
Abstract
Human embryonic stem cells (hESC) and induced pluripotent stem cells (iPSC) offer great hope for in vitro modeling of Parkinson's disease (PD), as well as for designing cell-replacement therapies. To realize these opportunities, there is an urgent need to develop efficient protocols for the directed differentiation of hESC/iPSC into dopamine (DA) neurons with the specific characteristics of the cell population lost to PD, i.e., A9-subtype ventral midbrain DA neurons. Here we use lentiviral vectors to drive the expression of LMX1A, which encodes a transcription factor critical for ventral midbrain identity, specifically in neural progenitor cells. We show that clonal lines of hESC engineered to contain one or two copies of this lentiviral vector retain long-term self-renewing ability and pluripotent differentiation capacity. Greater than 60% of all neurons generated from LMX1A-engineered hESC were ventral midbrain DA neurons of the A9 subtype, compared with ∼10% in green fluorescent protein–engineered controls, as judged by specific marker expression and functional analyses. Moreover, DA neuron precursors differentiated from LMX1A-engineered hESC were able to survive and differentiate when grafted into the brain of adult mice. Finally, we provide evidence that LMX1A overexpression similarly increases the yield of DA neuron differentiation from human iPSC. Taken together, our data show that stable genetic engineering of hESC/iPSC with lentiviral vectors driving controlled expression of LMX1A is an efficient way to generate enriched populations of human A9-subtype ventral midbrain DA neurons, which should prove useful for modeling PD and may be helpful for designing future cell-replacement strategies.
Introduction
The localized loss of a specialized neuronal cell population, paired with the urgent need for long-term treatment options, makes cell replacement therapy a promising approach to treat PD. Clinical trials involving grafting of embryonic ventral mesencephalic tissue in PD patients have provided proof-of-concept for the feasibility of cell therapy in this disease (Lindvall and Björklund, 2004). However, this approach is not widely applicable owing to the limited supply of such tissues and to ethical concerns.
Human pluripotent stem cells, such as human embryonic stem cells (hESC) (Thomson et al., 1998), have great potential regarding their therapeutic use for PD patients (Lee et al., 2000; Ying et al., 2003; Nat et al., 2007; Morizane et al., 2008). More recently, the generation of induced pluripotent stem cells (iPSC) by ectopic expression of a defined set of factors (Takahashi et al., 2007; Yu et al., 2007; Aasen et al., 2008; Lowry et al., 2008; Park et al., 2008b) has enabled the derivation of patient-specific pluripotent cells, providing valuable experimental platforms to model human disease (Dimos et al., 2008; Park et al., 2008a; Ebert et al., 2009; Soldner et al., 2009; Agarwal et al., 2010) and opening the possibility of implementing patient-matched cell-therapy applications (Hanna et al., 2007; Wernig et al., 2008; Raya et al., 2009). In addition, the possibility of genetically engineering human pluripotent stem cells to express potentially therapeutic molecules or to manipulate their differentiation toward a specific phenotype, which, for example, could be used to increase survival, differentiation, migration, and function of their progeny, creates new opportunities for the development of restorative treatment for PD patients.
The use of hESC/iPSC-based strategies to treat or model PD, however, is currently hampered by the lack of efficient protocols for the directed differentiation of human pluripotent stem cells into DA neurons with the appropriate characteristics of A9-subtype ventral midbrain neurons, the neuronal cell type lost to PD (for review, see Hwang et al., 2010). A deeper understanding of DA neuron differentiation, patterning, and specialization will surely facilitate the generation of an optimal cell source for cell replacement therapy and disease modeling for PD. During embryo development, midbrain DA neurons originate in the most ventral region of the midbrain and are patterned by local induction through the activity of signaling molecules such as sonic hedgehog (SHH), fibroblast growth factor 8 (FGF8), and wingless (WNT) (for review, see Smidt and Burbach, 2007). As a result of this, a characteristic region-specific makeup of transcription factor expression is established in neural progenitors. Among them, the LIM homeobox transcription factor 1 alpha (Lmx1a) is selectively expressed in the proliferating neural progenitors that give rise to ventral midbrain DA neurons in the mouse, and has been shown to play crucial roles in determining their fate (Andersson et al., 2006). Moreover, overexpression of Lmx1a in mouse embryonic stem cells greatly improved their differentiation toward ventral midbrain DA neurons (Andersson et al., 2006; Cai et al., 2009; Friling et al., 2009), prompting the possibility of using LMX1A modulation to similarly direct the fate of hESC/iPSC-derived neurons. In support of this possibility, LMX1A expression was found to be required for the midbrain DA neuron differentiation of hESC (Cai et al., 2009). In spite of these promising pieces of evidence, strategies for increasing DA neuron differentiation of human neural precursors or hESC by LMX1A overexpression have been met with limited success. Thus, transfection of LMX1A-expressing constructs in human neuroblastoma cell lines did not increase the numbers of tyrosine hydroxylase (TH)-positive neurons (Cai et al., 2009). Similarly, retroviral overexpression of LMX1A in immortalized human midbrain progenitors only resulted in very marginal increases in DA neuron differentiation (Roybon et al., 2008).
A possible caveat in previous attempts to overexpress LMX1A in human neural stem cells or hESC could be the low levels of transfection/transduction achieved in these notoriously hard-to-transfect cell lines. In this context, the use of lentiviral vectors (LVs) (Naldini et al., 1996), to obtain robust and sustained transgene expression in neural or pluripotent stem cells (Lois et al., 2002; Pfeifer et al., 2002; Ma et al., 2003; Consiglio et al., 2004), acquires particular relevance. Indeed, transduction of hESC-derived neural progenitors with LVs expressing LMX1A under the control of a constitutive phosphoglycerate kinase (PGK) promoter has been recently reported to promote the generation of ventral midbrain DA neurons (Friling et al., 2009).
In this study, we tested whether hESC/iPSC could be stably engineered using LVs so that they would overexpress LMX1A in neural progenitors when prompted to differentiate. We show that hESC/iPSC engineered in this way maintain their normal growth characteristics and retain their pluripotent differentiation ability, as judged by in vitro differentiation and teratoma formation assays. Notably, the levels of LMX1A overexpression achieved in neural progenitors differentiated from engineered hESC/iPSC are sufficient to boost the generation of ventral midbrain DA neurons. Analyses of the phenotypic and physiological properties of these cells, as well as the ability of grafted cells to survive and integrate into the striatum of adult mouse brains, showed that LMX1A-induced DA neurons differentiated from hESC/iPSC are essentially similar to authentic ventral midbrain DA neurons. To the best of our knowledge, this is the first demonstration that LVs can be exploited to stably engineer hESC/iPSC for their directed differentiation toward specific DA neuron phenotypes, thus making it possible to develop new strategies for modeling and treating neurodegenerative diseases.
Materials and Methods
Cell culture
The hESC lines (ES[2] and ES[4]) and the iPSC lines used in these studies have been previously described (Aasen et al., 2008; Raya et al., 2008, 2009). Human pluripotent stem cells were cultured on the top of irradiated (55 Gy) human foreskin fibroblasts (CCD1112SK; ATCC), seeded at a density of 7×104 cells/cm2 on 10-cm cell culture dishes coated with 0.1% gelatin (Chemicon, Millipore, Temecula, CA). The hESC and hiPSC were cultured at 37°C and 5% CO2 in hESC medium, consisting of KnockOut DMEM (Invitrogen, Paisley, UK) supplemented with 20% KnockOut Serum Replacement (Invitrogen), 0.5% human albumin (Grifols, Barcelona, Spain), 2 mM GlutaMAX (Invitrogen), 50 μM 2-mercaptoethanol (Invitrogen), nonessential amino acids (Cambrex, Walkersville, MD), and 10 ng/ml basic fibroblast growth factor (bFGF) (Peprotech, Rock Hill, NJ). Medium was changed every day. Individual hESC and iPSC colonies were passed by mechanical dissociation using a 150-μm-diameter plastic pipette (The Stripper; Midatlantic Diagnostics, Mount Laurel, NJ) and replated every 5–7 days. Karyotyping was performed by incubating colonies for 30 min in 2 μl/ml Colcemid (Invitrogen) by the G-banding method. Fifteen metaphases were analyzed for each line.
Plasmid construction, lentiviral production, and hESC and iPSC transduction
LMX1A or green fluorescent protein (GFP) expression was driven by the NESTIN enhancer (NesE) (1,852 bp) and the basic herpes virus thymidine kinase (HSV tk) promoter element (925 bp). This enhancer contains a sufficient cis-element that recapitulates endogenous NESTIN expression in the CNS (Lothian and Lendahl, 1997). The cassette containing the NesE, HSV tk promoter, and Lmx1a cDNA was cloned in the CSC.cPPT.hCMV.GFP.Wpre LV backbone previously described (Lie et al., 2005). High-titer vesicular stomatitis virus (VSV)–pseudotyped LV stocks were produced in 293T cells by calcium phosphate–mediated transient transfection of the transfer vector pRRL-SIN-PPT-NesE-Lmx1a-ires-GFP-WPRE, the late generation packaging construct pMDL, and the VSV envelope–expressing construct pMD2.G, and purified by ultracentrifugation as previously described (Consiglio et al., 2004). Expression titers, determined using HeLa cells by FACS analysis, were 5×108 to 1×109 transducing units (TU)/ml with an average HIV-1 p24 concentration of 100 μg/ml. The same day of transduction, 2×105 hESC (passage 15) were infected in suspension at multiplicity of infection (MOI) of 10 with LV.NES.LMX1A.GFP, LV.NES.GFP, or LV.PGK.GFP in the presence of 6 μg/ml Polybrene (Sigma, Sigma-Aldrich Co, St. Louis, MO). The day after transduction, the medium was changed and transduction efficiency was estimated by FACS analysis for GFP expression.
DA differentiation of hESC
For neural induction, embryoid bodies (EBs) were generated from large colony fragments obtained by mechanical splitting with a finely drawn Pasteur pipette and cultured in nonadherent dishes for 3–4 days in the presence of hES medium. At Stage 2, EBs were cultured 10 days in suspension in N2B27 medium consisting of DMEM/F12 medium (Invitrogen), Neurobasal medium (Invitrogen), 0.5× B-27 supplement (Invitrogen), 0.5× N2 supplement (Invitrogen), 2 mM GlutaMAX (Invitrogen), and penicillin–streptomycin (Invitrogen). In this stage, N2B27 medium was supplemented with 10 ng/ml bFGF, 100 ng/ml FGF8 (Peprotech), and 100 ng/ml SHH (Invitrogen). Finally, at Stage 3 for DA neuron maturation, the neural precursors cells (NPCs) were cocultured with PA6 for 3 weeks in N2B27 medium supplemented with FGF8 and SHH.
In vitro differentiation toward the embryonic germ layers
Differentiation toward endoderm, mesoderm, and neuroectoderm was carried out essentially as described (Raya et al., 2010).
Teratoma formation in nude mice
Severe combined immunodeficient (SCID) beige mice (Charles River Laboratories) were used to test the teratoma induction capacity of hES-LMX1A cells essentially as described (Raya et al., 2008). All animal experiments were conducted following experimental protocols previously approved by the Institutional Ethics Committee on Experimental Animals, in full compliance with Spanish and European laws and regulations. In brief, ∼1–3×106 hES-LMX1A cells were injected subcutaneously into the testis of SCID mice anesthetized with isoflurane. Five to 8 weeks after injection, teratomas were dissected, fixed overnight in 10% buffered formalin phosphate, and embedded in paraffin.
RT-PCR analysis
Total mRNA was isolated by guanidinium thiocynate–phenol–chloroform extraction (TRIzol; Invitrogen) and treated with DNase I. One microgram was used to synthesize cDNA by using the SuperScript III Reverse Transcriptase Synthesis Kit (Invitrogen). Quantitative PCR (qPCR) analysis was done in triplicate on 50 ng by using Platinum SYBER Green qPCR SuperMix (Invitrogen) in an ABI Prism 7000 thermocycler (Applied Biosystems). All results were normalized to the average expression of hypoxanthine phosphoribosyltransferase (HPRT) and B-2-MICROGLOBULIN. Results were obtained from three or four technical replicates of three or four independent biological samples at each data point. Transcript-specific primers used were as follows: B2M, 5’-GCCGTGTGAACCATGTGACT-3’ and 5’-GCTTACATGT CTCGATCCCACTT-3’; HPRT, 5’-TATGGACAGGACTGA ACG TCTTG-3’ and 5’-GCACACAGAGGGCTACAATGTG-3’; LMX1A, 5’-AGGAAGGCAA GGACCATAAGC-3’ and 5’-ATGCTCGCCTCTGTTGAGTTG-3’; PAX6, 5’-GCTTCAC CATGGCAAATAACC-3’ and 5’-GGCAGCATGCAGGAGTATGA-3’; SOX1, 5’-TACGT TTATTTCAGCAGCCTTAGG-3’ and 5’-TCCAGGACAAGGAAGGGTGTT-3’; NURR1, 5’-CGCCTGTAACTCGGCTGAAG-3’ and 5’-CTTGAGGCGAG GACCCATAC-3’; ALDH1A1, 5’-TGACAAGATCCAGGGC CGTACAAT-3’ and 5’-TGAGAGGAGTTTGC TCTGCTG GTT-3’; EN1, 5’-CAGACCCATAATCCTGCATTTCTC-3’ and 5’-TTTGGGA TGTAGGCAAATGGA-3’; OCT4, 5’-GGA GGAAGCTGACAACAATGAAA-3’ and 5’-GGCCTGCACG AGGGTTT-3’; NANOG, 5’-ACAACTGGCCGAAGAATA GCA-3’ and 5’-GGTTCCCAGTCGGGTTCAC-3’; TH, 5’-CCG AGCTGTGAAGGTGTTTGA-3’ and 5’-CGGGCCGGGTC TCTAGAT-3’; IRES, 5’-TGTGAGGGCCCGGAAAC-3’ and 5’-GGGAAAGACCCCTAGGAATGC-3’; UTR, 5’-CCTTGG CTCTTCCTTCCAACT-3’ and 5’-ATGGCTGCTGGGTCTTC TGT-3’.
DNA extraction, Southern, and integration analysis
DNA isolation was performed with the DNeasy Blood & Tissue kit (Invitrogen) according to the manufacturer's instructions. For Southern analysis, the genomic DNA was digested overnight with 40 U of PstI restriction enzyme (New England Biolabs, Ipswich, MA), electrophoresed on a 1% agarose gel, transferred to a neutral nylon membrane (Hybond-N; Amersham, GE Healthcare Limited, Buckingham-Shire) by capillary transfer, and hybridized with a GFP digoxigenin (DIG)–2’-deoxyuridine 5’-triphosphate–labeled probe generated by PCR using the PCR DIG Probe Synthesis Kit (Roche Diagnostics, GmbH, Mannheim, Germany). The probe was detected by an alkaline phosphatase–conjugated DIG-Antibody (Roche Diagnostics) using CDP-Star (Sigma-Aldrich) as a substrate for chemiluminescence. The GFP probe was generated using an eGFP template with the primers GFP-F 5’-CGCACCATCTT CTTCAAGGAC-3’ and GFP-R 5’-CATGCCGAGAGTGATC CCG-3’.
Quantification of transgene copy number per cell was analyzed by qPCR using Platinum SYBER Green qPCR SuperMix (Invitrogen) in an ABI Prism 7000 thermocycler (Applied Biosystems, Life Technologies, Paisley, UK). To determine the number of integrations, primers specific for the transgene sequence were generated: IRES-F, 5’-TGTGAGGGCCCGGAAAC-3’ and IRES-R 5’-GGGAAAGACCCCTAGGAATGC-3’. To quantify the number of cells, B-ACTIN primers were used: B-ACTIN-F 5’-ATTGGCAATGAGCGGTTCC-3’ and B-ACTIN-R 5’-ACA GTCTCCACTCACCCAG GA-3’. To measure the average proviral DNA per transduced cell, two standard curves were made: one employing a known amount of the LV (LV.NES.LMX1A.GFP) using the IRES primers, and a second curve employing a known number of cells and the B-ACTIN primers. Next, the average proviral number per cell was estimated by interpolation of the IRES-to-B-ACTIN ratio from each DNA sample in the two standard curves.
Immunochemistry and cell counts
hESC and iPSC were grown on plastic cover-slide chambers for immunostaining. Cells were fixed with 4% paraformaldehyde and then permeabilized with 0.5% Triton X-100 in Tris-buffered saline. Cells were then blocked in 0.5% Triton X-100 with 3% donkey serum for 2 hr before incubation with primary antibody overnight at 4°C. The following antibodies were used: mouse anti-Tra-1-60 (Chemicon; 1:100), mouse anti-Tra-1-81 (Chemicon; 1:100), rabbit anti-Sox2 (Chemicon; 1:500), rabbit anti-GAD (Chemicon; 1:1,000), mouse anti-HNA (Chemicon; 1:200), rabbit anti-calbindin (Chemicon; 1:1,000), rabbit anti-Nestin (Chemicon;1:200), rat anti-DAT (Millipore; 1:400, Hudson, WI), mouse anti-SSEA-4 (MC-813-70, 1:2), and mouse anti-SSEA-3 (MC-631, 1:2) (from the Developmental Studies Hybridoma Bank at the University of Iowa), rabbit anti-TH (Sigma; 1:1,000), rabbit anti-Girk2 (Sigma; 1:40), mouse anti-ACTA1 (Sigma; 1:400), mouse anti-SMA (Sigma; 1:400), rabbit anti-α-fetoprotein (1:400), rabbit anti-GFAP (Dako; 1:1,000, Dako, Denmark), mouse anti-Tuj1, (Covance; 1:500, Emeryville, California), mouse anti-Oct-3/4 (Santa Cruz; 1:100), rabbit anti-Gata4 (Santa Cruz; 1:50), goat anti-Nanog (Everest Biotech; 1:100, Oxford-Shire, UK), goat anti-FoxA2 (R&D Biosystems; 1:100), chicken anti-GFP (AveLabs; 1:200), rabbit anti-5-HT (Immunostar; 1:2,000), mouse anti-synaptophysin (Millipore; 1:500, Hudson, WI), rabbit anti-DBH (Chemicon; 1:200), and rabbit anti-Lmx1a (kindly provided by Dr. M.S. German, University of California, San Francisco Diabetes Center). Secondary antibodies used were all the Alexa Fluor Series from Invitrogen (all 1:500). Images were taken using a Leica SP5 confocal microscope. For quantification of stained cells, 300 cells per differentiated aggregate were randomly counted (average six to eight differentiated aggregates per experiment). Data points represent the average of at least three independent experiments.
Transplantation in immunodeficient mice
Twenty 6-week-old SCID beige mice were anesthetized and stereotaxically injected with 2×105 cells at Stage 3 in 2 μl using a Hamilton syringe in the striatum. The cells were injected at 1 μl/min rate. Mice were sacrificed at 1 month (n=11) and 5 months (n=8) after transplantation. Upon sacrifice, animals were perfused transcardially with paraformaldehyde (4%), and the brains sectioned at 20 μm on a freezing microtome and stained with antibodies as previously described. Brain sections were analyzed for the presence of immunoreactive cells along the entire length of the graft. All animal experiments were conducted following experimental protocols previously approved by the Institutional Ethics Committee on Experimental Animals, in full compliance with Spanish and European laws and regulations.
DA enzyme-linked immunosorbent assay (ELISA)
Levels of DA were quantified using an ELISA kit obtained from Rocky Mountain Diagnostics (Colorado Springs, CO;
Electrophysiology recordings
Electrophysiology studies were performed using E2 Technology (Aleria Biodevices, Barcelona, Spain). In brief, the cells are cultured in a plate composed of pairs of wells connected through a 1-mm-long microchannel, cells are seeded in the wells, and axons sprout all along the microchannel. For recording, one electrode is located in each well. NPCs were seeded in the wells on the top of PA6; 3 weeks later, recordings in loose-patch mode were obtained (Morales et al., 2008).
Statistical analysis
Data were statistically analyzed by Student's t test using Microsoft Excel software as indicated.
Results
In vitro differentiation of DA neurons from hESC/iPSC
In a first series of experiments, we used the ES[4] hESC line previously derived in our laboratory (Raya et al. 2008), maintained by mechanical passaging on top of feeder layers of irradiated human foreskin fibroblasts. To promote DA neuron differentiation, we implemented a three-stage protocol involving (1) hESC aggregation (via EB formation), (2) neural induction, and (3) DA neuron maturation using a PA6 feeder-based system (Kawasaki et al., 2002) (Fig. 1a). Specifically, neural induction was carried out by incubating EBs in suspension for 10 days in a defined medium supplemented with FGF2, SHH, and FGF8 (Ying et al., 2003; Roy et al., 2006) (Fig. 1b). After that, in Stage 2, NPCs positive for early neural markers, such as NESTIN and SOX2, started to become apparent in the dish (Fig. 1c and d). To achieve DA neuron maturation, NPCs were then treated in Stage 3 with FGF8/SHH and plated on top of PA6 cells. At this stage and under these conditions, almost 50% of the cells expressed the neuronal marker TUJ1, of which ∼50% were of DA lineage as judged by TH expression (Fig. 1e). In our hands, differentiation of hESC in monolayer (e.g., without EB formation) or in the absence of PA6 coculture resulted in poor DA neuron yield (<10% of the differentiated cells were neurons, of which <25% were DA neurons; Fig. 1f). Similarly, supplementation with FGF2 during the entire differentiation protocol resulted in fewer neurons and DA neurons compared with conditions where FGF2 was withdrawn during Stage 3 (Fig. 1f).
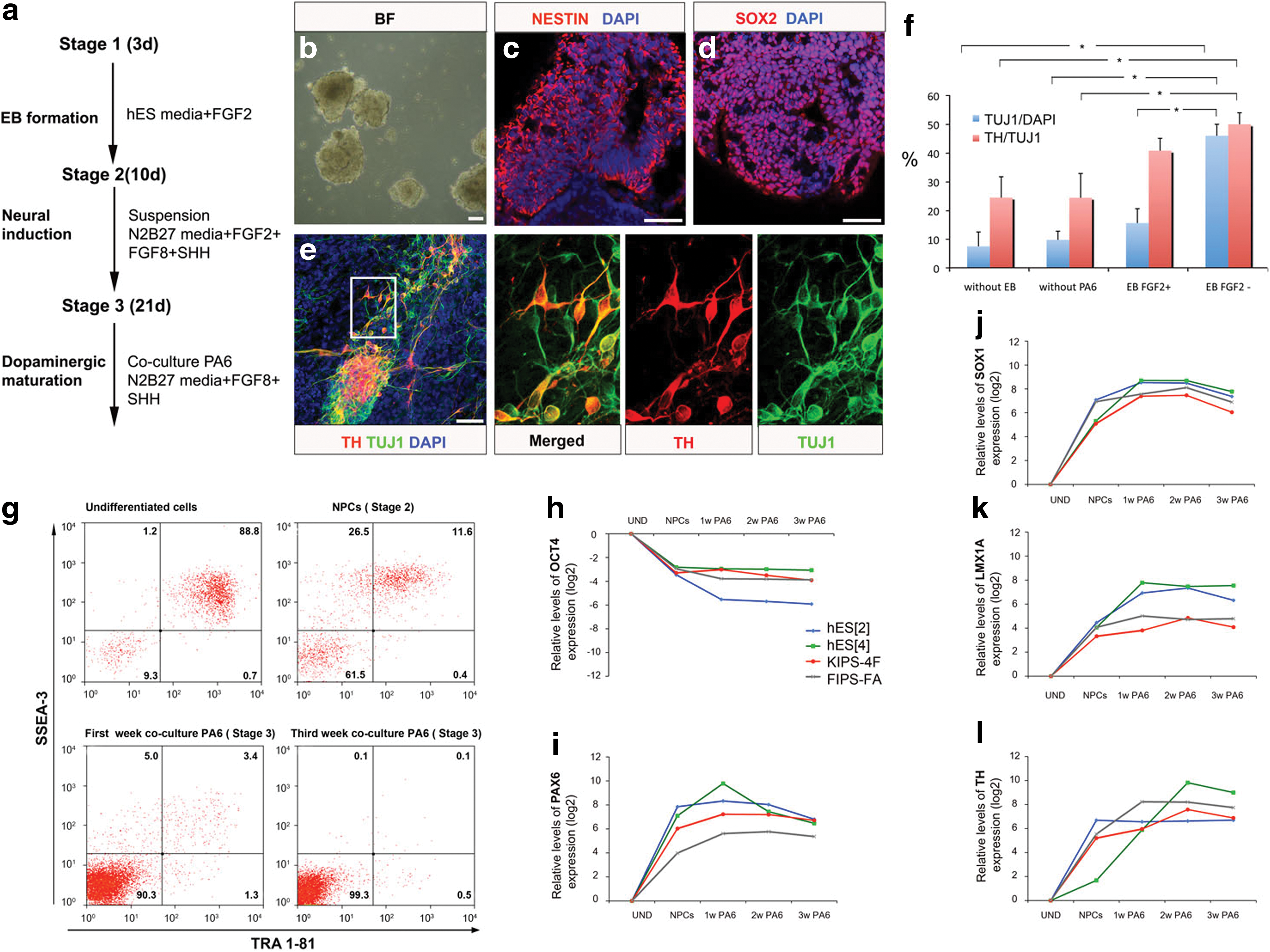
Differentiation protocol implemented for the generation of DA neurons and validation of specific expression of LMX1A in neural precursor cells.
As expected, the expression of SSEA-3 and TRA-1-81, markers of undifferentiated hESC, dramatically declined during the differentiation process, so that only 0.1% of the cells expressed both markers at the end of the protocol, as judged by flow cytometry (Fig. 1g). We confirmed these results by real-time quantitative RT-PCR analyses of OCT4 expression, which also showed a marked decrease as differentiation proceeded (Fig. 1h). To test whether our differentiation protocol could be applied to other lines of human pluripotent stem cells with comparable results, we subjected an additional hESC line {ES[2] (Raya et al., 2008)} and two independent lines of iPSC [KiPS4F#1 and cFA404FiPS4F#1 (Aasen et al., 2008; Raya et al., 2009, respectively)] to the same protocol and harvested samples at different time points for analyses of gene expression. The behavior of all four hESC/iPSC lines was similar in terms of the decrease in OCT4 expression and the up-regulation of neural and neuronal markers (Fig. 1h). Specifically, markers of neural precursors such as PAX6 and SOX1 were found up-regulated in all cell lines tested, peaking at the end of Stage 2 or the first week of Stage 3, whereas markers of DA neuron phenotype such as TH peaked at later time points. The expression of LMX1A, encoding a key transcription factor for midbrain identity (Andersson et al., 2006), showed an intermediate profile, with maximum levels reached between weeks 2 and 3 of Stage 3 (Fig. 1i–l). These results show that we have optimized a protocol for DA neuron differentiation from human pluripotent stem cells that is applicable to both hESC and iPSC, and that achieves DA neuron differentiation efficiencies in the range of previously published protocols (for review, see Hwang et al., 2010). However, and also in agreement with previous reports (for review, see Hwang et al., 2010), DA neurons generated with our protocol were not the majority of the specific subtype lost in PD (i.e., A9-subtype ventral midbrain DA neurons), as judged by the expression of G protein–activated inward rectifier potassium channel 2 (GIRK2; encoded by potassium channel, inwardly rectifying, subfamily j, member 6; KCNJ6) (Inanobe et al., 1999; Mendez et al., 2005) (see below).
Achieving neural-specific overexpression of LMX1A
Therefore, we next asked whether delivering the ventral midbrain DA lineage-associated transcription LMX1A to human pluripotent stem cells could provide appropriate regionalization cues during their differentiation. For this purpose, we generated an LV (LV.NES.LMX1A.GFP; Fig. 2a) in which LMX1A expression is driven by a neural NESTIN enhancer, active in neural progenitor cells (Lothian and Lendahl, 1997) and a minimal TK promoter. The vector also expressed GFP downstream of an IRES element, to allow monitoring of the transgenic expression of LMX1A during the differentiation process. After transduction of hESC with this LV at an MOI of 50, GFP expression was not observed in undifferentiated hESC, whereas it could be clearly detected by epifluorescence microscopy or flow cytometry during the early steps of neural differentiation (Fig. 2b and c). In contrast, >90% of hESC transduced in parallel with LVs expressing GFP from a constitutive PGK promoter (LV.PGK.GFP) displayed strong GFP expression in both the undifferentiated state and during neural differentiation (Fig. 2b and c). The specificity of the LV.NES.LMX1A.GFP construct in hESC undergoing neural differentiation was further confirmed by testing coimmunolocalization of GFP and NESTIN (Fig. 2d).
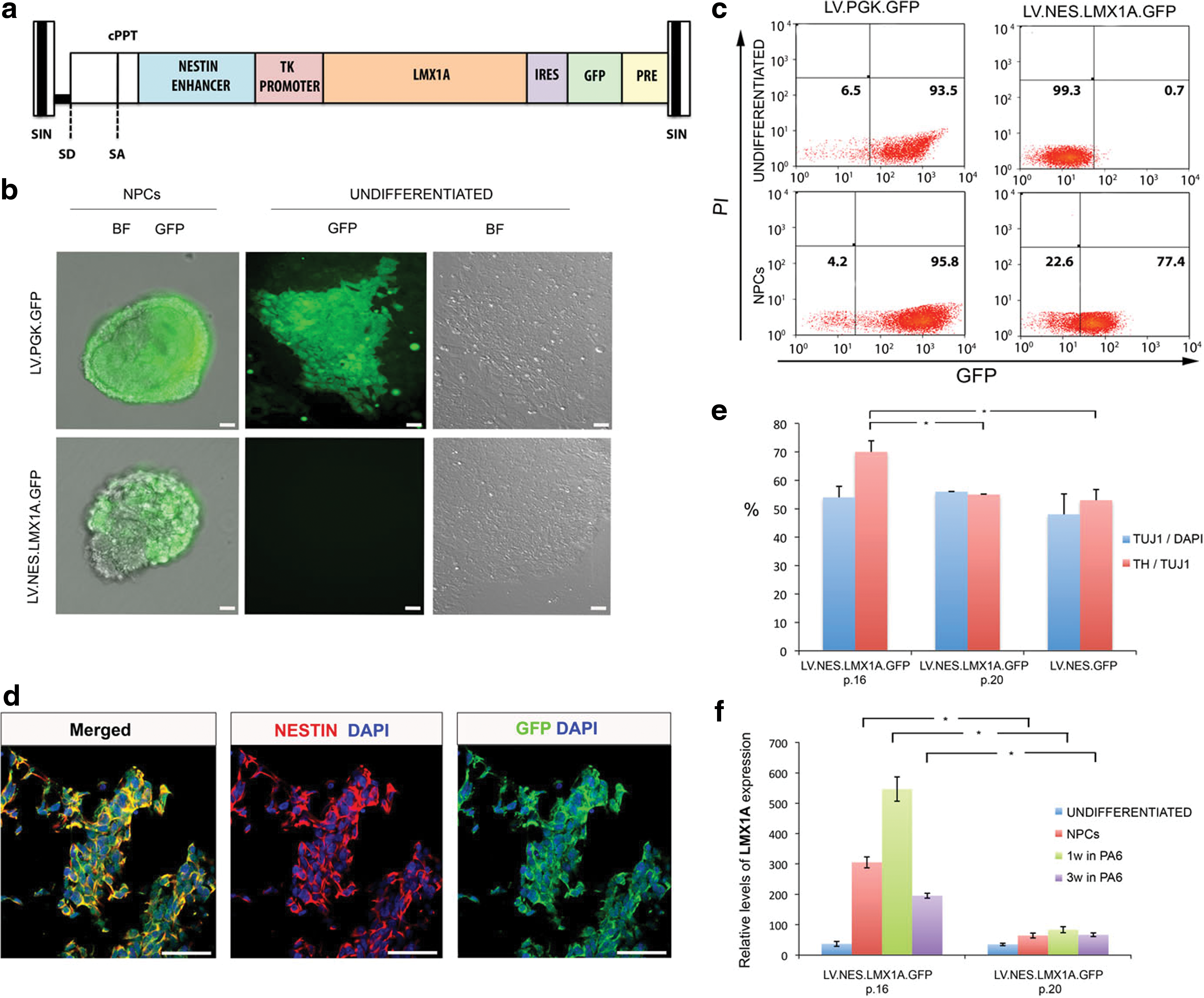
Ectopic LMX1A is specifically expressed in neural precursor cells
To investigate the effect of LMX1A overexpression during the differentiation of human pluripotent stem cells, we subjected hESC tranduced with LV.NES.LMX1A.GFP to our three-stage DA neuron differentiation protocol, and compared the overall yield of neurons and DA neurons with those of hESC transduced with a control (LV.NES.GFP) virus. Whereas no overt differences were found in the total number of neurons in each condition at the end of Stage 3, hESC overexpressing LMX1A generated a significantly higher proportion of DA neurons (Fig. 2e). However, the increased DA neuron differentiation of LV.NES.LMX1A.GFP-transduced hESC was progressively lost as cells were passaged (Fig. 2e), concomitant with a decrease in the levels of transgenic LMX1A overexpression (Fig. 2f), suggesting silencing of the LV transgenes and/or selection of untransduced hESC upon passaging.
Generation of hESC clones overexpressing LMX1A
To circumvent the loss of LMX1A expression in bulk-transduced hESC, we set out to generate clonal lines of LV.NES.LMX1A.GFP-transduced hESC (hES-LMX1A cells). As controls, stable cell lines transduced with LV.NES.GFP were generated (mock-hESC). In order to obtain hESC colonies derived from single cells, 5 days after infection (MOI 10) cells were trypsinized to single cells and serial dilutions plated on human feeder fibroblasts in six-well plates. Between 3 and 6 weeks after plating, colonies of tightly packed cells with a high nucleus-to-cytoplasm ratio and hESC-like morphology appeared in those cultures. We picked and expanded nine independent colonies from wells in which only two to four colonies had grown. The number of lentiviral integration(s) in each clonal line were first screened by qPCR on genomic DNA using primers specific to the IRES sequence and normalized for β-actin (see Materials and Methods for details). From the nine clones analyzed, five of them (clones A, C, G, H, and K) showed no lentiviral integrations, three of them (clones B, L, and I) contained a single lentiviral integration, and one (clone D) showed two integrations. The accuracy of this PCR-based screen was confirmed in selected clones by Southern blot hybridization (Fig. 3a). We selected clones D and L of transgenic hES-LMX1A cells for a thorough characterization and first tested whether the LV transduction could support undifferentiated hESC culture for extended periods of time. After 32 passages, or 8 months in continuous culture, transgenic hES-LMX1A cells maintained a normal 46-XY karyotype (Fig. 3b and data not shown) and remained undifferentiated and pluripotent, as judged by the expression of pluripotency-associated markers OCT4, NANOG, and SOX2, and surface markers SSEA-3, SSEA-4, TRA-1-81 (Fig. 3c–e and data not shown), and the ability to differentiate in vitro into derivatives of the three main embryonic germ layers (Fig. 3f–h and data not shown). In addition, injection of clone D hESC into immunocompromised mice resulted in complex teratomas containing derivatives from all three germ layers, further confirming that transgenic hES-LMX1A cells were pluripotent (Fig. 3i–k). Overall, these results show that genetic manipulation of hESC with LVs expressing LMX1A under the control of a neural NESTIN enhancer does not cause any conspicuous deleterious effect in their self-renewing or pluripotent differentiation abilities.

Generation of hESC clones overexpressing LMX1A.
Effect of neural-specific overexpression of LMX1A on DA neuron differentiation
Transgenic clones of hES-LMX1A cells showed a robust overexpression of LMX1A under DA neuron differentiation conditions, reaching levels of up to 10-fold higher than those of mock-hESC (Fig. 4a). During the course of DA neuron differentiation, both hES-LMX1A and mock-hESC showed a progressive decline in the expression of pluripotency-associated genes such as NANOG, until levels were barely detectable (Fig. 4b). However, the increase in the expression of genes associated with DA neuron differentiation, such as nuclear receptor subfamily 4, group a, member 2 [NR4A2, encoding nuclear receptor-related 1 (NURR1)], engrailed 1 (EN1), aldehyde dehydrogenase 1 (ALDH1A1), and TH, was significantly more pronounced in hES-LMX1A cells when compared with mock-hESC (Fig. 4c–f), suggesting that more cells were being specified to a DA neuron lineage. We directly addressed this possibility by analyzing the expression of relevant markers (Isacson et al., 2003) at various time points of the differentiation protocol by immunofluorescence (Fig. 4g–p). As expected, more LMX1A-positive cells were found in differentiating hES-LMX1A cells in comparison with mock-hESC at Stage 2 (Fig. 4g and h). Moreover, at the end of Stage 3, hES-LMX1A cells generated >25% more DA neurons than mock-hESC (Fig. 4i and j), as evaluated by quantifying the number of TH/TUJ1 double-positive cells (Fig. 4q). Importantly, the majority of DA neurons differentiated from hES-LMX1A cells, but not from mock-hESC, presented the typical bipolar morphology of mDA neurons, stained positive for FOXA2, coexpressed the A9-subtype marker GIRK2, and were mature, as judged by coexpression of DA transporter (DAT) (Fig. 4k–p). In contrast, DA neurons differentiated from hES-LMX1A cells did not express the A10-subtype marker calbindin, although some calbindin-expressing neurons were observed, whereas this marker was frequently coexpressed in mock-hESC-derived DA neurons (Fig. 4r and s). DA neurons differentiated from either hES-LMX1A cells or mock-hESC did not express the marker of adrenergic neurons dopamine-β-hydroxylase (DBH, Fig. 4t and u), or serotonin (5-HT), marker of serotonergic neurons (Fig. 4v and w).
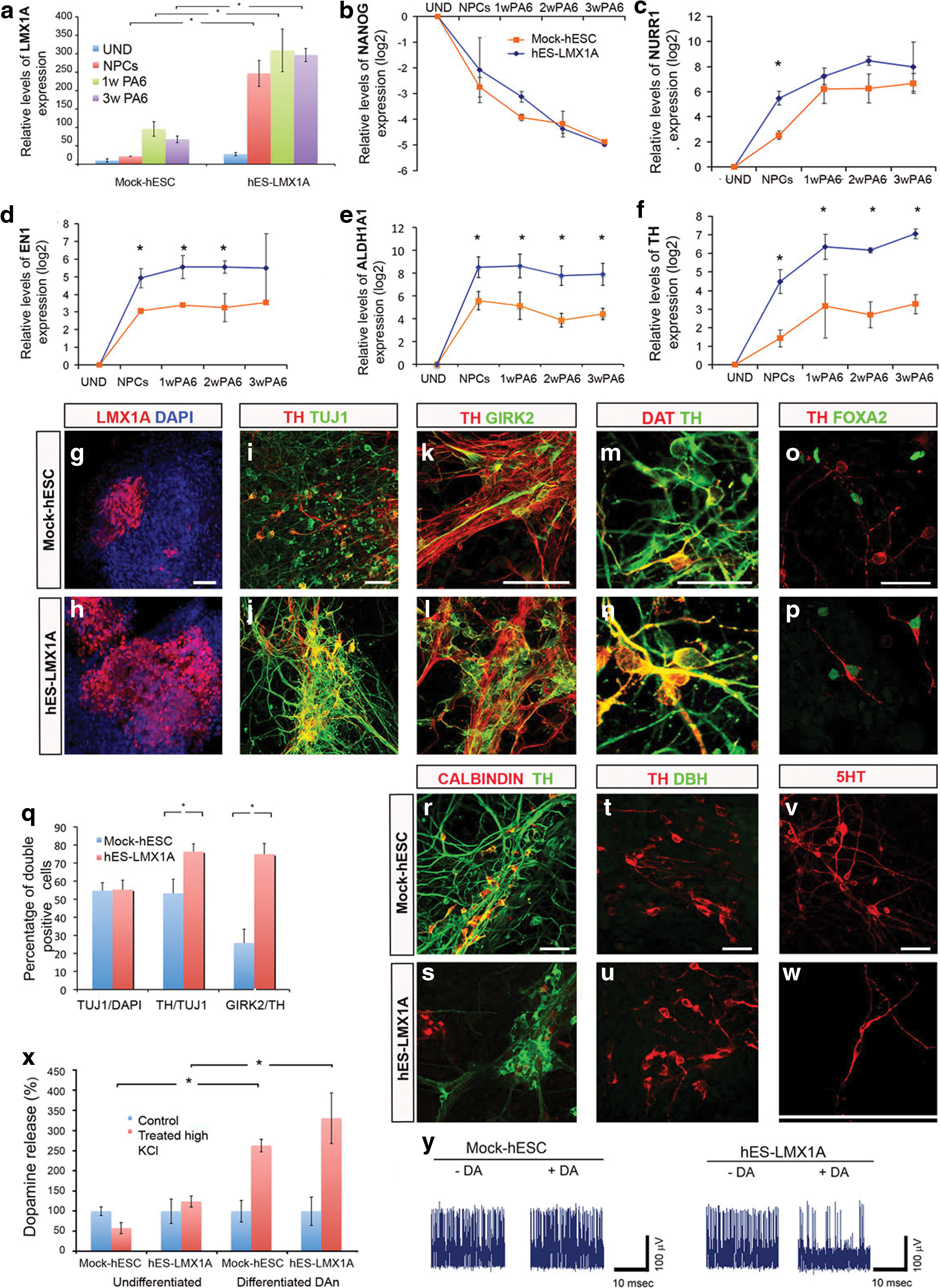
LMX1A enhances generation of midbrain DA neurons in hESC.
Functional characterization of DA neurons differentiated from hES-LMX1A cells
We next tested whether DA neurons allowed to differentiate fully in vitro (end of Stage 3) displayed the functional characteristics expected from bona fide DA neurons. For this purpose, we analyzed their ability to release DA in response to appropriate stimuli. Cells differentiated from either hES-LMX1A or mock-hESC (but not undifferentiated cells) showed robust DA release induced by a depolarizing solution of KCl (60 mM; see Fig. 4x). Next, we analyzed the electrophysiological properties of DA neurons through extracellular recordings using the E2 Technology, in which 10–50 axons grow within a microchannel and allow simultaneous recording of multiple neurons (Morales et al., 2008). These studies showed that cells differentiated from either hES-LMX1A or mock-hESC, but not undifferentiated cells, fired spontaneous action potentials, providing evidence that these cells are, indeed, functional neurons. Moreover, cells differentiated from hES-LMX1A, but not from mock-hESC, responded to the application of DA with a marked reduction in spontaneous firing activity (Fig. 4y), a behavior characteristic of midbrain DA neurons (Friling et al., 2009). Taken together, our results so far show that clones of hESC engineered to overexpress LMX1A in neural precursors, when subject to our optimized protocol for DA neuron differentiation, efficiently generate DA neurons that are molecularly and functionally of the ventral midbrain A9 subtype.
To investigate the functional properties of DA neurons differentiated from hES-LMX1A and mock-hESC, we first tested their ability to engraft and survive within the context of the adult brain. For this purpose, we transplanted 2×105 differentiating hESC at mid-Stage 3 (after 2 weeks of coculture with PA6 cells) directly into the striatum of unlesioned mice. One month after transplantation, grafted cells identified by GFP expression (Fig. 5a), immunofluorescence against TUJ1/TH (Fig. 5b), and antibodies against human nuclear antigen (HNA) (Fig. 5c) were found at the expected position in 10 of 11 mice. To analyze longer grafting times, we retained four mice injected with mock-hESC and four mice injected with hES-LMX1A cells for an additional 4 months. In all eight animals, we found viable grafts containing human TH-positive cells (Fig. 5d–f), although their numbers were much higher in those injected with hES-LMX1A cells (compare Fig. 5e and f). Moreover, confocal immunofluorescence microscopy showed that the vast majority of TH-positive cells found in the engrafted hES-LMX1A cells 5 months after transplantation were mature A9-subtype DA neurons, as shown by coexpression of GIRK2 and DAT (Fig. 5g). Consistent with this, we did not detect GABAergic or serotonergic neurons in the grafted area (Fig. 5h and i).
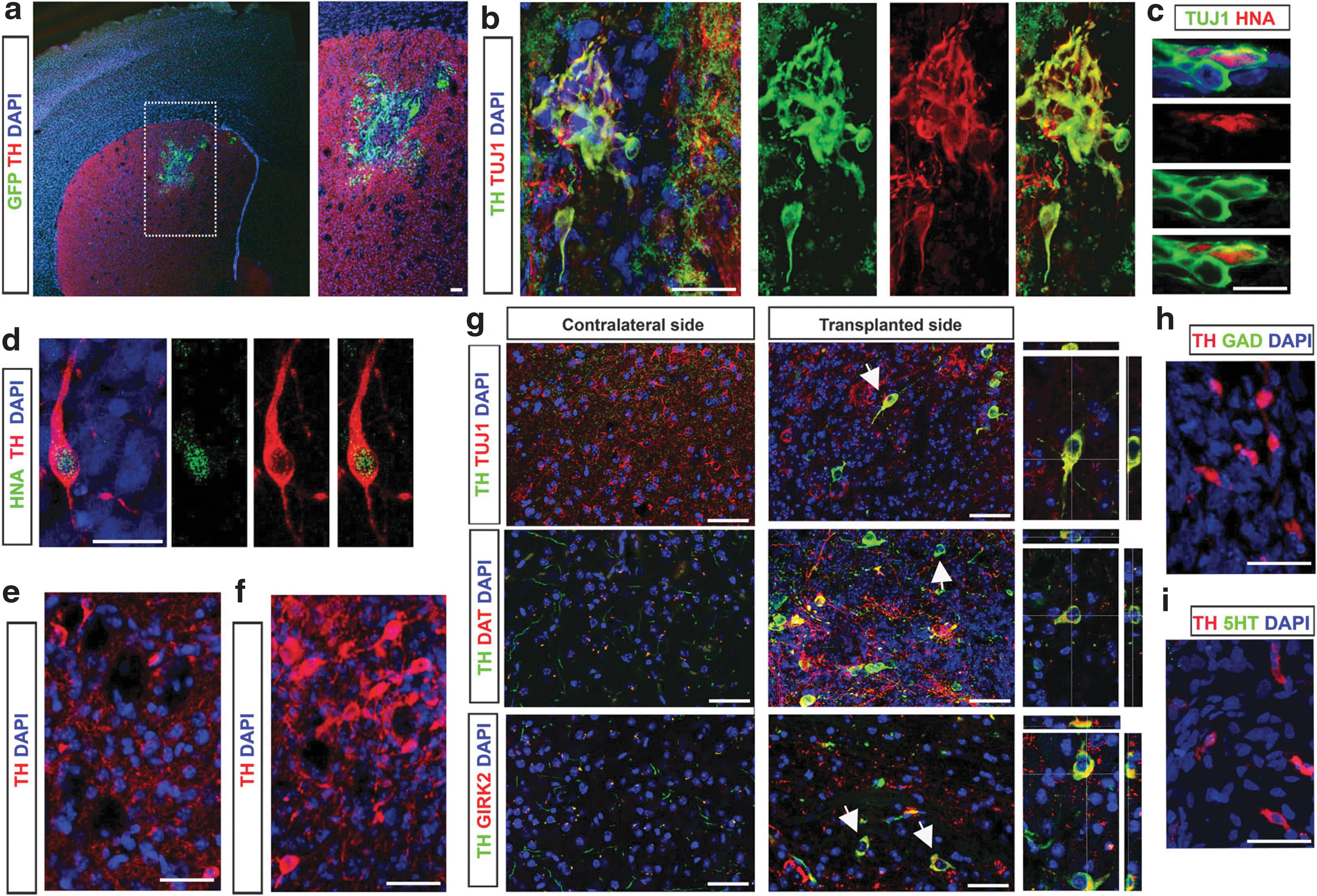
Functional characterization of the DA neurons generated.
Effect of LMX1A overexpression on DA neuron differentiation of iPSC
We next tested whether the strategy of overexpressing LMX1A under the neural NESTIN enhancer could promote appropriate DA neuron differentiation from human iPSC. For this purpose, we transduced iPSC with LV.NES.LMX1A.GFP or with control LVs. LMX1A overexpression was verified at the protein level by immunofluorescence at Stage 2 (Fig. 6a and g). Neuronal and DA neuron differentiation were evaluated at the end of Stage 3 by immunofluorescence analyses against TUJ1 and TH, respectively (Fig. 6b–e and h–k), and the maturation state of DA neurons by DAT expression (Fig. 6f and l). In addition to this, we followed by quantitative RT-PCR the expression of the pluripotency-associated gene NANOG, and those of the DA neuron differentiation markers NURR1, EN1, and TH (Fig. 6m and p). Overall, the results of these analyses were very similar to those of hESC and showed that overexpression of LMX1A did not significantly change the percentage of neurons differentiated from iPSC, but dramatically increased the proportion of neurons that are coaxed toward a DA neuron fate (Fig. 6q). Moreover, the presence of synaptic proteins such as synaptophysin was detected in discrete puncta that colocalized with TH immunoreactivity (Fig. 6r), suggesting the establishment of DA synaptic terminals. Finally, analyses of DA release confirmed the functionality of DA neurons differentiated from iPSC overexpressing LMX1A (Fig. 6s).
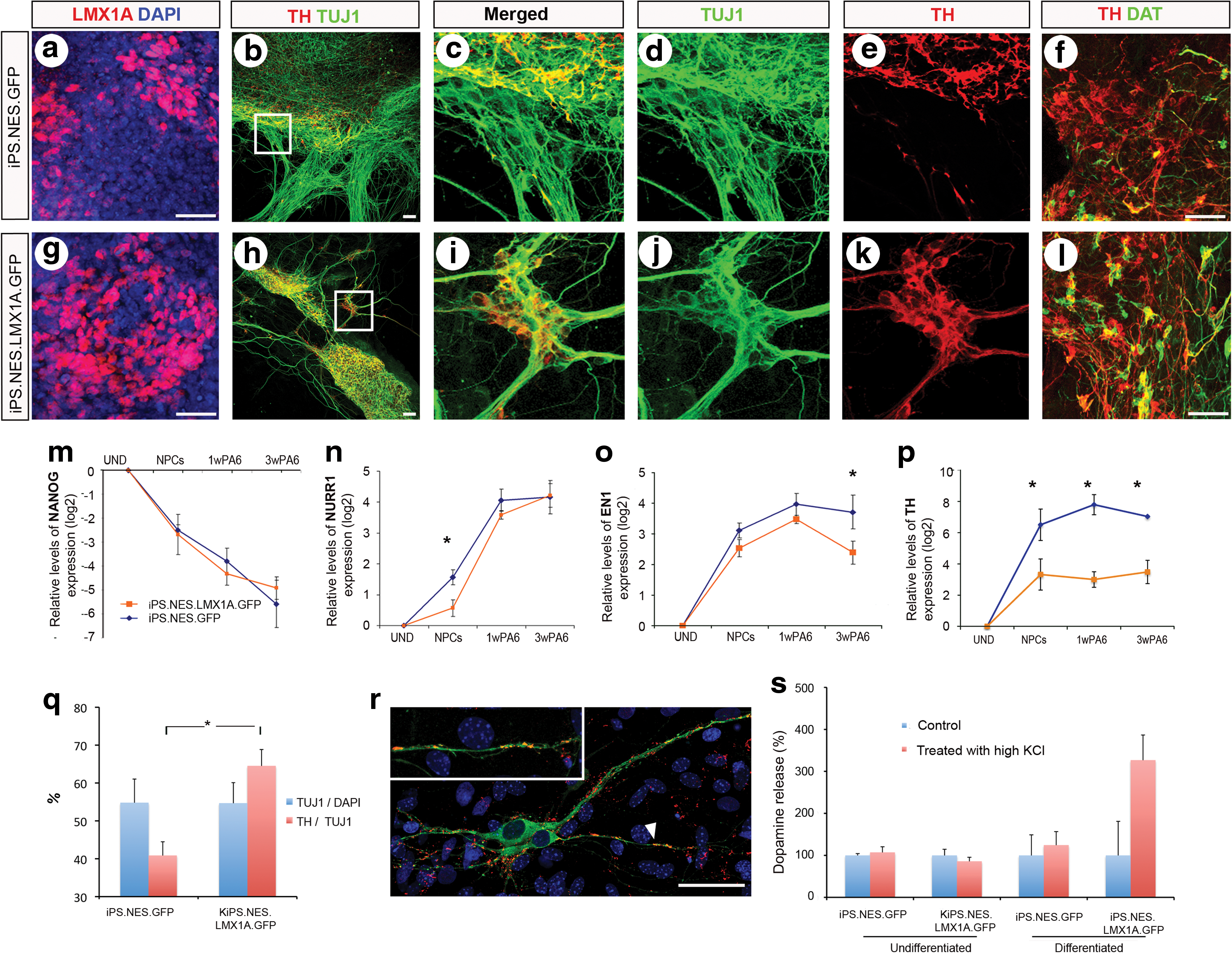
LMX1A enhances generation of midbrain DA neurons in iPSC.
Discussion
One of the major bottlenecks to the development of hESC/iPSC-based strategies for modeling and treating human disease is the relative lack of efficient directed differentiation protocols for generating the specific cell type(s) relevant to the disease. In the case of PD, a variety of protocols have been described that promote the differentiation of hESC/iPSC toward DA neurons (for review, see Hwang et al., 2010), but in most cases these were not appropriately patterned as A9-subtype ventral midbrain DA neurons, the relevant cell type for PD. Forcing the expression of patterning- and/or fate-associated transcription factors may help overcome this limitation, as evidenced by studies in which Lmx1a (Andersson et al., 2006; Friling et al., 2009) or a combination of Nurr1 and Pitx3 (Martinat et al., 2006) or Nurr1 and Foxa2 (Lee et al., 2010) was overexpressed in mouse neural precursor cells. Here, we decided to focus on LMX1A, because it was the only transcription factor the overexpression of which had been shown to clearly increase the percentage of ventral midbrain DA neurons differentiated from human neural progenitors (Friling et al., 2009). In this latter work, hESC-derived neural progenitors were transduced with LVs expressing Lmx1a under a constitutive PGK promoter. Building on these data, we decided to exploit the ability of LVs to provide long-term and robust transgene expression in pluripotent stem cells and neural progenitors (Lois et al., 2002; Pfeifer et al., 2002; Ma et al., 2003; Consiglio et al., 2004), as well as the selectivity of expression in neural progenitor cells conferred by the neural NESTIN enhancer (Lothian and Lendahl, 1997; Andersson et al., 2006), to address whether: (1) human pluripotent stem cells can be stably engineered to express LMX1A while maintaining their characteristic self-renewal and pluripotent differentiation abilities; and (2) expression of LMX1A in the context of engineered hESC/iPSC is sufficient to direct the fate of differentiating neural progenitors toward ventral midbrain DA neurons of the A9 subtype.
On the first issue, our results show that clonal lines of hESC containing one or two copies of the integrated LMX1A-expressing LVs retain the typical features of nonmanipulated hESC, including colony morphology and growth characteristics, long-term self-renewing ability while maintaining a stable karyotype, expression of pluripotency-associated transcription factors and surface markers, in vitro differentiation into derivatives of the three main embryo germ layers, and teratoma formation ability (Fig. 3). This is particularly important because, even though we confirmed the specificity of expression of the neural NESTIN enhancer in neural progenitors (Fig. 2), there was measurable expression of transgenic LMX1A in undifferentiated hESC (Fig. 4a), probably owing to leakage of our system. Therefore, it is important to note that the somewhat higher levels of LMX1A expression in undifferentiated hES-LMX1A cells, compared with mock-hESC, do not appear to be detrimental for their maintenance as undifferentiated, pluripotent stem cells, even upon long-term passaging.
However, when induced to differentiate toward neuronal fates, hES-LMX1A cells generated significantly higher numbers of DA neurons than mock-hESC (∼40% more, based on TH/TUJ1 double staining) and, more importantly, most of these DA neurons were of the ventral midbrain A9 subtype (up to 75% of them, compared with ∼25% in the case of mock-hESC, based on colabeling of TH and GIRK2). Consequently, of 100 hES-LMX1A cells, we obtained >30 A9-subtype DA neurons, versus approximately seven from mock-hESC, that is, close to a fivefold increase in the overall yield of the differentiation procedure. These differences are consistent with more differentiating cells being recruited into the ventral midbrain DA neuron lineage as transgenic LMX1A begins to be expressed in neural precursors, as evidenced by the increased numbers of LMX1A-positive cells in Stage 2 of differentiation.
Our results show that, under ventralizing conditions such as exposure to SHH, LMX1A is sufficient to direct the differentiation of human neural progenitors toward ventral midbrain DA neurons. This is in agreement with previous studies in the mouse, conducted by the Ericson laboratory (Andersson et al., 2006; Friling et al., 2009), and also in human cells (Friling et al., 2009). Therefore, the reported failure of other groups to induce human DA neuron differentiation by overexpressing LMX1A (Roybon et al., 2008; Cai et al., 2009) is likely the result of inadequate levels of expression of this factor, due either to low transfection efficiency (Cai et al., 2009) or to strong overexpression driven by retroviruses (Roybon et al., 2008). In this regard, it is important to note that the levels of LMX1A expression achieved in our clonal hES-LMX1A cell lines harboring one or two LV integrations are in the same range of endogenous LMX1A expression levels during DA differentiation. Thus, LMX1A expression in hES-LMX1A cells, at its peak in mid-Stage 3, is roughly only three times higher than that of mock-hESC (Fig. 4a). Taking into account that, as discussed above, the number of cells undergoing A9 DA neuron differentiation is about five times higher in hES-LMX1A cells than in mock-hESC, it follows that the levels of LMX1A expression per cell should be within the same order of magnitude in either case.
Our results also show that hES-LMX1A cells committed to DA lineages in vitro are able to complete differentiation to A9 ventral midbrain DA neurons upon grafting into the adult mouse brain. We transplanted differentiating cells at mid-Stage 3 (after 2 weeks of coculture with PA6 cells), because we surmised that cells at this stage of differentiation, at which they express high levels of NURR1, EN1, ALDH1A1, and TH (see Fig. 4c–f), would have become depleted of undifferentiated progenitors but were not yet fully differentiated, thus making them more likely to survive after grafting. Indeed, viable grafted cells were recovered 5 months after transplantation and shown to have completed differentiation in situ. Conversely, mock-hESC pretreated in vitro under the same conditions and transplanted into adult mouse brains differentiated very poorly in vivo, in agreement with previous reports that highlighted the inability of adult brain tissue to direct transplanted human neural progenitors to acquire the DA neuron fate (Ben-Hur et al., 2004). Therefore, in the absence of appropriate environmental cues, transgenic LMX1A expression appears to maintain a ventral midbrain DA neuron differentiation program in hES-LMX1A cells.
Our strategy of LV-mediated engineering of human pluripotent stem cells to overexpress LMX1A in neural progenitors provides an efficient way to generate enriched populations of neurons with the characteristics of A9 ventral midbrain DA neurons. As this strategy is applicable to both hESC and iPSC, it should be suitable for modeling PD pathogenesis in vitro, as well as worth considering when designing future cell-therapy applications for this disease.
Footnotes
Acknowledgments
We thank Teresa Lopez-Rovira, Senda Jiménez, and Alberto Garcia for excellent technical assistance, José Miguel Andrés Vaquero for assistance with flow cytometry, Mercé Martí and Lola Mulero Pérez for bioimaging assistance, and Federico Gonzalez for assistance with Southern blot analyses. We are thankful to Dr. M.S. German, University of California, San Francisco Diabetes Center, for providing the Lmx1a antibody. We are also grateful to Miguel A. Valverde and José Manuel Fernández for assistance in preliminary electrophysiological studies. A.S.-D. and I.R.-P. were partially supported by predoctoral fellowships from the Spanish Ministry of Science and Education (MEC). A.C. was supported in part by the Programa Ramon y Cajal from the Spanish Ministry of Science and Innovation (MICINN). Additional support was provided by grants from MICINN (BFU2009-13277, PLE2009-0144, and ACI2010-1117 to A.R.; BFU2010-21823 to A.C.), FIS (PI061897, CP05/00294), and Fondazione Guido Berlucchi 2010 (to A.C.). The work is also part of a CIBERNED Cooperative Project (to A.C., A.R., and M.V.). Work in the laboratory of J.C.I.B. was supported by MICINN, Fundacion Cellex, Tercel, Sanofi-Aventis, and the G. Harold and Leila Y. Mathers Charitable Foundation.
Author Disclosure Statement
All authors have nothing to disclose.