
Abstract
The neuronal ceroid lipofuscinoses (NCLs; Batten disease) are inherited neurodegenerative lysosomal storage diseases with common clinical features of blindness and seizures culminating in premature death. Gene-therapy strategies for these diseases depend on whether the missing activity is a secreted lysosomal protein taken up by neighboring cells, or an intramembrane protein that requires careful targeting. Therapies are best developed in animal models with large complex human-like brains. Lentiviral-mediated gene delivery to neural cell cultures from normal sheep and sheep affected with an NCL resulted in green fluorescent protein (GFP) expression in neurons and neuroblasts, more efficiently than in astrocytes. Similar transgene expression was obtained from two constitutive promoters, the viral MND promoter and the human EF1α promoter. In vivo studies showed stable and persistent GFP expression throughout the cell bodies, axons, and dendrites from intracortical injections and indicated ependymal and subependymal transduction. The sheep showed no ill effects from the injections. These data support continuing gene-therapy trials in the sheep models of Batten disease.
Introduction
Studies in human NCLs are very restricted, and progress to understand the biochemistry, pathogenesis, and therapeutic options depends largely on studying genetically defined animal models. Viral vector-mediated gene-therapy trials have been used to deliver palmitoyl protein thioesterase I (PPT1) and tripeptidyl peptidase I (TPP1), the enzymes deficient in infantile (CLN1) and late-infantile (CLN2) NCL, to knockout mouse models, and a clinical trial is ongoing in human patients with CLN2 mutations. Neonatal delivery of PPT1 partially reversed some pathological and behavioral characteristics of infantile NCL in mice, but did not change the age of death (Griffey et al., 2004, 2005, 2006). Delivering TPP1 to TPP1-deficient mice was more effective if given early in the disease progression (Sondhi et al., 2007, 2008). Viral-mediated gene transfer has also been a relative success in other lysosomal storage disorders caused by defects in soluble lysosomal proteins, most notably in both mouse and large-animal models of mucopolysaccharidoses (Haskins, 2009).
Scaling up from a small lissencephalic rodent brain to a large one replete with gyri and sulci presents a number of problems, and large-animal studies are particularly important for translating advances in viral-mediated gene therapy to the clinic. Issues to be resolved include ensuring that transduction is stable and that transduction and expression do not adversely affect the brain. As it is not practicable to transduce most cells in the brain, priority needs to be given to identify target cells that are accessible. The larger brain requires the development of much more sophisticated injection and surgical techniques than are necessary in mice.
Two naturally occurring forms of CLN5 and CLN6 NCL have been found in sheep, and well established flocks are available for model studies (Jolly et al., 2002; Tammen et al., 2006; Frugier et al., 2008). Both are classical subunit c of mitochondrial ATP synthase–storing forms of NCL (Chen et al., 2004; Frugier et al., 2008). The sheep brain has a similar complex structure and is more comparable in size to the human brain; also, sheep have a similar physiology and genetic organization. Neuropathology in these NCLs in both humans and sheep is dominated by cortical atrophy and neurodegeneration, and symptoms in the sheep closely parallel those in the human diseases. Although there is pathology in the human brainstem and thalamus at end-stage disease, no significant changes in these regions are seen in the sheep. This is possibly due to the sheep not being as intensively supported at end-stage disease, so that the degree of progression may not be as advanced as human disease. Even with this caveat, the similarities make the sheep particularly good experimental models of the human diseases.
Mutations in CLN5 result in the loss of function of an uncharacterized soluble mannose-6-phosphate–tagged lysosomal protein (Holmberg et al., 2004; Sleat et al., 2005, 2006). Along with CLN1, CLN2, and CLN10, which also encode soluble lysosomal proteins, this gene is an attractive gene-therapy candidate because of the phenomenon of cross-correction, where soluble lysosomal proteins can be secreted from cells and taken up by other cells via endocytosis. In contrast, the other form in sheep, CLN6, arises from mutations in a nonsecreted, endoplasmic reticulum membrane–associated protein (Gao et al., 2002; Wheeler et al., 2002; Heine et al., 2004; Mole et al., 2004). The products of CLN3, CLN7, and CLN8 are also membrane-associated intracellular proteins. These are likely to require careful targeting of a vector that spreads widely in the brain and has a high efficiency, unless the critical products of processing by these proteins are themselves secreted and endocytosed, such as neuroprotective factors that promote cell survival.
Lentiviruses are a family of complex retroviruses that, together with adeno-associated viruses (AAVs), are commonly used to derive vectors for gene therapy. Both these vectors have been tested previously in models of lysosomal storage disorders in mice. Lentiviruses were successfully used to treat a mouse model of mucopolysaccharidoses (Brooks et al., 2002) and to test the spread of virus and secretion of TPP1 in the mouse brain (Haskell et al., 2003).
As a prelude to gene-therapy trials in NCL sheep, we set out to determine whether recombinant lentiviral vectors would target sheep neural cells. Previously developed methods to culture neural cells isolated from fetal sheep result in a mixed population including neural precursors (neuroblasts), mature neurons, astrocytes, and microglia (Kay et al., 2006). These cultures were used to test the transduction and tropism (as measured by transgene expression) of lentiviral vectors for sheep neural cells. The efficiency of gene transfer between affected and normal unaffected sheep and the expression of transgenes from two different promoters, the myeloid proliferative U3 enhancer element (MND) (Challita et al., 1995; Kobayashi et al., 2005) and the human elongation factor 1α promoter (EF1α), were tested. MND is a strong constitutively active viral promoter previously tested in hematopoietic cells (Avilés Mendoza et al., 2001) and muscle (Richard et al., 2008). EF1α is also constitutively active and is commonly used in viral vectors for transgene expression in a variety of tissues, including the brain (Jakobsson et al., 2003). The lentiviral vectors were then used to test gene transfer in vivo, by direct stereotactic injection into the cerebrum or cerebrospinal fluid (CSF) of adult control and CLN6-affected sheep. The results of this study pave the way for future work aimed at targeting subsets of neural cells in NCL and other neurodegenerative disorders where sheep may be used as models of human disease.
Materials and Methods
Viral constructs and packaging
Approval for the use of recombinant lentiviral vectors was obtained from ERMA New Zealand (GMD03091). HIV-1–derived lentiviral plasmids (Meyerrose et al., 2008) expressing green fluorescent protein (GFP) under the myeloid proliferative U3 enhancer element (pLVMNDGFP) or human elongation factor 1α promoter (pLVEF1αGFP) (see Fig. 1a) were packaged using a third generation packaging system (Zufferey et al., 1998). In brief, 5 × 106 293FT cells (Invitrogen, Carlsbad, CA) in a T75 flask (Nunc A/S, Roskilde, Denmark) were transfected with 11.25 μg of pLV, 7.3 μg of pLP1, 2.8 μg of pLP2, and 3.95 μg of pVSV-G (Zufferey et al., 1998) in OptiMEM containing Lipofectamine-2000 (Invitrogen). Medium containing virus was recovered 48 and 72 hr post transfection and concentrated by ultracentrifugation in a Beckman SW28 rotor (Beckman Coulter, Brea, CA) at 112,500 g max for 90 min at 4°C, resuspended in phosphate-buffered saline (PBS) containing 40 g/L lactose, and stored at −80°C. Functional viral titers, determined by serial dilution on human HT1080 cells (ATCC, Manassas, VA), ranged from 1 × 109 to 1.3 × 1010 transducing units (TU)/ml. RNA genome titers were determined using a Quick Titer Lentivirus Quantitation kit (Cell Biolabs Inc., San Diego, CA) and were proportional to functional titers between constructs. For in vivo work, 8 μg/ml Polybrene (Sigma–Aldrich, Castle Hill, NSW, Australia) was added to the viral solution just prior to injection.

Lentiviral-mediated gene transfer to sheep neural cells in vitro.
Viral transduction of neural cell cultures
Frozen stocks of sheep fetal neural cells, isolated as previously described (Kay et al., 2006), were thawed rapidly, counted, and plated at 1 × 105 cells/cm2 on 10 μg/ml poly-L-lysine–coated 12-mm coverslips in 24-well plates (Nunc). Medium was Dulbecco's modified Eagle's medium/F12 (1:1) containing 2 mM KCl, 2 mM glutamine, penicillin/streptomycin, and 2% B27 supplement (Invitrogen). Half-volume medium changes were performed every 3 days. Cultures grown for 8–12 days were transduced with 2 × 105 viral particles/well. This resulted in a multiplicity of infection of 0.6 based on HT1080 titer, prevented saturating the cells with multiple viral particles per cell, and allowed determination of relative tropism for each cell type. Five days later, transduced cultures were either fixed with 4% paraformaldehyde (PFA) for immunocytochemistry or lysed in 125 mM Tris-HCl, pH 6.8, 2.5% sodium dodecyl sulfate, and 2.5% glycerol for western blotting. Protein concentrations were estimated using a Bio-Rad DC protein assay kit (Bio-Rad, Auckland, New Zealand) and bovine serum albumin as the standard.
Western blotting
To quantify GFP expression from the MND and EF1α promoters, lysates from transduced and control cells were separated on NuPAGE 4–20% gels (Invitrogen) before transfer to polyvinylidene fluoride membranes in a Bio-Rad Mini Trans-Blot at 100 V for 1 hr. Membranes were blocked with 5% nonfat milk powder in Tris-buffered saline, pH 7.6, with 1% Tween-20 (TBS-T) for 1 hr before overnight incubation with antibodies against GFP (rabbit anti-GFP; Abcam, Cambridge, UK; ab290; 1:10,000) in 1% nonfat milk powder in TBS-T. After washing, the membranes were incubated in TBS-T containing 1% nonfat milk powder and goat anti-rabbit horseradish peroxidase (1:10,000; GE Healthcare, Amersham, UK), then detected using ECL-Plus reagents (GE Healthcare) and visualized using a Fujifilm Las Imager 3000. Membranes were then rinsed in TBS-T, reblocked, and probed with an anti-actin antibody (rabbit anti-actin 20–33; Sigma–Aldrich, A5060; 1:5,000), used to normalize loadings.
Immunocytochemistry
Cells were washed briefly in PBS, before blocking in 3% normal goat serum. Astrocytes were recognized by reaction to mouse anti-glial fibrillary acidic protein (GFAP) (Sigma–Aldrich, G3803; 1:500), neurons by reaction to mouse anti-microtubule-associated protein 2 (MAP2) (Millipore, Billerica, MA, MAB3518; 1:500), and neuroblasts by reaction to mouse anti-polysialated neuronal cell adhesion molecule (PSA-NCAM) (Millipore, MAB5324; 1:1,000). GFP expression was detected with rabbit anti-GFP (Abcam, ab290; 1:20,000) that was preincubated on normal nontransduced cells to remove nonspecific binding. Secondary antibodies were goat anti-mouse Alexa 568 (Invitrogen, A-11031) or goat anti-rabbit 488 (Invitrogen, A-11008). Following immunolabeling, cells were incubated in 4′,6-diamidino-2-phenylindole (DAPI; Sigma–Aldrich; 100 ng/ml), for 5 min at room temperature, to detect nuclei and determine total cell number, and then mounted on glass slides in ProLong Gold antifade (Invitrogen) for imaging. For each well, 20 fields at 400 × magnification were imaged and the proportions of GFP-positive cells and total cells of each type calculated.
Statistical analyses
Statistical analyses were performed using the R statistical software (
Animals and in vivo viral injections
Sheep
All animal work was approved by the Lincoln University Animal Ethics Committee and complied with the New Zealand Animal Welfare Act, 1999. Homozygous affected South Hampshire lambs were generated by mating heterozygous ewes to homozygous affected rams, and the genotype was determined using a discriminatory c.822G>A polymorphism in the CLN6 gene (Tammen et al., 2006). At the time of vector injection, the sheep were 8 months of age and in good health. Normal age-matched sheep were treated as controls.
In vivo viral injections
Sheep were fasted overnight. Anesthesia was induced with a mixture of ketamine (7.5 mg/kg live weight) and diazepam (0.3 mg/kg live weight) given intravenously. Then the sheep were intubated (9.5-mm cuffed endotracheal tube) and maintained on a mixture of halothane (2–4%) and oxygen, within a closed circuit system.
Viral vector was injected intracortically into two affected and two control sheep. With the sheep in the prone position, the head was secured for injection in a stereotactic frame (Kopf, model 1630; David Kopf Instruments, Tujunga, CA), and the surgical site was clipped and prepared for surgery by repeated scrubs with 4% chlorhexidine gluconate and polyvinylpyrrolidone iodine followed by draping. Following a medial skin incision and retraction of underlying musculature and fascia, two 3-mm holes were drilled through the frontal bone 5 mm either side of the midline and 47 mm rostral to the occipital ridge. To establish the depth to the tissue-ventricle interface, a 20-gauge needle was attached to a fine tube containing a column of sterile saline trypan blue solution and lowered into the brain using a stereotaxic manipulator. The ventricular interface was indicated by the positive flow of CSF once the ventricle was reached. The needle was then withdrawn 5 mm, and a 10-μl Hamilton syringe with a 26-gauge needle was threaded down the 20 gauge to be 4 mm proud of the end and 1 mm superficial to the tissue-ventricle interface. One microliter of viral vector solution containing 1.3 × 108 TU in 10 μl was infused over 1 min. The complete needle unit was then withdrawn simultaneously 1 mm over a minute before the next 1 μl of virus was discharged, until all 10 μl of the viral solution was infused. The procedure was repeated on the other side of the brain. The wound was closed with a continuous subcuticular suture followed by interrupted skin sutures that were removed subsequently. An analgesic (buprenorphine HCl) was administered intramuscularly (330 μg/animal), as was a mixture of procaine and benzathine penicillin (12,000 IU/kg), and the animals were observed until full recovery.
An additional affected and control animal pair were injected with 1 × 109 TU in 100 μl directly into the CSF at the cisterna magnum.
Treated animals were housed indoors, fed a mixed ration of lucerne chaff and balanced sheep nuts, and monitored daily for any adverse reactions.
Immunohistochemistry
Brains were perfusion-fixed at postmortem via the carotid artery with 10% formalin in 0.9% NaCl after they were flushed with warm 0.9% NaCl. They were left in fixative for 7 days, then equilibrated over 7 days in cryoprotectant, 20% sucrose and 10% ethylene glycol in 0.9% NaCl, and stored frozen at −80°C until sectioned. Serial sagittal sections (50 μm) were cut on a freezing sliding microtome (Microm, Walldorf, Germany) and then stored at −20°C in cryoprotective solution in 96-well plates.
All antibodies were diluted in 10% normal goat serum in PBS containing 0.3% Triton X-100. To identify the gene injection site and spread of the virus, every 12th section was stained for GFP. Sections were thawed and blocked in 15% normal goat serum for 60 min, and incubated overnight at 4°C, in rabbit polyclonal anti-GFP (Abcam, ab290; 1:20,000) that had been incubated with normal sheep brain sections for 1 hr to remove nonspecific labeling. Immunoreactivity was detected with fluorescent Alexa-488 goat anti-rabbit IgG (Invitrogen, A-11008; 1:2,000) for 4 hr at room temperature. In other cases, the secondary antibody was biotinylated goat anti-rabbit IgG (Sigma–Aldrich, B7389; 1:1,000) for 4 hr at room temperature, followed by ExtrAvidin peroxidase (Sigma–Aldrich, E2886) for 4 hr at room temperature, which was detected with a solution of 0.5 mg/ml 3,3′-diaminobenzidine (DAB; Sigma–Aldrich, D5637) and 0.01% H2O2 in PBS.
Parallel series of sections, identified following initial screening of sections around the approximate injection site, were double-labeled with GFP and either GFAP or the fluorescent Nissl dye, NeuroTrace (Invitrogen, N21482), as markers of transduced glial cells and neurons, respectively. A standard cresyl violet Nissl stain (Oswald et al., 2005) was applied to some sections from around the injection site, which were examined for any signs of injection-related pathology. For GFAP and GFP immunofluorescence, sections were blocked in 15% normal goat serum for 60 min, then incubated in rabbit anti-cow GFAP (DAKO, Denmark, Z0334; 1:1,000) overnight at 4°C. The secondary antibody, Alexa-594 goat anti-rabbit IgG (Invitrogen, A-11012; 1:1,000), was applied for 4 hr at room temperature. Sections were then fixed in 4% PFA for 20 min. GFP was detected using rabbit anti-GFP Alexa-488 conjugated antibody (Invitrogen, A-21311; 1:500), overnight at 4°C.
Parallel series of sections were stained for GFP with rabbit polyclonal anti-GFP, 1:2,000 (as above), and NeuroTrace, 1:150 in PBS, for 60 min at room temperature.
Sections were mounted in a solution of 0.5% gelatin and 0.05% chromium potassium sulfate on glass slides, air-dried, and coverslipped using glycerol. Negative control sections, in which either the primary or secondary antibody was omitted, were included in all staining runs. No immunostaining was observed in any of the negative control sections.
Results
Lentiviral vectors transduce sheep neural cells in vitro
Before embarking on in vivo studies, transduction, cell tropism, and transgene expression from two lentiviral constructs were tested in both control and affected sheep fetal neuronal cultures. These mixed cultures contained the target cells of interest; neurons, neuroblasts, and astrocytes. Eight to twelve days after plating, neural cultures were transduced with lentiviral vectors encoding GFP under the control of either MND, a viral promoter (Fig. 1a and b), or EF1α, a constitutive human promoter (Fig. 1a and f) and double-labeled for GFP and either MAP2 to identify neurons (Fig. 1c and g), PSA-NCAM to identify neuroblasts (Fig. 1d and h), or GFAP to identify astrocytes (Fig. 1e and i). Transgene expression was observed in all three cell types in cultures from both promoters, and there was no significant difference in the proportion of cells of each type expressing GFP between promoters (p = 0.69). That there was little difference in overall GFP expression from either promoter was confirmed by western blotting of cells from two independent control preparations transduced with an equivalent number of viral particles (Fig. 1j).
As in normal cultures, transduction of affected cultures resulted in transgene expression in neurons (Fig. 2a), neuroblasts (Fig. 2b), and astrocytes (Fig. 2c). On average, 20% neurons, 24% neuroblasts, and 18% astrocytes were identified in cultures (Table 1), and there was no significant difference (p > 0.05) between the proportions of these cell types in the affected and normal cultures.
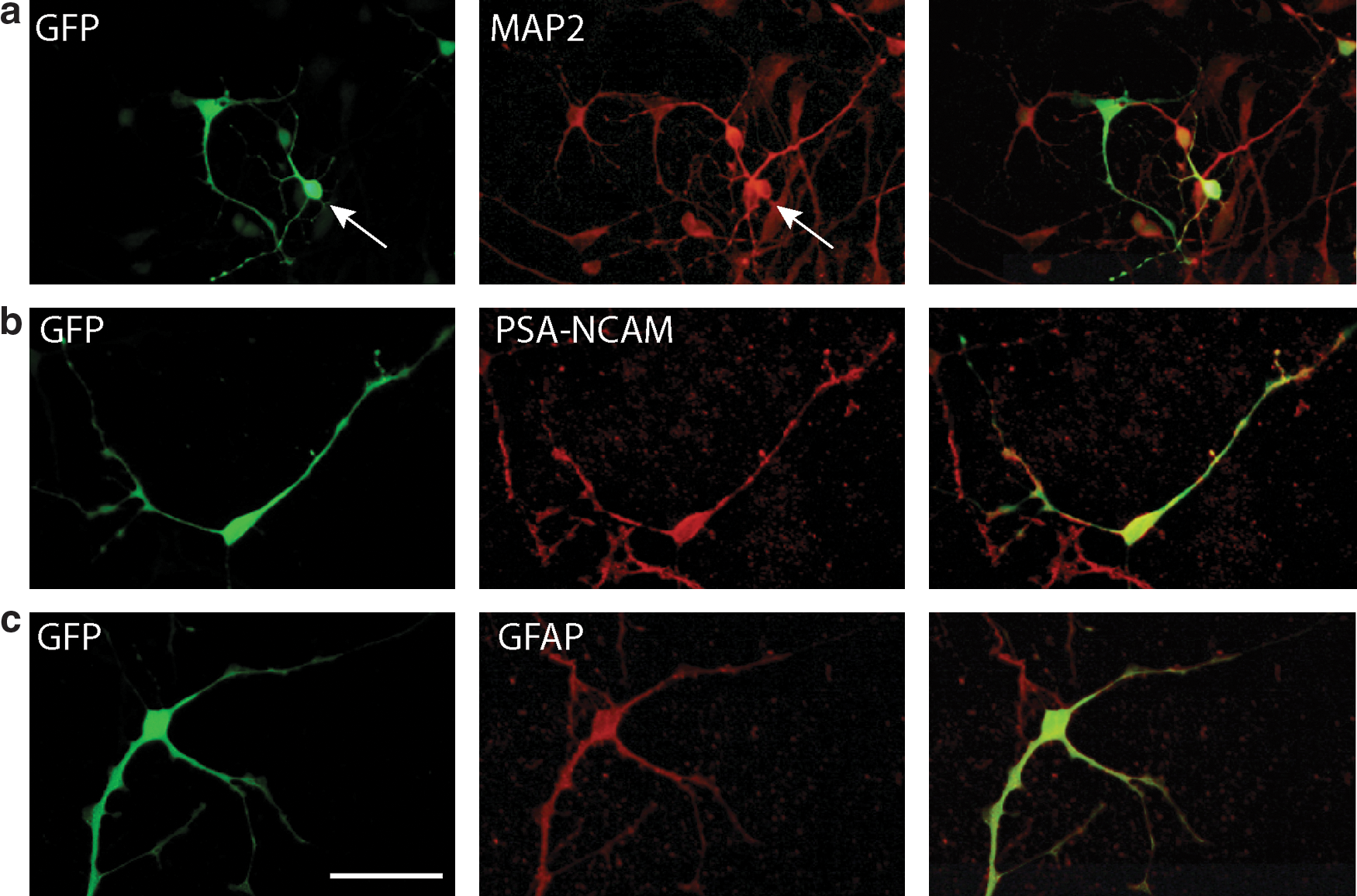
Lentiviral-mediated gene transfer to affected neural cells in vitro transduced with lentivirus-GFP (green) and labeled to detect
Embryonic day 60 neural cells were cultured from three normal and three affected sheep and transduced with lentivirus expressing GFP. The proportion of each cell type in cultures was determined as a percentage of total DAPI-positive cells. The proportion of GFP-positive cells of each cell type was also calculated. The proportions of neurons and neuroblasts transduced were significantly higher than expected in comparison with their presence in cultures. Data presented are means ± SEM.
If the virus transduced all cell types equally (without tropism for one cell type over another), the percentage of transduced GFP-positive cells of each cell type should be similar to the percentage of that cell type in culture (Table 1). However, the percentages of transduced cells identified as neurons or neuroblasts in both affected and normal cultures were significantly greater (p = 5.9 × 10–12 and 9.7 × 10–11, respectively) than expected compared with their abundance in the cultures. In addition, there was a suggestion that the affected cultures were more efficiently transduced than controls (p = 0.029), driven largely by a difference observed in the transduction of affected astrocytes (Table 1).
Lentiviral-mediated gene transfer to adult sheep brain
As there were no significant differences between the performance and expression from the two promoters being tested, the MND vector was chosen as representative for in vivo studies. Lentiviral particles were injected bilaterally into the brain of two affected and two normal 8-month-old animals, rostral to the occipital ridge, aiming for just dorsal to the ventricle (Fig. 3a). Transduction of cells via injection directly into the CSF at the cisterna magnum was also explored in an affected and a control animal.
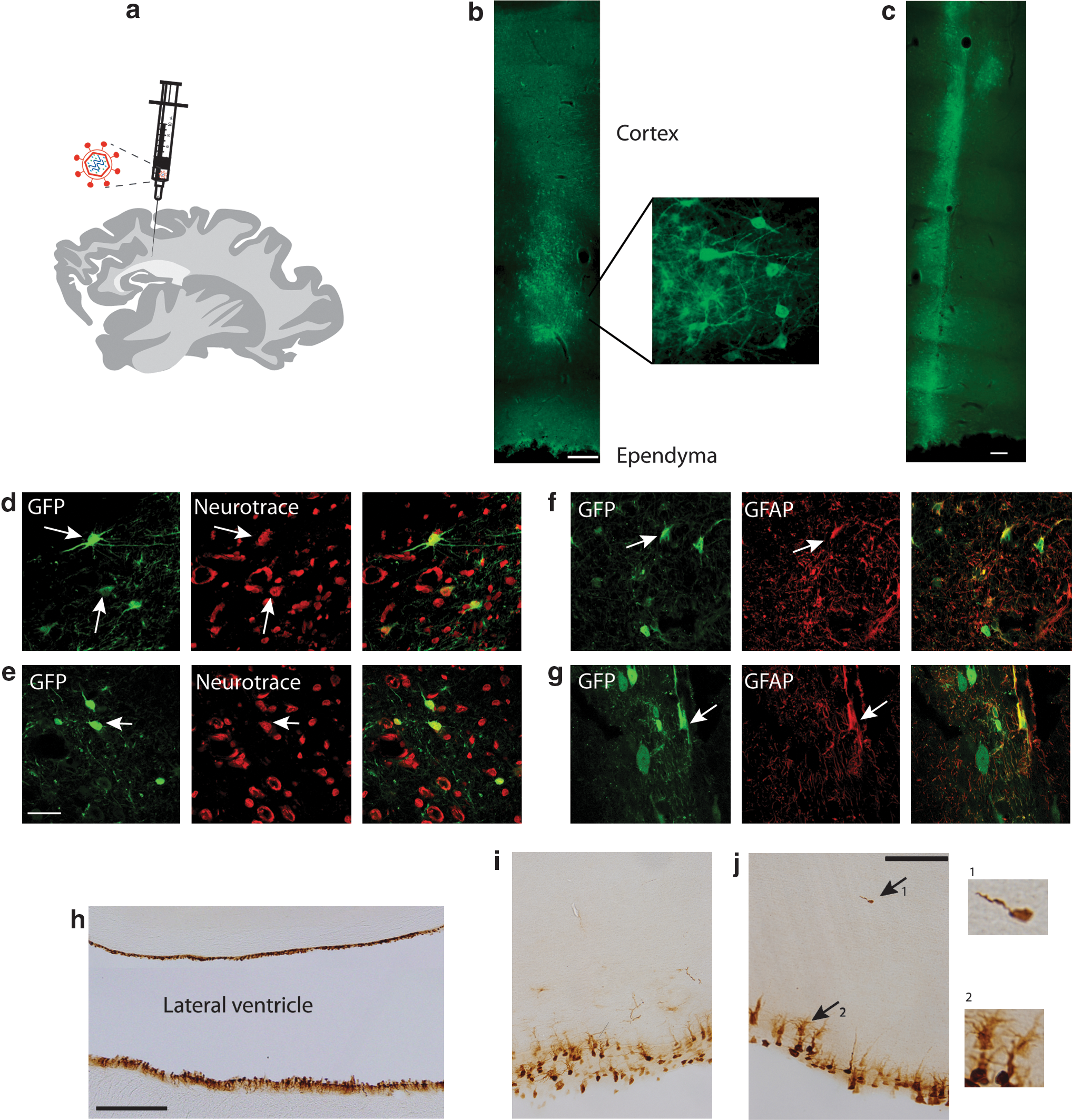
Lentiviral-mediated gene transfer to the sheep brain.
None of the sheep showed ill effects from the injections. Recovery from the anesthesia was uneventful, and all the animals resumed normal eating within 24 hr. Rectal temperatures remained normal throughout the 3 weeks they were monitored after the operations. Growth rates and general condition remained the same as those of the uninjected controls.
Brains were taken from a control and an affected animal 40 days after the intracerebral injections and from the other pair after 80 days. Immunohistochemistry revealed GFP expression in cells along the extent of the needle track through the line of the injection (Fig. 3b and c) from all four animals. The intense fluorescence and high density of expressing cells at the injection site suggested potent lentivirus transduction, which declined further away from the center of injection. GFP-positive cells were detected in the brain parenchyma up to 2.5 mm rostral-caudal and lateral to the injection site. Both neurons (Fig. 3d and e) and astrocytes (Fig. 3f and g) were similarly transduced in affected and normal sheep brain.
A heterogeneous population of neurons was transduced, with numerous multipolar and bipolar neurons displaying GFP immunoreactivity. Multipolar neurons with polygonal cell soma were colabeled with NeuroTrace (Fig. 3d). Neurons with spheroid cell soma and bipolar projections exhibited GFP expression throughout the axon hillock and up to 150 μm along axonal and dendritic extensions (Fig. 3e). GFP expression was also evident within stellate cell bodies, characteristic of mature astrocytes. These were confirmed to be astrocytes by colabeling with GFAP (Fig. 3f and g). Both transduced neurons and astrocytes were evident along the extent of the injection track.
Control and affected animals showed no discernible difference in the number of transduced cells, staining intensity, or cell types transduced. Transduction and expression were stable with no differences in GFP expression being noted between the animals analyzed 40 and 80 days post injection. Examination of Nissl-stained sections confirmed that there were no signs of inflammation or other pathology around the injection sites detectable in Nissl-stained sections (data not shown), and the GFP-expressing cells retained healthy morphologies typical of their type.
Transduced cells were also revealed along the entire rostral-caudal extent of the ependyma lining the lateral ventricle in the subventricular region. This was particularly evident in one affected animal that also displayed a smaller and less intensely stained region of transduced cells corresponding to the injection site within the brain parenchyma (Fig. 3h). The injection protocol was designed to inject the vector into the subventricular zone (SVZ); it was anticipated that the hole from the needle would close sufficiently to block leakage into the ventricle, but it is not clear how much of the injection volume was retained in the cerebrum. In the case of this animal, the widespread gene transfer along the rostral, caudal, and lateral extent of the ependyma of the lateral ventricle and lesser staining within the brain parenchyma indicate that there may have been considerable leakage into the ventricle. GFP was strongly expressed within the cell soma of cuboidal ependymal cells arranged in a single layer lining the lateral ventricle (Fig. 3h and i). In one affected animal, transgene expression was also observed in subependymal cell bodies in the region 30 μm from the ventricular surface. This region contained transduced cells with large cell soma and multiple GFP-expressing processes extending vertically above the soma or radiating laterally, and which extended at least another 50 μm into the SVZ region (Fig. 3j). These cells may be typical of type B astrocytes, believed to represent the neural stem cells of the SVZ. Additionally, occasional cells with a characteristic, unipolar morphology consistent with migrating neuroblasts were seen in this affected animal deeper within the subependymal region, at a distance of up to 200 μm from the ventricular surface (Fig. 3j, arrow). They were not evident along the entire extent of the SVZ, the few revealed by immunohistochemistry being present at more rostral regions.
A thorough examination of sagittal sections across the mediolateral extent of each brain hemisphere revealed no transduced cells 40 days post injection in the two animals in which the vector was injected directly into the CSF at the cisterna magnum.
Discussion
Here, we report the first demonstration of lentiviral-mediated gene transfer to sheep neural cells and transgenic protein production in vivo. Transductions in the sheep brain were stable, with GFP-expressing cells equally evident 40 and 80 days post injection. The cell types transduced in vivo correlated with the in vitro studies. Neither the vector nor the expression of GFP caused observable pathology to transduced or neighboring cells. Although expression from the intracortical injections was most intense along the line of the injection site, some cells were specifically labeled up to 2.5 mm from this site, indicating either migration of transduced cells or spread of the viral particles from the sites of injection. Furthermore, transgene expression was noted at considerable distances along the axons/dendrites, an important factor for secretion of soluble transgene products to distal regions of the brain. Because of the leakage into the ventricle, noted by labeling of the ventricular surface, it is not possible to know what the optimal distribution from intracortical injections might be, and future experiments are required to address this.
The transduction of cells along the extent of the ventricular surface (Fig. 3h–j) probably results from this leakage. It has been suggested that subsets of ependymal cells function as multipotent neural stem cells in adult mice (Johansson et al., 1999); however, most studies suggest that type B astrocytes in the SVZ are the bona fide adult neural stem cells (Chiasson et al., 1999; Doetsch et al., 1999a; Laywell et al., 2000). Although type B astrocytic cells have their cell bodies located at the apical edge of the SVZ, a subpopulation may send occasional processes through the ependymal layer to contact the ventricle (Doetsch et al., 1999b; Conover et al., 2000), and thus provide physical access to neural stem cells for vectors present in the lateral ventricle. The identity of these stem cells has been the subject of intense research, and it is now well accepted that some of the GFAP-expressing cells are stem cells in rodent and human SVZs. Confirmation of stem cell transduction would provide an additional route for spread of the gene product via migration of stem cell progeny into the degenerating brain. The few cells identified with neuroblast morphology in vivo (Fig. 3j) and the tropism for neuroblasts in vitro suggest that lentiviruses may be targeted to neural progenitor cells in the sheep brain. The dogma in LSD therapeutics is that 5% of normal enzyme activity is sufficient to restore normal cellular function. Expression from lentiviral vectors can provide supraconcentrations of secreted protein (e.g., Brooks et al., 2002; Haskell et al., 2003), which spreads from the transduced cell into the parenchyma. Therefore, transduction of a large number of neuroblasts and migration to the degenerating cerebral cortex may provide a wider distribution of the soluble lysosomal protein than possible without multiple injections directly into affected regions of the brain. It remains to be seen whether this is a viable therapeutic target. Further advances and testing of other vector pseudotypes and injection protocols targeting multiple regions are required to improve gene transfer to this potential therapeutic target. These results also indicate that intraventricular injection may be a route to the transduction of ependymal cells and secretion of soluble gene products into the CSF and surrounding parenchyma cells.
Our study injected a relatively small volume and number of particles in comparison with recent studies in monkey and humans. In a human trial in CLN2-deficient patients, 1012 AAV particles were injected into 12 sites across the rostral-caudal extent of the cerebral cortex (Worgall et al., 2008). This compares with the 108 particles injected here into the sheep brain, which is only an order of magnitude smaller. In a recent monkey study, between 33 and 199 μl of AAV was injected into the brainstem or thalamus of four cynomologus monkeys, which have a brain size similar to that of sheep (Aguilar Salegio et al., 2010). Interestingly, this study showed that vector spread was directly proportional to the injection volume albeit that the larger injections also contained more viral particles. Larger injections did not cause significant adverse mechanical injury. These and other studies using lentivirus (Kitagawa et al., 2007; Ahmed et al., 2010) suggest that vector spread could be improved by increasing injection volumes.
The lack of transduction of the ventricular cells or any other following injection into the cisterna magnum, even though the vector number and injection volume was 10 times that of the cortical injections, demonstrates that this is not a viable route to expose this vector to the ventricular surface or other parts of a larger brain, despite positive indications in rodent studies. CSF from the lateral ventricles travels to the cisterna magnum and, from this large reservoir, bathes the spinal cord and then the brain before being returned to the circulation via the arachnoid villi. Based on the extent of incidental labeling of ependymal and subependymal cells lining the lateral ventricles resulting from our parenchymal injections, it is likely that virus transduced ependyma adjacent to the cisterna magnum injection site, and there was probably too little vector left to be effective by the time the CSF had completed its return journey to the brain. This lack of ventricular transduction by this route could be a feature of the size of the CNS of the species used, as well as the particular vector. A study of intrathecal delivery of lentiviral vectors into neonatal mice reported patchy transduction of meninges and secretion of a soluble enzyme into the CSF (Fedorova et al., 2006), and intrathecal delivery of AAV1 vectors into a mouse model of metachromatic leukodystropy resulted in widespread transduction within the brain (Iwamoto et al., 2009). Injecting microgram quantities of α-L-iduronidase weekly into the cisterna magnum of a canine model of mucopolysaccharidosis-I achieved functional concentrations in the brain and associated membranes (Kakkis et al., 2004). Evidently, size of the rodent CNS is not a hindrance to the transduction of cells via the intrathecal delivery of vectors, whereas it is in sheep. This indicates that reports of positive transgene expression via this route in rodent models of disease do not ensure that human studies will be successful. Testing therapies in large-animal models with large and complicated CNS is a valuable prerequisite for recognizing and overcoming potential hurdles.
Only one previous study has tested viral gene transfer in sheep, targeting RNA interference to the hypothalamus using AAV vectors (Dufourny et al., 2008). In addition to utility in the naturally occurring genetic models of NCL developed in sheep (Tammen et al., 2006; Frugier et al., 2008), their similar brain structure and size to humans makes them useful as large-animal models in other diseases. For instance, sheep models have also been developed for conditions such as perinatal hypoxia–ischemia (Dean et al., 2008), Tay-Sachs disease (Torres et al., 2010), and Huntington's disease (Jacobsen et al., 2010), which may be amenable to gene therapy.
Lentiviral vectors pseudotyped with vesicular stomatitis virus glycoprotein (VSV-G) have been extensively characterized in rodents (Brooks et al., 2002; Watson et al., 2002; Jakobsson et al., 2003). Tropism in these cases and in the current study is measured by transgene expression, assuming that the promoters are ubiquitously expressed in all cell types. However, as in other model organisms, VSV-G–pseudotyped lentiviral vectors under ubiquitous promoters favored transgene expression in neurons rather than glial cells (Desmaris et al., 2001; Jakobsson et al., 2003). Our in vitro studies confirm this transgene expression bias in sheep neural cultures, establishing cell-culture–based analysis as a valuable tool for determination of vector tropism, viral vector function, and promoter activity prior to time-consuming in vivo studies. In rodent brain, pseudotyping with a lyssaviral envelope enhances axonal uptake and retrograde transport to distal neurons (Desmaris et al., 2001), which could be explored in sheep. The persistence of neural progenitor cells in the SVZ of rodents and humans (Reynolds and Weiss, 1992; Eriksson et al., 1998; Curtis et al., 2003) suggests this zone as a target. The incidental transduction of this region via leakage into the ventricles (Fig. 3h–j) suggests intraventricular injection as a route.
Three naturally occurring sheep NCL mutations have been identified in orthologues of the genes CLN5 and CLN6 representing NCLs associated with those affecting soluble lysosomal proteins and those associated with membrane-associated proteins, respectively, and well established flocks are available (Tammen et al., 2006; Frugier et al., 2008). These subgroups may require different treatment strategies, the former being amenable to cross-correction from transduced cells supplying the missing protein to surrounding parenchyma, whereas delivery of corrected gene products for the membrane-associated NCLs will require a vector that spreads widely in the brain at high efficiency. This may require transduction of targeted stem cells in vivo that can replace compromised cells, unless the membrane-associated protein processes some factor that is secreted and taken up by other cells.
Although few clinical trials have been approved using lentiviral vectors, gene-therapy trials have begun for one form of Batten disease in humans, CLN2 deficiency (late-infantile NCL) using AAV vectors (Worgall et al., 2008). The well-developed sheep models provide an ideal resource to study vector and gene product spread, optimal timing of interventions, and injection-site targets to optimize functional effects.
In summary, this study provides the first evidence of lentiviral-mediated gene transfer to the sheep brain, and an approach with which to test further gene-therapy strategies and vectors in NCLs.
Footnotes
Acknowledgments
The authors thank Nigel Jay (Lincoln University) for expert technical assistance and Bronwyn Carlisle (University of Otago) for graphics assistance. This work was supported by grants from the U.S. National Institutes of Health (NS053559: D.N.P., G.W.K., L.A.B., N.L.M., R.G.M.; and NS043205: M.S.S.), the University of Otago School of Medical Sciences Deans Bequest (K.S.L., S.M.H.), and the Neurological Foundation of New Zealand (D.N.P., G.W.K., K.S.L., L.A.B., N.L.M., S.M.H.).
Author Disclosure Statement
No competing financial interests exist.