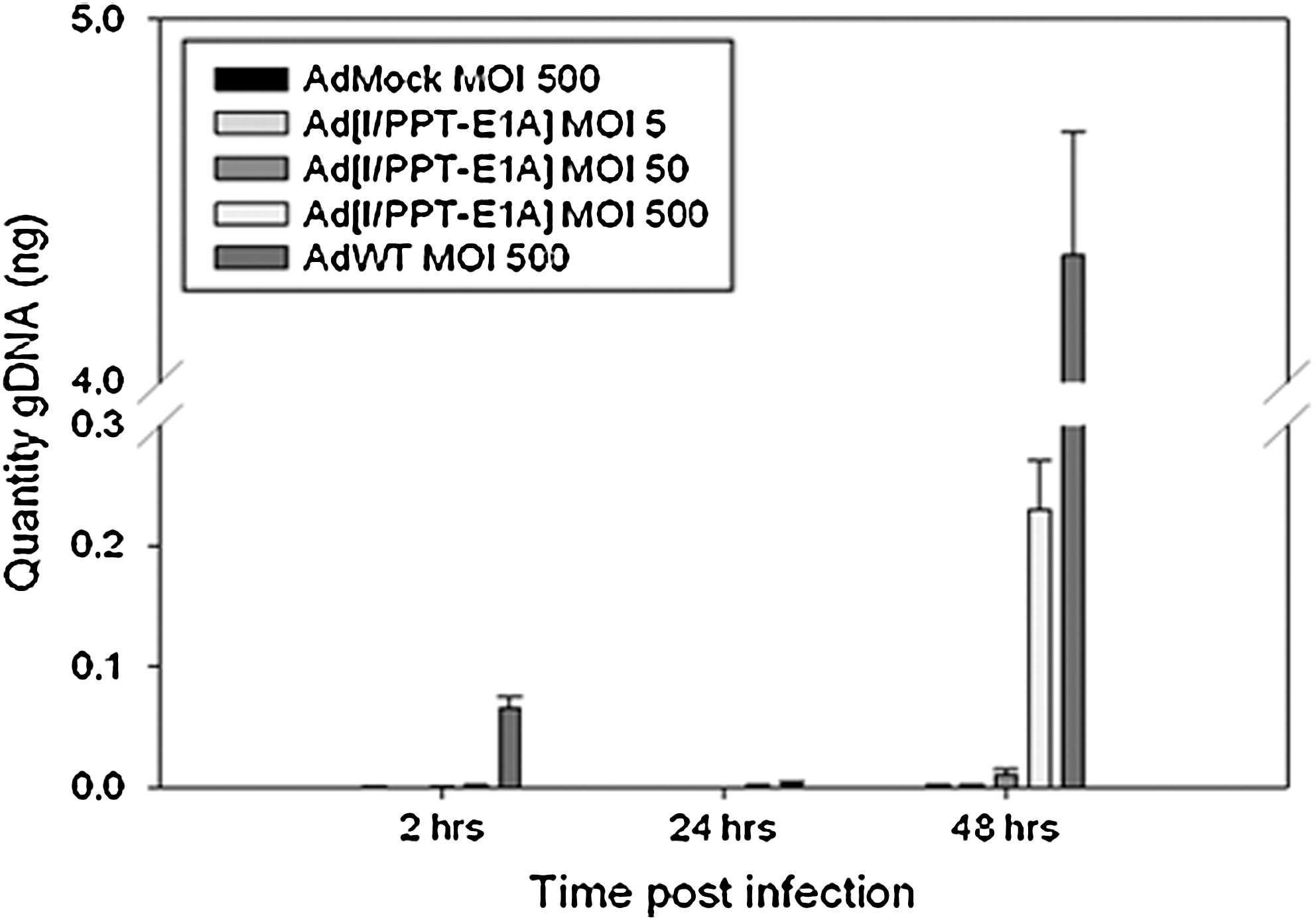
Adenoviral genome replication bar graph for human hepatocytes, measured by qPCR. Hepatocyte cells were infected in triplicate with AdMock or AdWT at MOI 500 and with Ad[I/PPT-E1A] at MOI 5, 50, and 500. Cells were harvested at 2, 24, and 48 hr, and qPCR to detect viral DNA quantities was performed using a probe/primer mix targeted to an 84-bp fragment of the adenovirus fiber gene. The means±SEM are shown for triplicate wells.
Intracellular localization of Ad[I/PPT-E1A] particles in human primary hepatocytes by TEM and immunogold TEM
We examined the location of adenovirus particles in infected basal prostate cells and hepatocytes initially by TEM. TEM images showed replicated AdWT in “rafts” within the nuclei of both primary prostate cells and primary hepatocytes by 60 hr post infection (Fig. 6A and C). We also observed replicated Ad[I/PPT-E1A] in similar rafts in the nuclei of primary prostate cells (Fig. 6B). Replication rafts were not observed in the nuclei of primary hepatocytes with either Ad[I/PPT-E1A] or AdMock virus. However, there were large numbers of Ad[I/PPT-E1A] and AdMock virus particles sequestered, only in the hepatocyte cytoplasm, indicating no evidence for viral replication in these cells. (Fig. 6D and E). To validate that the particles observed within the cell cytoplasm, as well as the raft formations in the nuclei, were adenovirus, we performed immunogold labeling of the hexon protein located in the viral capsid, using a rabbit polyclonal antibody linked to immunogold nanoparticles. In primary prostate cells, the virus particles situated in the nuclear rafts colocalized with the hexon immunogold nanoparticles (Fig. 6F and G).
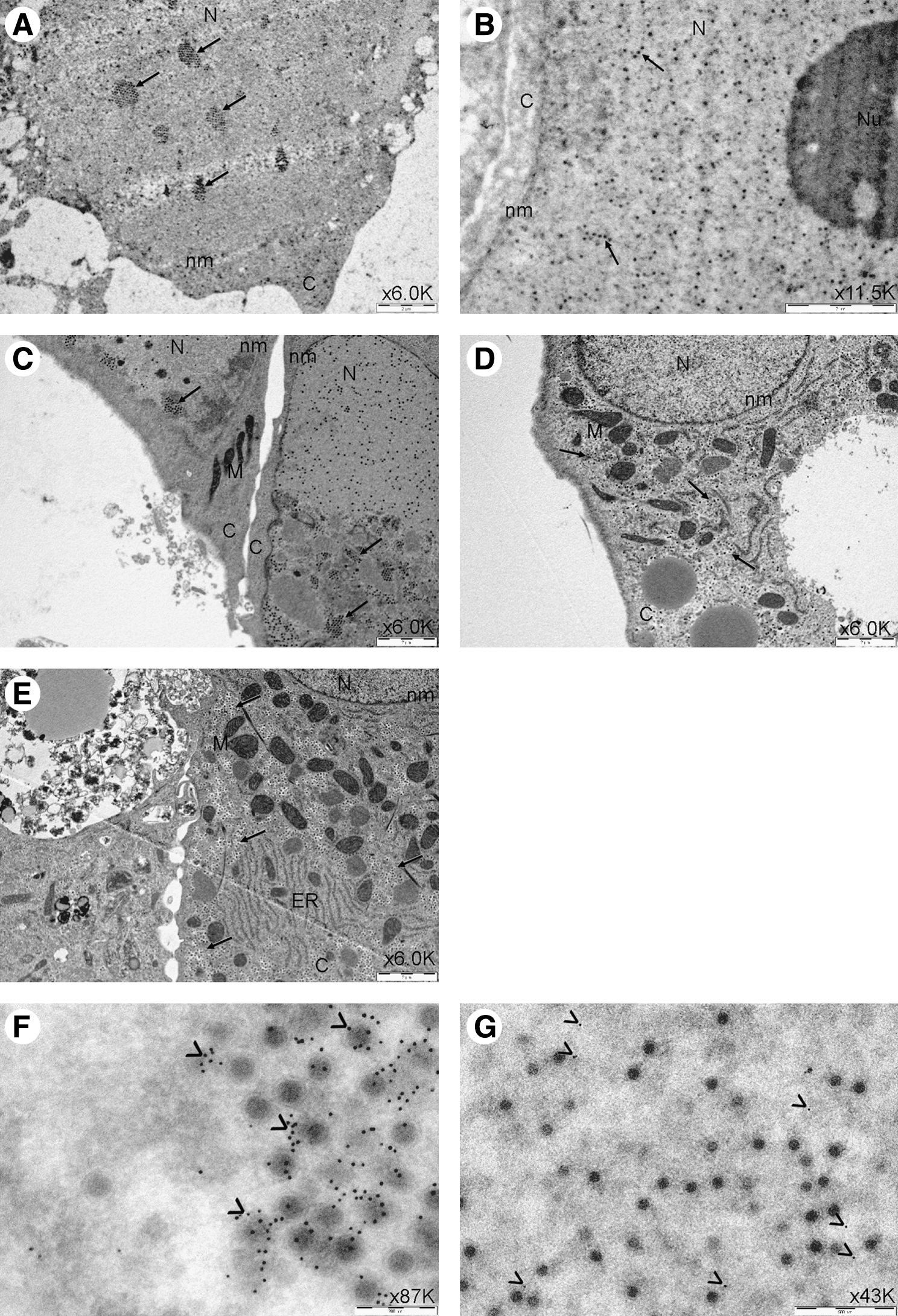
TEM images of human primary prostate cells
CAR levels in nonprostate primary cells
Finally, the protein levels of the primary adenovirus receptor CAR (green) were measured by immunocytochemistry in primary nonprostate bronchial epithelial cells (HBEC), vascular endothelial cells (HUVEC), and hepatocytes and compared with those of the prostate cell line LNCaP and the adenoviral replication-competent HEK293 cells. Figure 7 confirms that CAR is expressed at high levels in HEK293 cells and LNCaP cells, particularly at lateral cell junctions where it colocalizes (yellow) with actin (red). In contrast, very little CAR was detected in the bronchial epithelial cells and vascular endothelial cells. Interestingly, higher CAR levels were seen in the cytoplasm and at the cell surface of primary hepatocytes, suggesting a possible route of viral entry into these cells.
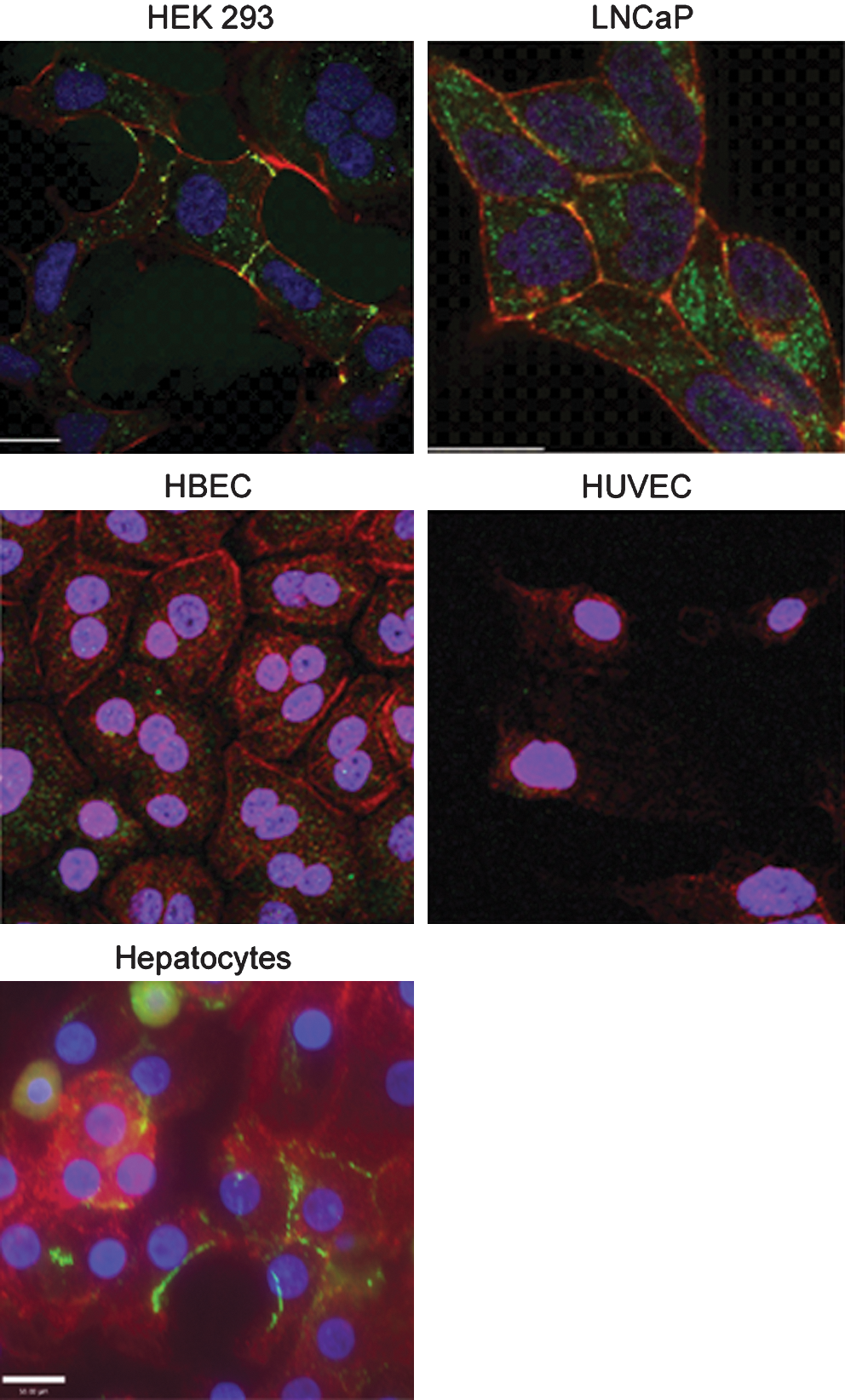
Immunocytochemical detection of CAR protein (green) in HEK293 cells, LNCaP cells, primary human bronchial cells (HBEC), primary human umbilical vein endothelial cells (HUVEC), and human hepatocytes. Actin (red) stained for cell morphology, and DAPI (blue) was used to visualize the nuclei. Images were taken at ×60 magnification, under oil.
Discussion
We have applied in vitro cell culture techniques as a preclinical testing approach to examine the ability of Ad[I/PPT-EA], a human prostate-specific conditionally replicating adenovirus, to infect, replicate, and produce cytotoxic effects in nonprostate primary cells from humans. Initially, we examined the cytotoxic effects of Ad[I/PPT-E1A] at a range of MOI values in primary bronchial epithelial cells, urothelial cells, vascular endothelial cells, and hepatocytes using an MTS assay. We compared these data with viral cytotoxicity in two permissive cell types: the LNCaP prostate cell line and primary prostate cells isolated from a prostate cancer patient. The data revealed high levels of cytotoxicity of Ad[I/PPT-E1A] in both prostate cell types studied, and confirmed that no significant cytotoxic effects were observed in all the nonprostate cells, apart from the human hepatocytes (Fig. 1F). This effect could not be explained by active replication of Ad[I/PPT-E1A] virus particles, leading to viral burst and subsequent death of infected hepatocytes, because replication was not detected in these cells using the hexon immunostaining assay, which stains for the capsid protein (see below). Equally, I/PPT promoter activity was not observed in primary hepatocytes infected with an AdI/PPT-gfp virus (Fig. 4). In contrast, AdWT induced cytotoxicity of human primary hepatocytes even at MOI 5 (Fig. 1G), implying at least 100-fold higher activity toward these cells compared with Ad[I/PPT-E1A]. Furthermore, AdWT was confirmed to replicate in primary hepatocytes.
Viral replication assays were carried out to examine the replication activity of Ad[I/PPT-E1A] in the nonprostate primary cell types. By infecting the primary cells with Ad[I/PPT-E1A] at MOI 500 and titering the medium samples collected at different time points by hexon titer assays, the data clearly showed that Ad[I/PPT-E1A] did not actively replicate in any of the nonprostate primary cell types studied, including hepatocytes. In contrast, highly active replication was observed in the positive control cells, i.e., the LNCaP prostate cell line and the primary prostate cells. Interestingly, primary prostate cells induced to terminally differentiate and form cellular multilayers in the presence of serum and 10 nM dihydrotestosterone showed delayed viral replication with Ad[I/PPT-E1A], indicative of attenuated viral replication. This may be because superficial differentiated luminal-like cells can mask the CAR located on the underlying basal cell surface, which is required for adenoviral infection. Evidence for a reduction in CAR expression in differentiated primary prostate tissue has been previously reported by IF studies (Maitland et al., 2010). This was not observed for the AdWT virus; however, at high viral titers, AdWT may also transduce cells via other surface receptors and by endocytosis mechanisms (Carlisle et al., 2009).
The transcriptional activity of the prostate-specific I/PPT promoter was examined in primary hepatocytes using a gfp-reporter adenovirus (Fig. 4). After 48 hr, no evident gfp expression was observed in hepatocytes infected with MOI 500 AdI/PPT-gfp. However, the I/PPT promoter was transcriptionally active in primary prostate cells by this time.
To examine this further, we used a highly sensitive qPCR assay to quantify Ad[I/PPT-E1A] genome replication in hepatocytes at MOI 5, 50, and 500 and compared the levels with those of AdMock and AdWT at MOI 500. At MOI < 500, the quantity of Ad[I/PPT-E1A] genomic DNA was comparable to that of the replication-incompetent AdMock. Only at the highest MOI used (MOI 500), was there an increase in genome quantity, indicative of genome replication. This may be due to “leaky” TARP promoter activity within the adenovirus genome. In this context, the TARP promoter is surrounded by less condensed chromatin compared with the human genome context (on chromosome 7), where highly condensed chromatin tightly regulates TARP gene expression. However, the hepatocyte replication data indicated that active viral replication due to viral burst, cell death, and reinfection was not occurring in these cells, suggesting that these genomes were not packaged into infective, replication-competent adenovirus. Electron microscopy images were taken of AdWT-, AdMock-, and Ad[I/PPT-E1A]-infected prostate cells and human hepatocytes. The data confirmed the hypothesis that Ad[I/PPT-E1A] virus did not actively replicate in hepatocytes, but was sequestered in large numbers in the cell cytoplasm and was not present in the nucleus. This observation was confirmed with the replication-incompetent virus, which was also present in high numbers in the hepatocyte cytoplasm. In contrast, AdWT was organized in rafts in the nuclei of both primary prostate cells and primary hepatocytes, which is indicative of replication.
Taking the above observations into consideration, the replication data (Fig. 3) for hexon immunostaining, the adenoviral-gfp reporter assay, and the outcome of the electron microscopy analyses, all indicate that Ad[I/PPT-E1A] does not actively replicate in human primary hepatocytes. The cytotoxic effect observed in these cells upon treatment with Ad[I/PPT-E1A] (Fig. 1F and G) might be explained by cytoplasmic sequestration of viral particles in combination with a fragile condition and short life span of human primary hepatocytes in in vitro culture (Fig. 2). The increased CAR levels observed in the hepatocyte cytoplasm and on the surface membranes (Fig. 7), when compared with bronchial epithelial cells and vascular endothelial cells, could clearly result in higher levels of adenovirus being sequestered within the hepatocyte cytoplasm.
It is well known that culture conditions, including seeding density, matrix conditions, and medium supplements, affect the morphological development, expression of metabolic enzymes, response to drugs, and survival of cultured human primary hepatocytes. To the best of our knowledge, the effect of oncolytic adenoviruses on cultured human primary hepatocytes has not been reported. Therefore, it remains to be clarified whether this phenomenon is an artifact of the model system used. When an extrapolation of the in vitro conditions in which cytotoxicity in cultured human primary hepatocytes, as observed here, is made to the in vivo situation, and assuming a worst case scenario of maximal viral progeny generation within the prostate, substantial leakage of virus from the prostate into the circulation, and lack of any neutralization of systemic virus in the circulation, it is clear that the in vitro upper safety margin threshold (MOI 50) will never be achieved in a liver of a patient treated locally with Ad[I/PPT-E1A] in the planned clinical trial. It has to be taken into account that the human body is capable of efficient neutralization of systemic adenovirus by binding to erythrocytes via the complement receptor CR1 and CAR expressed on the surface of these cells (Carlisle et al., 2009) and by the immune system. These neutralization effects are the most likely explanation for the fact that systemic adenovirus infection in immunocompetent individuals, such as the patients to be recruited in the planned trial, is not associated with serious effects, and that to date, oncolytic adenoviruses have been well tolerated by patients even upon systemic administration. For example, intravenous administration of CG7870, a PSA-selective oncolytic adenovirus expressing the E3 region proteins that are involved in controlling the host immune response, was well tolerated up to 6.0×1012 VP in patients with hormone-refractory prostate cancer (Small et al., 2006). In addition, only 20% of the patients with metastatic colorectal cancer treated with up to eight infusions in the hepatic artery from 2.0×108 to 2.0×1012 VP of Onyx-015 experienced transient grade I/II hepatic toxicity (Au et al., 2007). Finally, mild and transient dose-dependent transaminitis was observed after repeated intravenous injections with Onyx-015 at doses of 2.0×1012 VP and higher in 10 patients with metastatic solid tumors (Nemunaitis et al., 2001). The systemic levels in these three trials are comparable to the levels that will be locally administered in our trial (1.0×1011 to 5.0×1012 VP). Thus, if a part of the locally administered Ad[I/PPT-E1A] does leak into the circulation, no adverse effects are to be expected. Based on results from previous prostate cancer trials on oncolytic adenoviruses, leakage of Ad[I/PPT-E1A] to the circulation upon intraprostatic administration is expected to be minimal. DeWeese et al. (2001) showed by qPCR that after a single intraprostatic treatment at 20–80 deposits up to 1.0×1013 VP with the prostate-specific oncolytic adenovirus CV706, 0.007–0.129% of the administered dose could be detected in blood after 30 min in the majority of the patients. This was followed by a second viral DNA peak in 13 out of 16 patients between days 2 and 8. In four patients, this peak was higher than the first peak with the highest peak at <2% of dose. It was not reported if the PCR signal was derived from viable virus. In the trials conducted by Freytag et al. (2002, 2003), using a single intraprostatic injection up to 1.0×1012 VP with the nonprostate-specific oncolytic adenovirus Ad5-CD/TKrep, viral DNA was found in blood up to day 76, but infectious virus could not be demonstrated.
To reduce hepatotoxicity further, the use of microRNA-regulated gene expression systems to enhance the safety and efficacy of viral gene-therapy approaches has been described (Sakurai et al., 2011). By inserting four copies of the hepatocyte-specific microRNA, mir122, into the 3’ untranslated region of the adenoviral E1A transcription cassette, Cawood et al. (2011) were able to genetically reduce hepatic adenovirus replication and liver pathology. If required, such an approach could be applied to further enhance Ad[I/PPT-E1A] in future clinical trials.
The data presented here confirm the specificity of Ad[I/PPT-E1A] and are part of the clinical dossier submitted for regulatory approval. The risk of adverse events in patients treated with Ad[I/PPT-E1A] related to the in vitro observation of cytotoxicity of cultured human primary hepatocytes by clinically unrealistically high amounts of Ad[I/PPT-E1A] is minimal and will be controlled by recruiting only immunocompetent patients. As a prelude to clinical trials, the use of multiple primary cultures from a variety of normal human cell types should provide a convenient assay for testing oncolytic adenovirus specificity in vivo, to limit and predict any undesired off-target effects.
Footnotes
Acknowledgments
We thank Mr. Mike Stower (York District Hospital, York, UK) and Mr. Matthew Simms (Castle Hill Hospital, Hull, UK) for providing prostate tissue samples; Professor Jenny Southgate (Jack Birch Unit, University of York, York, UK) for donating primary human urothelial cells; Berith Nilsson for provision of AdWT genomic DNA; Meg Stark for technical assistance with electron microscopy; and members of the Yorkshire Cancer Research Unit for their technical advice. This work was supported by the European Union through the Sixth Framework Programme Integrated Project GIANT (contract no. LSHB-CT-2004-512087). Norman Maitland also received invaluable core support from Yorkshire Cancer Research.
Author Disclosure Statement
Rachel Adamson, April Frazier, Helen Evans, Karen Chance, Ellen Schenk, Magnus Essand, Richard Birnie, Ragai Mitry, Anil Dhawan, and Norman Maitland declare no competing financial interests.