
Abstract
The lentiviral vector system is used extensively in gene therapy trials for various neurological and neurodegenerative disorders. The vector system permits efficient and sustained gene expression in many cell types through integration of the transgene into the host cell genome. However, there is a significant issue concerning the therapeutic use of lentiviral vectors, that transgene insertion may lead to tumorigenesis by altering the expression of proto-oncogenes adjacent to the integration sites. One useful approach for improving safety is to restrict vector transduction to neuronal cells. We have reported the use of human immunodeficiency virus type 1 (HIV-1)-based vectors for efficient retrograde transport by pseudotyping with rabies virus glycoprotein (RV-G) or fusion glycoprotein B type, in which the cytoplasmic domain of RV-G was substituted with the counterpart of vesicular stomatitis virus glycoprotein (VSV-G). Here we developed a novel vector system for neuron-specific retrograde gene transfer (termed NeuRet) by pseudotyping the HIV-1 vector with fusion glycoprotein C type (FuG-C), in which a short C-terminal segment of the extracellular domain and the transmembrane/cytoplasmic domains of RV-G were replaced with the corresponding regions of VSV-G. FuG-C pseudotyping caused efficient gene transfer, mainly through retrograde transport, into neuronal cells in diverse brain regions, whereas the pseudotyping resulted in less efficiency for the transduction of glial and neural stem/progenitor cells. Our NeuRet vector system achieves efficient retrograde gene delivery for therapeutic trials and improves their safety by greatly reducing the risk of gene transduction of dividing cells in the brain.
Introduction
One significant issue in the use of integrative viral vectors for gene therapy concerns transgene insertion into the genome of the host cells, which may alter the expression of proto-oncogenes adjacent to the integration sites, resulting in tumorigenesis. The risk of gene transfer with a gammaretroviral vector has been highlighted by the induction of malignancy in mouse models (Li et al., 2002) and the development of lymphoproliferative disease in gene therapy for X-linked severe combined immunodeficiency (Hacein-Bey-Abina et al., 2003). Use of an HIV-1-based lentiviral vector, in comparison with a gammaretroviral vector, reduces the frequency of interaction with cellular promoters around the integration sites in primary hematopoietic cells (De Palma et al., 2005) and results in lower genotoxicity in a tumor-prone mouse model (Montini et al., 2006). However, the in utero and neonatal gene transfer of a nonprimate lentiviral vector is reported to generate liver and lung tumors (Themis et al., 2005). Transduction of repopulating cells with an HIV-1 vector results in frequent integration of the vector surrounding the proto-oncogene loci (Beard et al., 2007). In addition, activation of oncogenic pathways in the brain of mouse models causes the development of glial tumors (Holland et al., 2000; Dai et al., 2001; Marumoto et al., 2009). Because lentiviral vectors can transduce a wide range of host cells including neuronal and glial cells (Desmaris et al., 2001; Baekelandt et al., 2002; Watson et al., 2002; Duale et al., 2005; Kitagawa et al., 2007), as well as neural stem/progenitor cells (Englund et al., 2002; Ostenfeld et al., 2002; Consiglio et al., 2004; Geraerts et al., 2006; Capowski et al., 2007), there is concern that vector integration into dividing cells may lead to tumorigenesis in the brain. One useful approach for protecting against this possibility is to restrict vector transduction to neuronal cells.
Retrograde axonal transport of some viral vectors confers an advantage for the delivery of genes into neuronal cell bodies situated in regions remote from the injection site (Baumgartner and Shine, 1998; Perrelet et al., 2000; Zheng et al., 2005; Barkats et al., 2006). Pseudotyping of equine infectious anemia virus- and HIV-1-based vectors with selective variants of rabies virus glycoprotein (RV-G) increases the efficiency of retrograde gene transfer in the CNS of rodents and nonhuman primates (Mazarakis et al., 2001; Azzouz et al., 2004b; Mentis et al., 2006; Kato et al., 2007; Federici et al., 2009; Jarraya et al., 2009). More recently, we have provided a vector with highly efficient retrograde gene transfer by pseudotyping an HIV-1-based vector with fusion glycoprotein B type (FuG-B), in which the cytoplasmic domain of RV-G was replaced with the corresponding part of VSV-G (Kato et al., 2011). The HIV-1-based vector pseudotyped with RV-G or FuG-B transduces both neuronal and glial cells at the injection site (Kato et al., 2007, 2011).
In the present study, we developed a novel vector system for neuron-specific retrograde gene transfer (termed NeuRet) by pseudotyping the HIV-1 vector with fusion glycoprotein C type (FuG-C), in which a short C-terminal segment of the extracellular domain and the transmembrane/cytoplasmic domains of RV-G were substituted with the corresponding regions of VSV-G. The NeuRet vector indeed exhibited efficient gene delivery into various brain regions, mainly through retrograde transport, whereas the vector displayed less efficiency in the transduction of glial and neural stem/progenitor cells. Therefore, our new vector system greatly reduces the risk of vector transduction of dividing cells in the CNS and improves the safety of future gene therapy trials for neurological and neurodegenerative disorders.
Materials and Methods
Plasmid construction
The envelope plasmids (pCAGGS-VSV-G and pCAGGS-RV-G) contained VSV-G and RV-G cDNAs under the control of the cytomegalovirus enhancer/chicken β-actin (CAGGS) promoter (Niwa et al., 1991). RV-G cDNA was isolated from the challenge virus standard strain of rabies virus passaged in suckling mouse brain (Morimoto et al., 1998). Envelope plasmids encoding two kinds of fusion glycoproteins (FuG-C and FuG-D) were constructed. The domain structures of VSV-G and RV-G were predicted by a computer algorithm (Rose et al., 1982). For construction of the FuG-C envelope plasmid (pCAGGS-FuG-C), a 1367-bp DNA fragment containing the N-terminal region of the extracellular domain of RV-G (439 amino acids) was amplified by PCR with forward primer 5′-ATTTTGGCAAAGAATTCGCCCTTAGGAAAGATGGTTC-3′ and reverse primer 5′-AGCTCGATTGGATTTTTGGAGAGGTGAACTTCAACAAAATC-3′, and a 339-bp DNA fragment containing the C-terminal region of the extracellular domain (16 amino acids) and transmembrane/cytoplasmic domains of VSV-G was amplified by PCR with forward primer 5′-TCCAAAAATCCAATCGAGCTTG-3′ and reverse primer 5′-TTGTTGGGGGCTGCAGGAATTCCGTTTTTTTTTTTTTTTTTCATAAAAATTAAAAAC-3′. For construction of the FuG-D envelope plasmid (pCAGGS-FuG-D), a 1,281-bp DNA fragment containing the N-terminal region of the extracellular domain of RV-G (414 amino acids) was amplified by PCR with forward primer 5′-TTTTGGCAAAGAATTCGCCCTTAGGAAAGA-3′ and reverse primer 5′-GAGGATGTTCCAGGGGGATAACTGAAGATT-3′, and a 418-bp DNA fragment containing the C-terminal region of the extracellular domain (41 amino acids) and transmembrane/cytoplasmic domains of VSV-G was amplified by PCR with forward primer 5′-TATCCCCCTGGAACATCCTCACATTCAAGA-3′ and reverse primer 5′-GGGGCTGCAGGAATTCCGTTTTTTTTTTTT-3′. A cDNA fragment encoding FuG-C or FuG-D was connected downstream of the CAGGS promoter of the envelope plasmid by using an In-Fusion advantage PCR cloning kit (Clontech, Mountain View, CA). The nucleotide sequences of pCAGGS-FuG-C and pCAGGS-FuG-D were confirmed by DNA sequencing analysis.
Viral vector production
DNA transfection and viral vector preparation were performed as described previously (Hanawa et al., 2002, 2004) with some modifications. The transfer plasmid (pCL20c-MSCV-GFP) carried the cDNA encoding enhanced green fluorescent protein (GFP) downstream of the murine stem cell virus promoter. HEK293T cells (CRL-11268; American Type Culture Collection [ATCC], Manassas, VA) were transfected with transfer, envelope, and packaging plasmids by the calcium phosphate precipitation method. Viral vector particles were pelleted by centrifugation at 10,000×g for 16–18 hr and resuspended in phosphate-buffered saline (PBS). For injection, vector particles were applied to a Sepharose Q FF ion-exchange column (GE Healthcare, Buckinghamshire, UK) in PBS and eluted with a linear 0.0–1.5 M NaCl gradient. Fractions were monitored by absorbance at 260 and 280 nm. Peak fractions containing the particles were collected and concentrated by centrifugation through a Vivaspin filter (Vivascience, Lincoln, UK).
For measurement of functional titer, HEK293T, Neuro2A (CCL-131; ATCC), and N1E-115 (CRL-2263; ATCC) cells were seeded into 10-cm tissue culture dishes and transduced with proper concentrations of viral vectors, and the functional titer was estimated by flow cytometry (FACSCalibur, Becton Dickinson, Tokyo, Japan). For measuring RNA titer, viral RNA in vector stock solution was isolated with a NucleoSpin RNA virus kit (Clontech), and the copy number of the RNA genome was determined with a Lenti-X qRT-PCR titration kit (Clontech). PCR amplification was performed on duplicate samples, using the StepOne real-time PCR system (Applied Biosystems/Invitrogen, Foster City, CA) under the following conditions: 1 cycle of 95°C for 3 min; and 40 cycles of 95°C for 15 sec and 54°C for 1 min.
Stereotaxic surgery
Animal care and handling procedures were conducted according to the guidelines established by the Animal Research Committee of Fukushima Medical University (Fukushima, Japan), and by the Animal Care and Use Committee of the Primate Research Institute of Kyoto University (Inuyama, Japan). Seventy-nine C57BL/6J mice (12 weeks old) and 6 crab-eating monkeys (Macaca fascicularis, 4.0–5.3 kg) were used for the present study.
For injection of viral vectors, mice were anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneal) and vectors were introduced into the dorsal region of the striatum (1.0 μl/site, two sites on a track) or the subventricular zone (SVZ) (1.0 μl/site, one site) through a glass microinjection capillary connected to a microinfusion pump. The anteroposterior, mediolateral, and dorsoventral coordinates (mm) from bregma and dura were 0.5/2.0/2.5 and 0.5/2.0/3.2 for the dorsal striatum and 0.8/1.3/2.0 for the SVZ according to an atlas of the mouse brain (Paxinos and Franklin, 2001). For labeling of dividing cells in the brain, mice that had received vector injection were treated intraperitoneally with 5-bromo-2′-deoxyuridine (BrdU, 80 mg/kg; Roche Diagnostics, Tokyo, Japan) daily for 7 consecutive days.
Monkeys were sedated with ketamine hydrochloride (5 mg/kg, intramuscular) and xylazine hydrochloride (0.5 mg/kg, intramuscular), and then anesthetized with sodium pentobarbital (20 mg/kg, intravenous). By the aid of a magnetic resonance imaging-guided navigation system (StealthStation TRIA [Medtronic, Minneapolis, MN] and Brainsight Primate [Regue Research, Montreal, PQ, Canada]), viral vectors were injected through a 50-μl Hamilton microsyringe into the striatum (5.0 μl/site, two sites per track, five tracks per monkey: two tracks for the caudate nucleus and three tracks for the putamen).
Histological analysis
Animals were anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneal) and perfused transcardially with 4% paraformaldehyde in 0.1 M phosphate buffer (PB; pH 7.4) for mice or with a mixture of 10% formalin and 15% picric acid in 0.1 M PB for monkeys. Perfusion was carried out 4 weeks (for detection of neuronal and glial cells) or 1 week (for detection of neural stem/progenitor cells) after injection of the lentiviral vectors. For immunostaining by the avidin–biotin–peroxidase complex method, transverse sections (30-μm thickness for mice and 60-μm thickness for monkeys) were incubated with rabbit polyclonal antibody for GFP (Molecular Probes/Invitrogen, Eugene, OR) at a 1:2000 dilution, and then with biotinylated goat anti-rabbit IgG antibody (Vector Laboratories, Burlingame, CA) at a 1:500 dilution. Immunoreactive signals were visualized with a VECTASTAIN Elite ABC kit (Vector Laboratories). For double-immunofluorescence histochemistry, sections were incubated with rabbit polyclonal anti-GFP antibody (1:2000 dilution) and one of mouse monoclonal antibodies for neuronal nuclei (NeuN, 1:400 dilution; Chemicon, Temecula, CA), glial fibrillary acidic protein (GFAP, 1:400 dilution; Sigma-Aldrich, St. Louis, MO), Olig2 (1:500 dilution; Millipore, Temecula, CA), Iba1 (1:200 dilution; Abcam, Tokyo, Japan), nestin (1:500 dilution; Millipore), platelet-derived growth factor receptor-α (PDGFRα, 1:200 dilution; Abcam), SOX2 (1:500 dilution; R&D Systems, Minneapolis, MN), or tyrosine hydroxylase (TH, 1:100 dilution; Chemicon). Sections were then incubated with fluorescein isothiocyanate-conjugated goat anti-rabbit IgG and cyanine 3 (Cy3)-conjugated donkey anti-mouse IgG (1:500 dilution; Jackson ImmunoResearch Laboratories, West Grove, PA). For double-immunofluorescence histochemistry for GFP and BrdU, sections were incubated with rabbit polyclonal anti-GFP antibody (1:2000 dilution) and then with 4% paraformaldehyde in 0.1 M PB. Sections were treated with 2 N HCl for 10 min and 0.1 M boric acid buffer (pH 8.5) for 10 min, and sequentially incubated with mouse monoclonal anti-BrdU antibody (1:200 dilution; Becton Dickinson) and the aforementioned secondary IgGs. Fluorescence images were taken under a confocal laser-scanning microscope (LSM510; Zeiss, Thornwood, NY) equipped with proper filter cube specifications for fluorescein isothiocyanate and Cy3 fluorescence channels.
For cell counts, a series of sections through the forebrain and midbrain was used for immunostaining by the avidin–biotin–peroxidase complex method. The number of immunostained cells in each brain region was counted with a computer-assisted imaging program (NIH Image 1.62; National Institutes of Health, Bethesda, MD). For characterization of striatal and SVZ cells, representative sections were used for double-immunofluorescence histochemistry. In individual animals, the number of immunostained cells in the regions of interest (0.2×0.2 mm) was counted. Eight to 10 sections obtained from each brain region of individual animals were used for cell counts, and the average per section was calculated.
Neurosphere-forming assay
Primary cultures of neural stem/progenitor cells and the assay for neurosphere formation were carried out as described previously (Higashi et al., 2008). Mice were anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneal), their brains were removed, and the SVZ was dissected from both hemispheres. Tissues were enzymatically and mechanically dissociated, after which cells were resuspended in serum-free Dulbecco's modified Eagle's medium (DMEM)–F12 medium. For the neurosphere-forming assay, cells were seeded into 6-well tissue culture dishes, and cultured in medium containing epidermal growth factor (20 ng/ml; Sigma-Aldrich), fibroblast growth factor-2 (10 ng/ml; Sigma-Aldrich), and heparin (2 μg/ml; Sigma-Aldrich). After having been cultured for 6 days, floating spheres were counted under a fluorescence stereomicroscope (DP71; Olympus, Tokyo, Japan).
Statistical analysis
Analysis of variance (ANOVA), post hoc Tukey's highly significant difference (HSD) test, and Student t test were used for statistical comparisons. Values are expressed as means±SEM of the data.
Results
Creation of lentiviral vectors pseudotyped with novel fusion glycoproteins
In our previous study, we tested two kinds of fusion glycoproteins: FuG-A, which contained the extracellular domain of RV-G fused to the transmembrane and cytoplasmic domains of VSV-G; and FuG-B, which contained the extracellular and transmembrane domains of RV-G fused to the cytoplasmic domain of VSV-G, for production of the HIV-1-based lentiviral vector (Kato et al., 2011). The vector pseudotyped with FuG-B displayed greater retrograde gene transfer in the brains of rodents and nonhuman primates as compared with the RV-G pseudotype, whereas the transduction property of the FuG-A-pseudotyped vector resembled that of the RV-G pseudotype. These data suggested a possibility that different combinations of RV-G and VSV-G segments may alter the transduction property of the pseudotyped vectors. Because the extracellular domain of viral glycoproteins is considered to be involved mainly in the interaction of the virus with host cells, substitution of part of this domain may shift the cell type specificity for vector transduction. In the present study, for the development of the NeuRet vector we constructed two different types of fusion glycoproteins (designated FuG-C and FuG-D), which are composed of distinct combinations of the N-terminal and C-terminal segments of the extracellular domain from RV-G and VSV-G, fused to the transmembrane/cytoplasmic domains of VSV-G (see Fig. 1). The extracellular domain of FuG-C contained the N-terminal region of the RV-G extracellular domain (439 amino acids) and the C-terminal region of the VSV-G extracellular domain (16 amino acids), whereas the extracellular domain of FuG-D contained the N-terminal region of the RV-G extracellular domain (414 amino acids) and the C-terminal region of the VSV-G extracellular domain (41 amino acids).
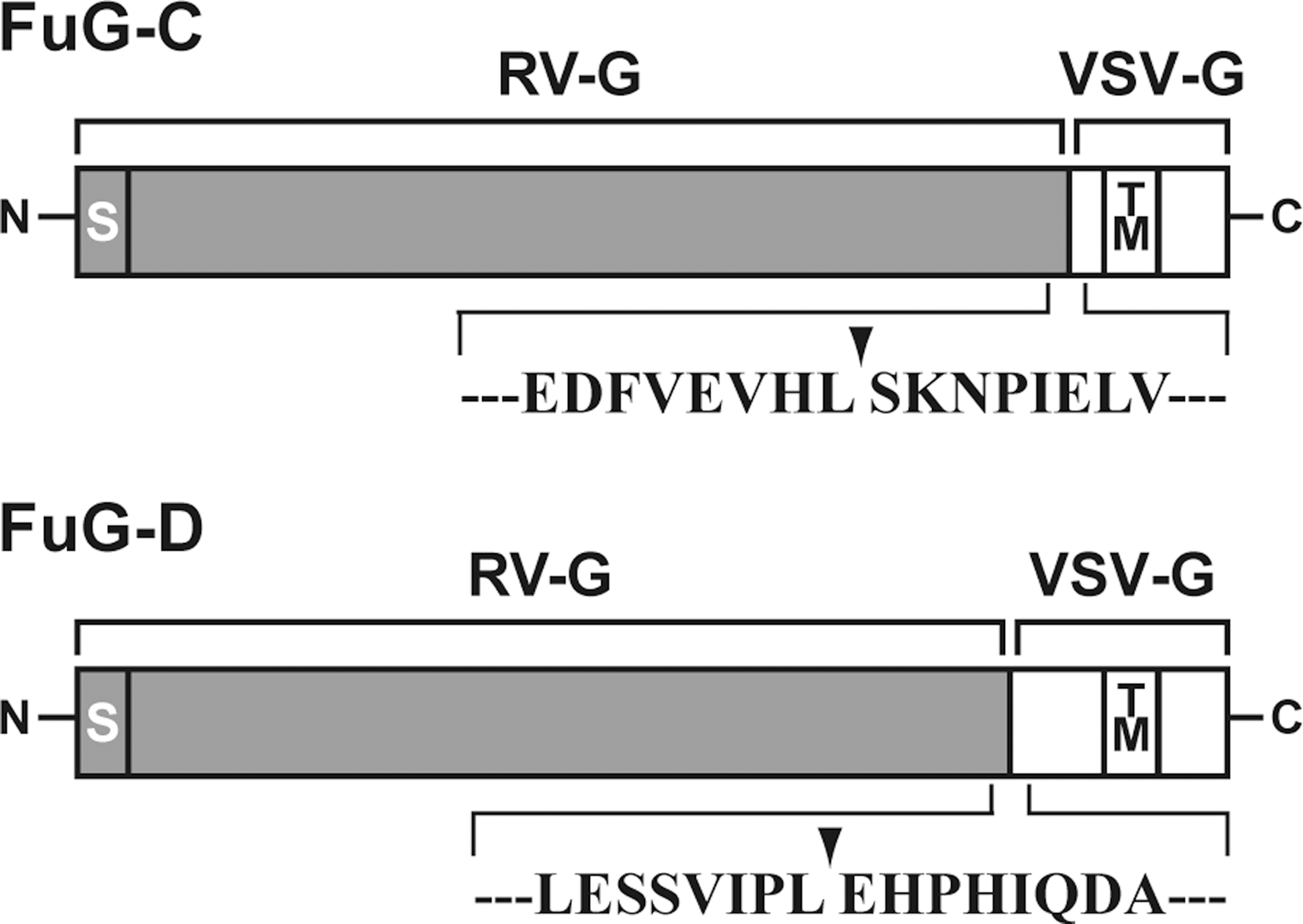
Structure of fusion glycoproteins used for lentiviral vector pseudotyping. FuG-C and FuG-D are composed of different combinations of N-terminal and C-terminal segments of the extracellular domain from RV-G and VSV-G, fused to the transmembrane (TM) and cytoplasmic domains of VSV-G. Amino acid sequences around the junction (arrowhead) between the RV-G and VSV-G segments are shown. S, signal peptide.
We produced HIV-1-based lentiviral vectors carrying the GFP transgene by pseudotyping with distinct glycoproteins (VSV-G, RV-G, FuG-C, or FuG-D), transduced three different cell lines (HEK293T, Neuro2A, and N1E-115) with these vector preparations, and then measured the functional titer (transducing units) by flow cytometry (Table 1). A functional titer of the FuG-C pseudotype was detected only in Neuro2A cells, but the value was significantly lower than that for each of the VSV-G and RV-G titers (ANOVA, Tukey's HSD test, p<0.01). A functional titer of the FuG-D pseudotype was not detected in any of these cell lines. To estimate the concentration of physical particles of the vector, we determined the copy number of viral RNA in the vector preparations by quantitative reverse transcription-PCR (Table 1). The RNA copy number in the FuG-C pseudotype was comparable to the value for the VSV-G or RV-G pseudotype, whereas the copy number of the FuG-D pseudotype was markedly reduced as compared with that of the other pseudotypes (ANOVA, Tukey's HSD test, p<0.01).
HEK293T cells in eighteen 10-cm tissue culture dishes were transfected with the envelope plasmid encoding VSV-G, RV-G, FuG-C, or FuG-D together with the transfer and packaging plasmids. Conditioned medium was collected and centrifuged, and the resultant supernatant was further centrifuged. Vector particles were resuspended in 1 ml of PBS. HEK293T, Neuro2A, and N1E-115 cells were transduced with proper concentrations of viral vectors, and the functional titer (transduction units/ml) was analyzed by flow cytometry. For RNA titer (copies/ml), viral RNA in the vector stock was isolated, and the copy number of the RNA genome was determined by quantitative reverse transcription-PCR. Values were obtained from four independent experiments.
FuG-C, fusion glycoprotein C type; FuG-D, fusion glycoprotein D type; ND, not detected; RV-G, rabies virus glycoprotein; VSV-G, vesicular stomatitis virus glycoprotein.
p<0.01, significant difference from VSV-G or RV-G pseudotype (ANOVA, Tukey's HSD test).
p<0.01, significant difference from VSV-G, RV-G or FuG-C pseudotype (ANOVA, Tukey's HSD test).
Retrograde gene transfer by viral vectors pseudotyped with fusion glycoproteins
We first tested the efficiency of gene transfer through retrograde transport by the pseudotyped lentiviral vectors. Because the efficiency of retrograde gene transfer of the VSV-G-pseudotyped vector is less efficient (Kato et al., 2007), we injected vector pseudotyped with RV-G, FuG-C, or FuG-D with an equivalent RNA titer of 4.8×1010 copies/ml (1.0 μl×two sites) into the dorsal striatum of mice. Brain sections prepared from the injected mice were immunostained with anti-GFP antibody for histological examination (Fig. 2A). Strong signals indicating GFP immunoreactivity were found in a large area of the dorsal striatum in RV-G vector-injected mice, whereas the signals in FuG-C vector-injected animals existed broadly but weakly in the striatum (see below for detailed analysis of gene transduction around the injected sites). In FuG-D vector-injected animals, there was no visible immunoreactivity in the corresponding region. To characterize transgene expression through retrograde axonal transport, we examined sections prepared through the primary motor cortex (M1), primary somatosensory cortex (S1), parafascicular thalamic nucleus (PF), and substantia nigra pars compacta (SNc), each of which innervates the dorsal striatum. As shown in Fig. 2B, in the M1 and S1 areas the injection of RV-G-pseudotyped vector generated some GFP-positive cells on the side ipsilateral to the injection site; and a few immunopositive cells were seen on the contralateral side. Injection of FuG-C-pseudotyped vector produced a larger number of immunopositive cells on the ipsilateral and contralateral sides in the cortical areas than on the corresponding sides in RV-G vector-injected mice. Transgene expression in both hemispheres supports previous anatomical data showing the ipsilateral and contralateral projections of the corticostriatal pathways (McGeorge and Faull, 1989; Gabbott et al., 2005). We also found a larger number of cells in the PF and SNc on the ipsilateral side in FuG-C vector-injected mice than in RV-G vector-injected animals. The number of GFP-positive cells in these four brain regions was counted (Table 2). The cell number in the four regions (on both sides in the M1 and S1, and on the ipsilateral side in the PF and SNc) of FuG-C vector-injected mice was significantly greater than in the corresponding regions in RV-G vector-injected animals (Student t test, p<0.01 for M1, S1, and SNc; p<0.05 for PF). On the other hand, injection of the FuG-D pseudotype resulted in no immunopositive cells in these brain regions (Fig. 2C). Our results suggest that the FuG-C-pseudotyped vector enhanced retrograde gene transfer into the brain regions innervating the dorsal striatum and that the FuG-D pseudotype showed a considerable reduction in transduction activity.
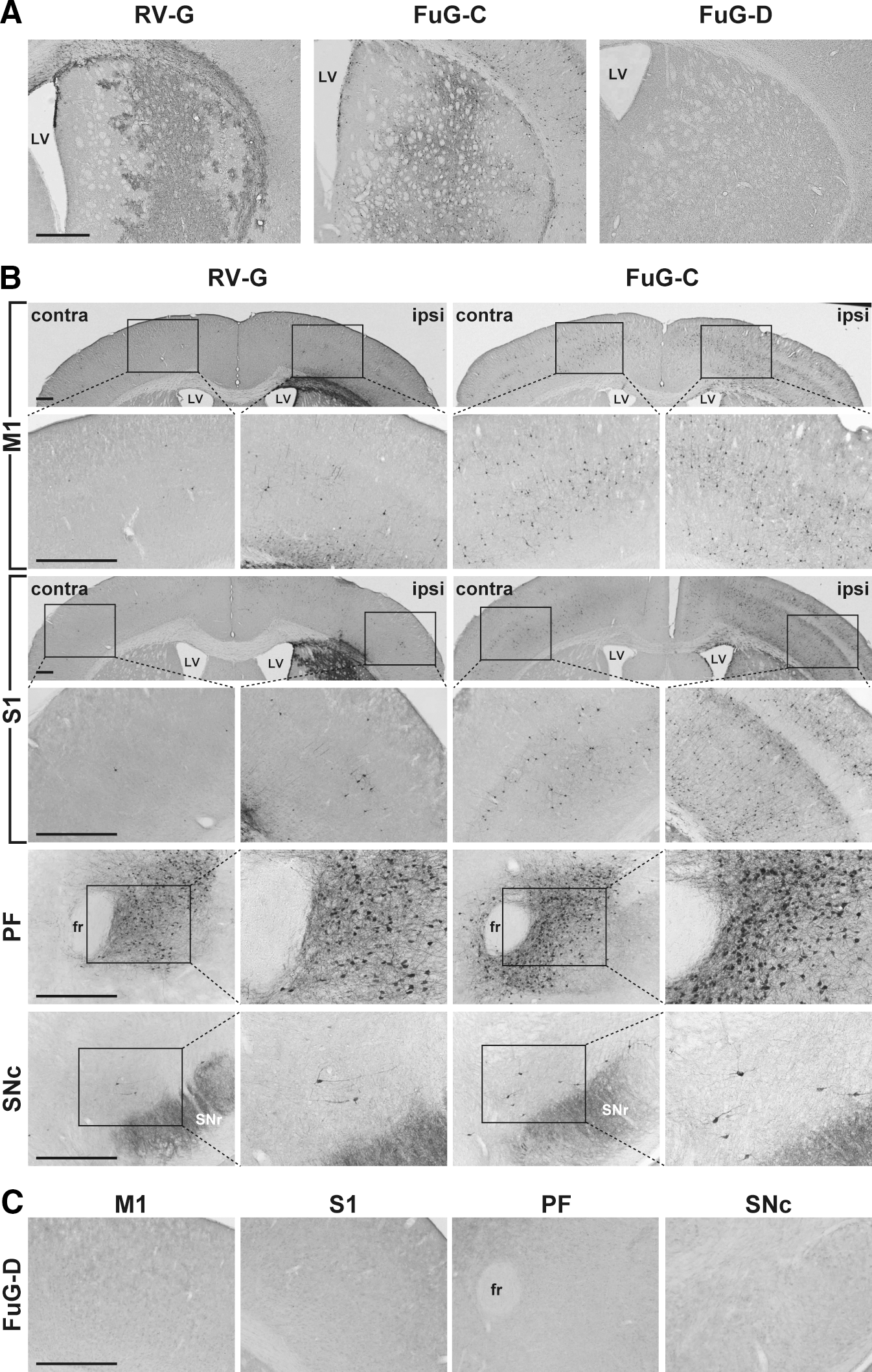
Transgene expression patterns in brain regions after injection of pseudotyped lentiviral vectors into the dorsal striatum in mice. Vector pseudotyped with RV-G, FuG-C, or FuG-D with equivalent copy numbers of viral RNA (4.8×1010 copies/ml, 1.0 μl×two sites) was unilaterally injected into the dorsal striatum, and 4 weeks later a series of sections was prepared for immunohistochemistry with anti-GFP antibody.
Lentiviral vector pseudotyped with RV-G or FuG-C and with equivalent copy numbers of viral RNA (4.8×1010 copies/ml, 1.0 μl×two sites) was unilaterally injected into the dorsal striatum of mice (n=4), and a series of sections through the primary motor cortex (M1), primary somatosensory cortex (S1), parafascicular nucleus of the thalamus (PF), and substantia nigra pars compacta (SNc) were immunostained with anti-GFP antibody. The number of GFP-positive cells per section on the ipsilateral (ipsi) and contralateral (contra) sides of the M1/S1 and on the ipsilateral side of the PF/SNc to the injection site is expressed.
FuG-C, fusion glycoprotein C type; RV-G, rabies virus glycoprotein.
p<0.01, significant difference from RV-G pseudotype (Student t test).
p<0.05, significant difference from RV-G pseudotype (Student t test).
To further characterize the enhanced retrograde gene transfer of the FuG-C pseudotype, we injected various titers of viral vectors pseudotyped with RV-G or FuG-C (3.0×109 to 4.8×1010 copies/ml) into the mouse striatum. Sections through the M1, S1, PF, and SNc were stained with anti-GFP antibody, and GFP-positive cells on the side ipsilateral to the injection site were counted (Fig. 3). Representative microscopy images of the immunohistochemical data are shown in Supplementary Fig. S1 (supplementary data are available online at
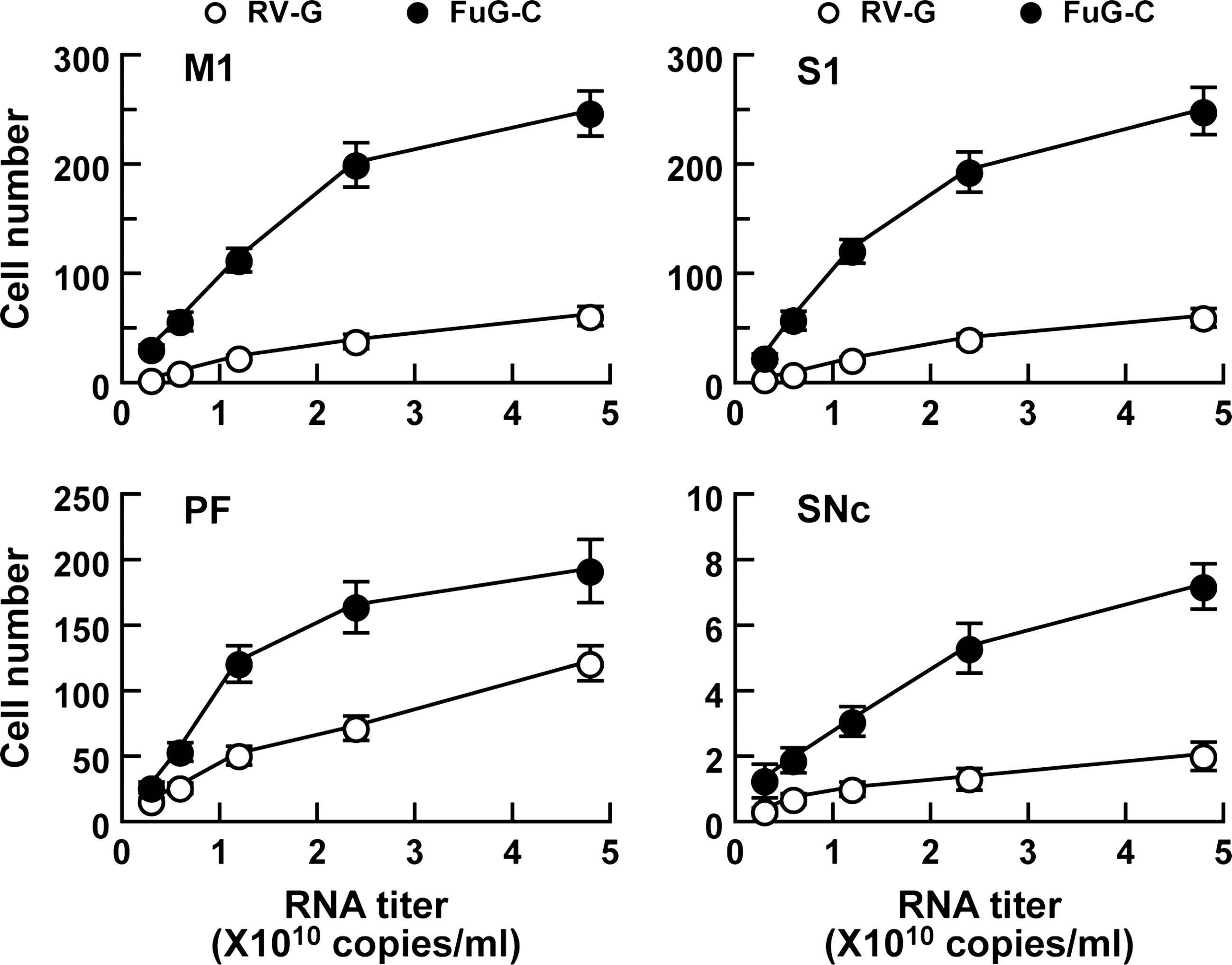
Efficiency of gene transfer through retrograde transport of pseudotyped lentiviral vectors. RV-G- or FuG-C-pseudotyped vector of various RNA titers (3.0×109 to 4.8×1010 copies/ml) was unilaterally injected into the dorsal striatum of mice (n=4), and a series of sections through M1, S1, PF, and SNc was used for GFP immunostaining. The number of GFP-positive cells per section on the side ipsilateral to the injection site is plotted. Data obtained from injection of vectors with 4.8×1010 copies/ml were taken from the experiment indicated in Table 2. Representative microscopy images of the staining obtained from the vector injection are shown in Supplementary Fig. 1 (Supplementary data are available at
Property of gene transduction around injection sites by pseudotyped lentiviral vectors
To examine the property of gene transduction around the injection sites of the FuG-C-pseudotyped vector, we introduced vector pseudotyped with VSV-G, RV-G, or FuG-C (1.2×1010 copies/ml) into the dorsal striatum. Striatal sections were stained by double-immunofluorescence histochemistry for GFP and the neuronal marker NeuN or for GFP and the astrocytic marker GFAP (Fig. 4). To evaluate the efficiency of gene transfer into neuronal and astrocytic cells in the striatum, we normalized the numbers of GFP+/NeuN+ and GFP+/GFAP+ cells by the total numbers of NeuN+ and GFAP+ cells, respectively. The ratio of GFP+/NeuN+ relative to total NeuN+ cell number was 81.7±2.9, 21.4±1.8, and 6.2±1.4% for VSV-G-, RV-G-, and FuG-C-pseudotyped vectors (n=4), respectively. The efficiency of gene transfer into neuronal cells around the injection sites was thus reduced in the case of the FuG-C pseudotype, being lower than that for the VSV-G or RV-G pseudotype (ANOVA, Tukey's HSD test: p<0.001 vs. VSV-G, p<0.01 vs. RV-G). The ratio of GFP+/GFAP+ relative to total GFAP+ cell number was 5.9±0.7, 71.5±3.6, and 0.3±0.03% for VSV-G, RV-G, and FuG-C vectors (n=4), respectively. Transfer of the GFP gene into astrocytes in the striatum was less efficient for the FuG-C pseudotype (ANOVA, Tukey's HSD test: p<0.001 vs. RV-G). In addition, double-immunofluorescence histochemistry for GFP and the oligodendrocytic marker Olig2 or the microglial marker Iba1 revealed the inefficient transduction of the FuG-C pseudotype into the other glial cell types in the striatum (see Supplementary Fig. S2). These data suggest that FuG-C pseudotyping altered the property of gene transduction of lentiviral vectors surrounding the injection sites.
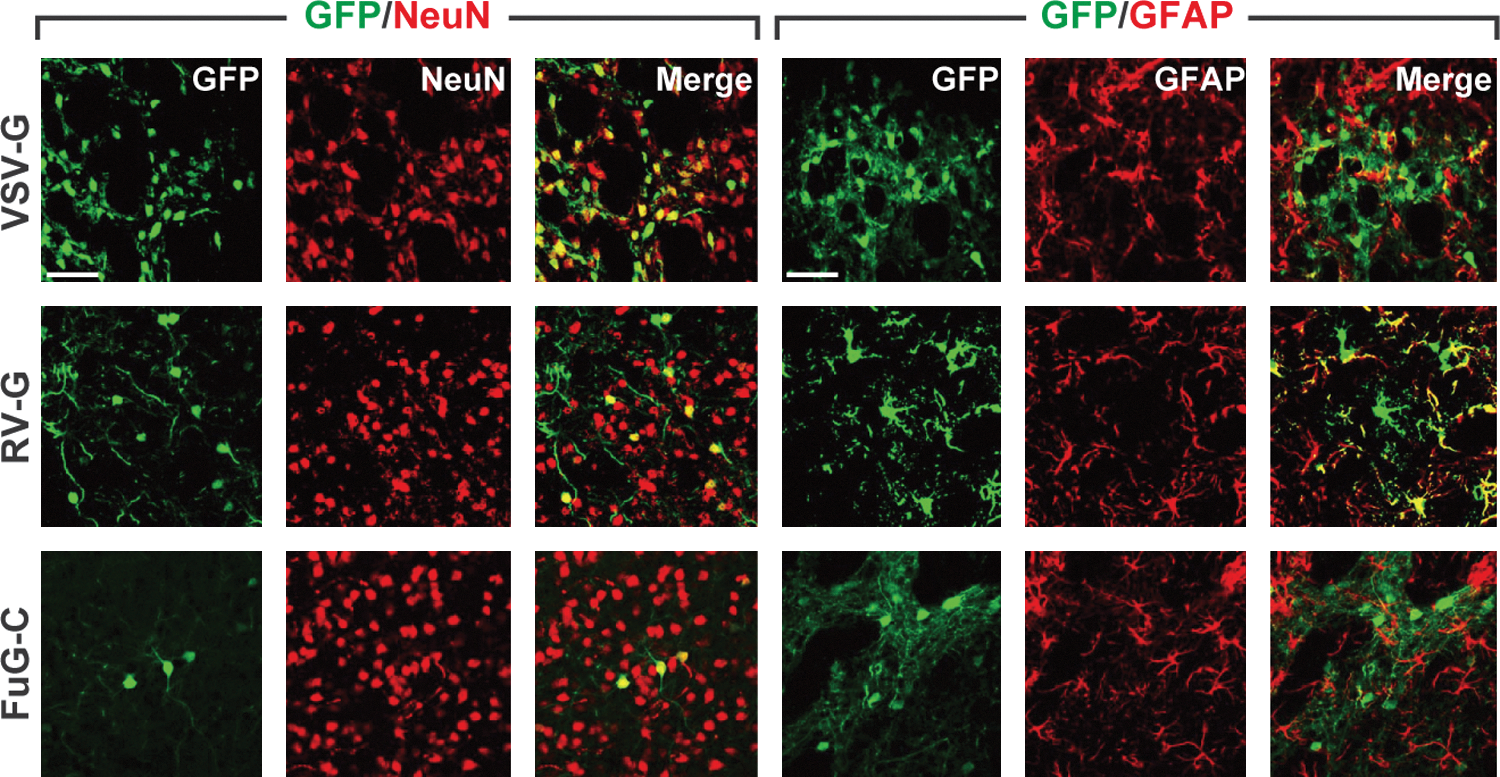
Gene transduction around injection sites of pseudotyped vectors. Vector pseudotyped with VSV-G, RV-G, or FuG-C (1.2×1010 copies/ml) was injected into the dorsal striatum of mice, and sections through the striatum were stained by double-immunofluorescence histochemistry for GFP/NeuN or for GFP/GFAP. Confocal microscopy images of double immunostaining are shown. GFP-positive signals, NeuN- or GFAP-positive signals, and merged images are shown in green, red, and yellow, respectively. Scale bars: 50 μm.
Gene delivery into neural stem/progenitor cells by pseudotyped vectors
Previous studies have shown the efficient gene delivery of the VSV-G-pseudotyped HIV-1 vector into neural stem/progenitor cells (Englund et al., 2002; Ostenfeld et al., 2002; Consiglio et al., 2004; Geraerts et al., 2006; Capowski et al., 2007). We first tested the transduction of the pseudotyped vectors into dividing cells in the SVZ. Vector pseudotyped with VSV-G, RV-G, or FuG-C (1.2×1010 copies/ml) was injected into the SVZ of mice, and then BrdU was injected to label dividing cells in the brain. Sections were stained by double-immunofluorescence histochemistry for GFP and BrdU (Fig. 5A). We found many double-labeled cells in the SVZ in VSV-G and RV-G vector-injected mice, but only a few in FuG-C vector-injected animals. To validate the efficacy of gene transduction into dividing cells, we calculated the ratio of the number of GFP+/BrdU+ cells relative to the total number of BrdU+ cells. The relative ratio was 33.6±3.3, 34.4±2.8, and 1.5±0.3% for VSV-G, RV-G, and FuG-C vectors (n=4), respectively. This parameter for FuG-C vector-injected mice was markedly lower than that for the control animals (ANOVA, Tukey's HSD, p<0.001 vs. VSV-G or RV-G vector). For investigation of the transduction of pseudotyped vectors into neural stem/progenitor cells, brain sections were stained by double-immunofluorescence histochemistry for GFP and nestin, which is a neural stem/progenitor cell marker (Fig. 5B). Injection of VSV-G and RV-G pseudotypes caused GFP expression in a large number of nestin-positive cells in the SVZ, whereas injection of the FuG-C pseudotype resulted in transduction of only a small number of SVZ cells; and these GFP-positive cells did not appear to express nestin. Double-immunofluorescence histochemistry for GFP and another neural stem/progenitor cell marker, PDGFRα or SOX2, confirmed the low efficiency of FuG-C vector transduction of these stem cells (Fig. 5C and D). Furthermore, gene transduction of the pseudotyped vectors into neural stem/progenitor cells was confirmed by the results of the neurosphere-forming assay. Vector pseudotyped with VSV-G or FuG-C was injected into the SVZ of mice, the tissues were dissected from the SVZ, and cells were dissociated from them and cultured for neurosphere formation (Fig. 5E). The ratio of GFP-expressing neurospheres to the total number of spheres formed was calculated. The ratio was 43.5±2.1 and 3.3±0.6% for VSV-G- and FuG-C-pseudotyped vectors (n=4), respectively (Student t test, p<0.001). These data indicate that the FuG-C-pseudotyped vector displayed less efficacy of gene transduction of neurosphere-forming cells, which include stem and transit-amplifying cells (Reynolds and Rietze, 2005; Gritti et al., 2009; Pastrana et al., 2011).
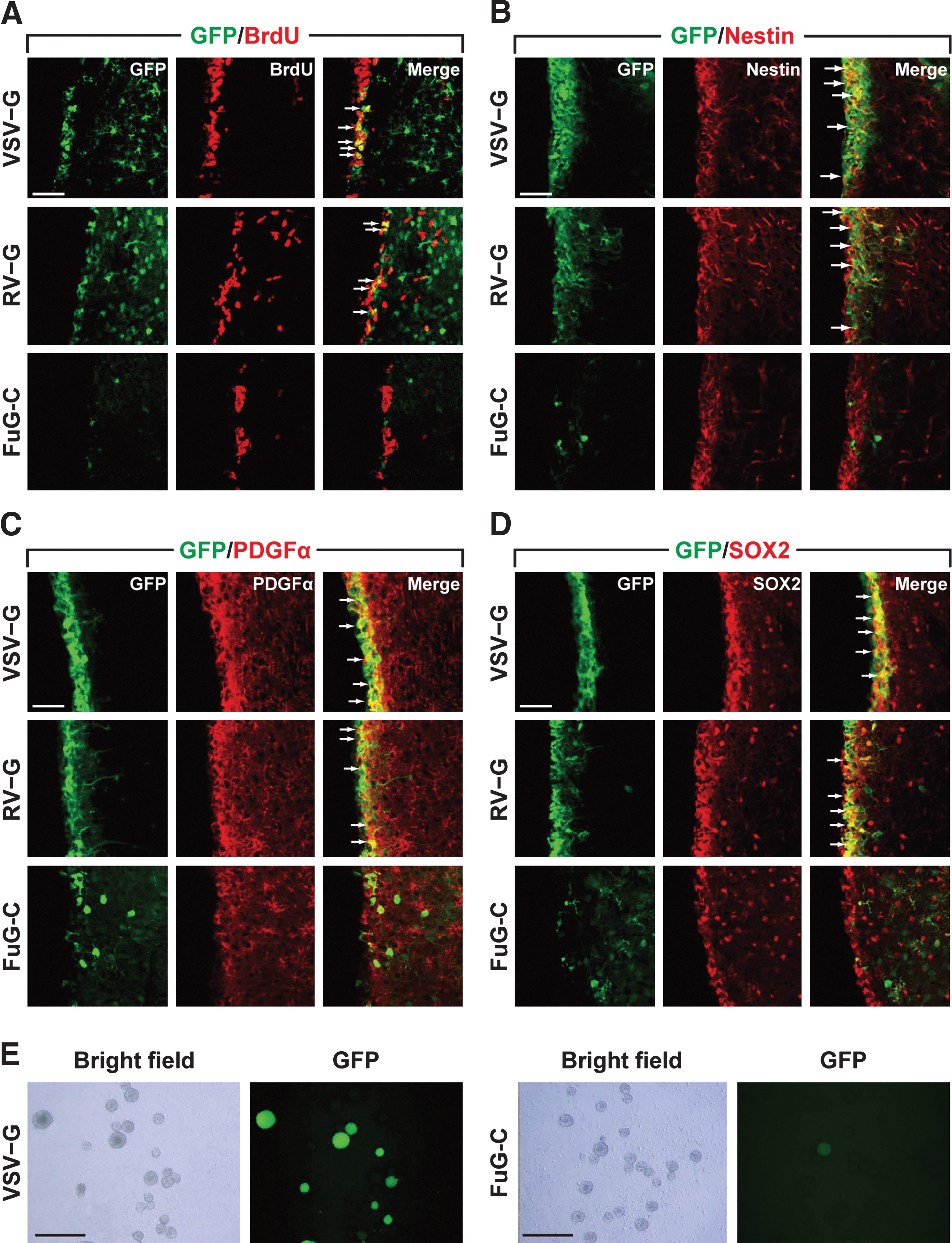
Gene delivery into neural stem/progenitor cells by pseudotyped vectors.
Gene transfer into monkey brain by pseudotyped vectors
The nigrostriatal dopamine system is a major target for the gene therapy of Parkinson's disease. To verify the capability of FuG-C-pseudotyped vector for efficient gene transfer via retrograde transport in the nigrostriatal dopamine system in primates, we injected vector pseudotyped with RV-G or FuG-C into the striatum (caudate nucleus and putamen) of crab-eating monkeys (1.2×1010 copies/ml, 5.0 μl×10 sites). Intrastriatal injections of the vector pseudotypes caused transduction at the injection sites, although the signal intensity in FuG-C vector-injected striatum appeared to be much weaker than that in RV-G vector-injected striatum (Fig. 6A). For characterization of retrograde gene transfer, a series of SNc sections was analyzed by immunohistochemistry with anti-GFP antibody. Injection of FuG-C-pseudotyped vector transduced a larger number of GFP-positive neurons in the SNc compared with that of RV-G-pseudotyped vector (Fig. 6B). The number of GFP-positive SNc cells in RV-G and FuG-C vector-injected monkeys was 129.1 and 523.2 cells per section (n=2), respectively. SNc sections prepared from FuG-C vector-injected animals were stained by double-immunofluorescence histochemistry for GFP and the dopamine neuron marker TH. The GFP transgene was expressed in a majority of TH-immunoreactive neurons (69.1%, n=2; Fig. 6C). These data show that the FuG-C pseudotype indeed achieved efficient gene transfer through retrograde transport into the monkey nigrostriatal dopamine system.
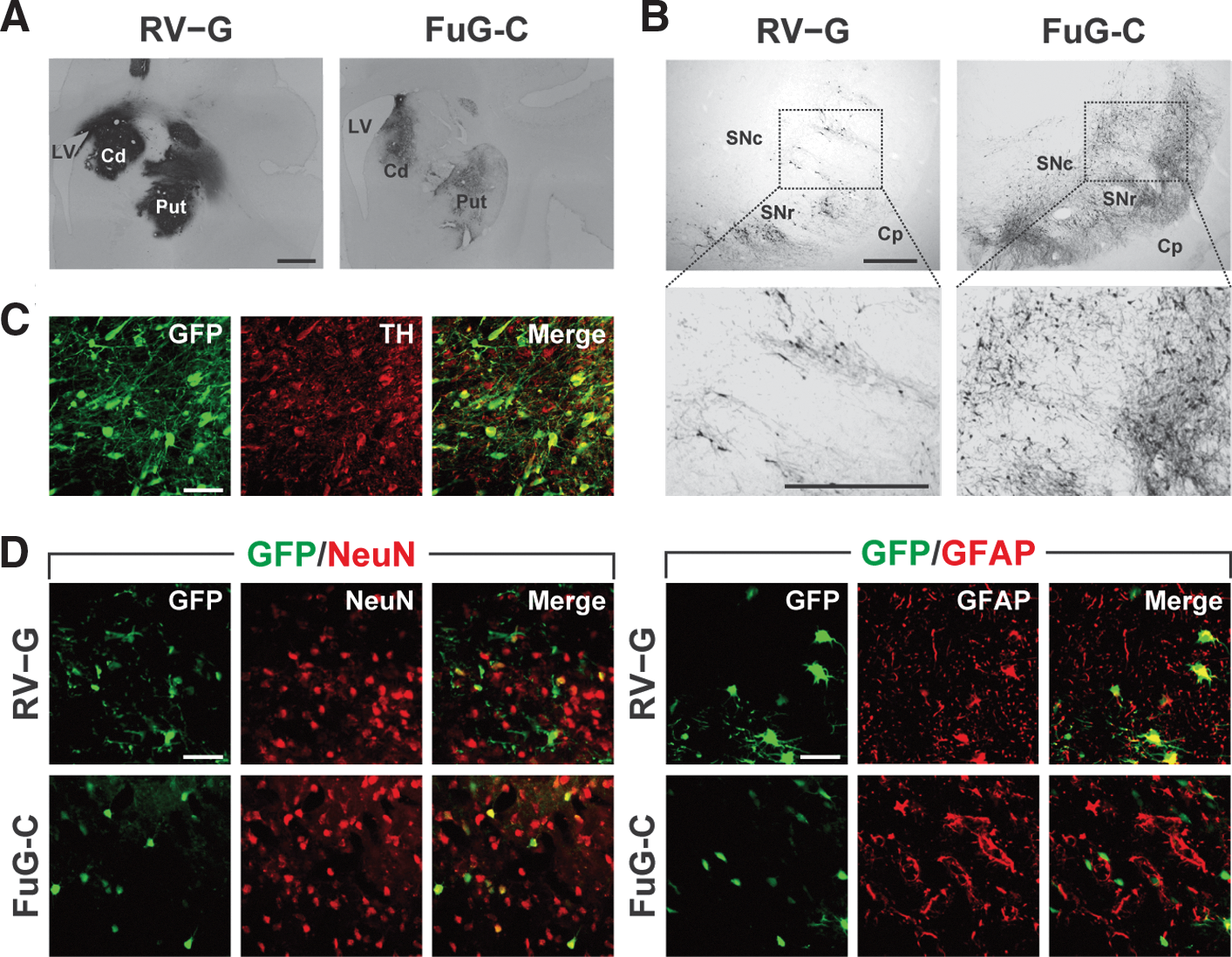
Transgene expression in the nigrostriatal dopamine system after injections of pseudotyped vectors into monkey striatum. RV-G or FuG-C vector (1.2×1010 copies/ml, 5.0 μl×10 sites) was stereotaxically injected into the caudate nucleus (Cd) and the putamen (Put), and histological analysis was performed on brains fixed 4 weeks postinjection.
To compare the efficiency of retrograde gene transfer of the FuG-C pseudotype with that of our original retrograde gene transfer vector pseudotyped with FuG-B (Kato et al., 2011), we injected the FuG-B pseudotype into the striatum of crab-eating monkeys by the same experimental procedures as used for FuG-C vector injection and stained SNc sections for GFP immunohistochemistry (see Supplementary Fig. S3A). The number of GFP-positive SNc cells in FuG-B vector-injected monkeys (n=2) was 549.6 cells per section. When the sections were used for double-immunofluorescence histochemistry for GFP and TH, the transgene was expressed in a majority of the TH-containing neurons (70.2%, n=2; Supplementary Fig. S3B). Comparison of the data obtained from the analysis of monkeys injected with FuG-C- and FuG-B-pseudotyped vectors indicates that the FuG-C vector achieved a high level of retrograde gene transfer into the nigrostriatal dopamine neurons, which was comparable to the gene transfer efficiency of the FuG-B pseudotype.
To assess the property of gene transduction around the injection sites in monkey brain by the FuG-C-pseudotyped vector, we prepared sections through the striatum of crab-eating monkeys that received an intrastriatal injection of vector pseudotyped with RV-G- or FuG-C, and stained the sections by double-immunofluorescence histochemistry for GFP/NeuN or GFP/GFAP (Fig. 6D). The ratio of GFP+/NeuN+ cells to total NeuN+ cells was 7.2 and 12.8% for RV-G and FuG-C vectors (n=2), respectively; and that of GFP+/GFAP+ cells to total GFAP+ cells was 62.2 and 0.6% for RV-G and FuG-C vectors (n=2), respectively. The FuG-C pseudotype thus displayed a low efficiency of gene transfer into neuronal cells around the injection sites, and vector transduction into glial cells was also inefficient in monkey striatum.
Our in vivo study demonstrated that pseudotyping of the HIV-1-based lentiviral vector with FuG-C greatly promoted the efficiency of gene transfer into neuronal populations through retrograde axonal transport in the brain regions of both rodents and nonhuman primates. In contrast, the FuG-C-pseudotyped vector generated less efficient gene transduction into glial cells in the striatum and neural stem/progenitor cells in the SVZ. The transduction property of the FuG-C-pseudotyped vector, in comparison with the RV-G-pseudotyped vector, shifted particularly in the extent of retrograde gene transfer and the cell type preference for transduction. On the basis of these data, we defined the FuG-C lentiviral pseudotype as the NeuRet vector. Our NeuRet vector system provides a powerful strategy for neuron-specific gene delivery, mainly through retrograde transport in the brain.
Discussion
In the present study, we successfully developed the NeuRet vector system by pseudotyping the HIV-1-based lentiviral vector with FuG-C, which is composed of the respective N-terminal and C-terminal segments of the extracellular domain from RV-G and VSV-G (439 and 16 amino acids, respectively), connected to the transmembrane/cytoplasmic domains of VSV-G. FuG-C pseudotyping of the NeuRet vector greatly enhanced the efficiency of retrograde gene transfer into neuronal populations, whereas it resulted in less efficiency of gene transduction into glial and neural stem/progenitor cells. These results demonstrate that the NeuRet vector system offers a promising approach for gene therapy trials for neurological and neurodegenerative diseases through enhanced retrograde transport and that the system improves the safety of gene therapy by profoundly suppressing the efficacy of gene transduction into proliferating cells in the CNS.
In our previous study, we reported efficient gene transfer through retrograde transport into brain regions innervating the striatum by an HIV-1-based vector pseudotyped with FuG-B, in which only the cytoplasmic domain of RV-G was replaced with the VSV-G counterpart (Kato et al., 2011). In the present study, pseudotyping with FuG-C also achieved efficient retrograde gene delivery into the corresponding brain regions. In particular, the gene transfer efficiency of FuG-C-pseudotyped vector into the monkey nigrostriatal dopamine system was comparable to that of the FuG-B pseudotype. These results suggest that the N-terminal region of the RV-G extracellular domain of 439 amino acids used for FuG-C construction is involved in the interaction with synaptic terminals required for retrograde transport. Amino acid residues that are reported to be required for rabies virus virulence exist in the RV-G-derived extracellular domain in FuG-C (Prehaud et al., 1988; Coulon et al., 1998). However, the vector pseudotyped with FuG-D, which contained a shorter N-terminal region (414 amino acids) of the RV-G extracellular domain, was unable to transduce any cells in the in vitro and in vivo experiments, and the RNA titer of the FuG-D vector was much lower than that of the other pseudotypes. The data suggest that FuG-D pseudotyping may impair the formation of vector particles, probably through inefficient incorporation of the envelope glycoprotein into the particles.
FuG-C pseudotyping in the NeuRet vector resulted in less capability to transduce glial and neural stem/progenitor cells in the brain. On the other hand, our previous study shows that the FuG-B pseudotype transduces both neuronal and glial cells surrounding the injection sites to a similar extent as the RV-G pseudotype and that the FuG-A pseudotype, in which the transmembrane and cytoplasmic domains of RV-G were substituted with the corresponding domains of VSV-G, also transduces both cell types in the injection area (Kato et al., 2011), although gene transduction into neural stem/progenitor cells of these two pseudotypes has not been tested. Comparison of the structures of these chimeric envelope glycoproteins suggests that the C-terminal part of 16 amino acids in the extracellular domain may be implicated in determining the host range of vector transduction. Although the C-terminal part of the extracellular domain of VSV-G, together with the transmembrane and cytoplasmic domains, is reported to mediate the process of membrane fusion of the viral envelope (Robison and Whitt, 2000; Jeetendra et al., 2002), the role of this part in the specificity of vector transduction has not been proved. The mechanism by which the structure of the extracellular domain C-terminal part influences the cell type specificity of vector transduction remains to be clarified. Previous studies show that RV-G interacts with certain neuronal receptors, such as the nicotinic acetylcholine receptor α-subunit, low-affinity nerve growth factor receptor, and neural cell adhesion molecule (Hanham et al., 1993; Gastka et al., 1996; Thoulouze et al., 1998; Tuffereau et al., 1998). Identification of the receptors involved in binding of FuG-C will be necessary to address the molecular mechanism that underlies the cell type specificity of vector transduction.
For gene therapy trials with lentiviral vectors, there is the important issue that vector insertion into the host genome may lead to tumorigenesis by altering the expression of cellular oncogenes surrounding the integration sites. One possible approach for overcoming this issue has been the development of lentiviral vectors with defective integrase (Philippe et al., 2006; Yáñez-Muñoz et al., 2006; Philpott and Thrasher, 2007; Banasik and McCay, 2010). Integrase-defective vectors produce increased levels of circular episomes in transduced cells and induce transient expression of transgenes, minimizing the risk of insertional mutagenesis. Another approach involves modification of the integration-site selection of lentiviral vectors. The use of zinc-finger nucleases in the integrase-defective vector system increases the frequency of transgene integration into specific target sites (Lombardo et al., 2007). Expression of a recombinant protein composed of the chromodomain of heterochromatin protein-1α fused to the HIV integrase-binding domain of LEGF/p75 alters target site preferences and decreases the rate of integration events that occur within transcriptional units (Silvers et al., 2010). In this study, we developed the NeuRet vector, which confers neuron-specific gene transduction mainly through retrograde axonal transport. For therapeutic trials for neurological and neurodegenerative disorders, this vector system will provide an additional strategy to improve the safety of integrating lentiviral vectors by greatly reducing the efficiency of transgene integration into dividing cells.
Retrograde axonal transport of viral vectors provides great advantages in model experiments of gene therapy for neurological and neurodegenerative disorders. Injections of adenoviral and RV-G-pseudotyped equine infectious anemia viral vectors into the striatum allow the delivery of genes into the nigrostriatal dopamine system in animal models of Parkinson's disease (Zheng et al., 2005; Barkats et al., 2006; Jarraya et al., 2009). Intramuscular injection of these vectors permits gene delivery into spinal cord neurons in models of motor neuron diseases (Baumgartner and Shine, 1998; Perrelet et al., 2000; Azzouz et al., 2004b). The FuG-B-pseudotyped HIV-1 vector displays highly efficient retrograde gene transfer in the nigrostriatal dopamine system in nonhuman primates (Kato et al., 2011). Here, we demonstrated that the NeuRet vector achieves highly efficient and selective retrograde gene delivery into neurons in brain regions innervating the striatum, in particular into the nigrostriatal dopamine neurons in monkeys. This NeuRet vector is an improvement in protecting against vector integration into proliferative cells in the brain and thus minimizes the risk of tumorigenesis. In addition, this vector excludes the possibility of nonspecific biological reactions of transgene products in other cell types. Our newly developed gene transfer system will warrant a safer strategy with lentiviral vectors for gene therapy trials for various neurological and neurodegenerative disorders.
Footnotes
Acknowledgments
This work was supported by a grant-in-aid from the Core Research for Evolutional Science and Technology of Japan Science and Technology Agency. Part of this work was supported by Highly Creative Animal Model Development for Brain Sciences, carried out under the Strategic Research Program for Brain Sciences by the Ministry of Education, Culture, Sports, and Technology of Japan. The authors thank St. Jude Children's Research Hospital (Dr. A. Nienhuis) and George Washington University for providing the HIV-1-based vector system. The authors are grateful to M. Kikuchi, N. Sato, M. Watanabe, and T. Kobayashi for technical support in the animal experiments and to T. Kuroda for conducting the immunohistochemical staining.
Author Disclosure Statement
K. Kobayashi holds a provisional patent for the NeuRet vector with a novel type of fusion glycoprotein, and the other coauthors have no competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.