
Abstract
Intact p53 function is essential for responsiveness to cancer therapy. However, p53 activity is attenuated by the proto-oncoprotein Mdm2, the adenovirus protein E1B 55kD, and the p53 C-terminal domain. To confer resistance to Mdm2, E1B 55kD, and C-terminal negative regulation, we generated a p53 variant (p53VPΔ30) by deleting the N-terminal and C-terminal regions of wild-type p53 and inserting the transcriptional activation domain of herpes simplex virus VP16 protein. The oncolytic adenovirus vector Ad-mΔ19 expressing p53VPΔ30 (Ad-mΔ19/p53VPΔ30) showed greater cytotoxicity than Ad-mΔ19 expressing wild-type p53 or other p53 variants in human cancer cell lines. We found that Ad-mΔ19/p53VPΔ30 induced apoptosis through accumulation of p53VPΔ30, regardless of endogenous p53 and Mdm2 status. Moreover, Ad-mΔ19/p53VPΔ30 showed a greater antitumor effect and increased survival rates of mice with U343 brain cancer xenografts that expressed wild-type p53 and high Mdm2 levels. To our knowledge, this is the first study reporting a p53 variant modified at the N terminus and C terminus that shows resistance to degradation by Mdm2 and E1B 55kD, as well as negative regulation by the p53 C terminus, without decreased trans-activation activity. Taken together, these results indicate that Ad-mΔ19/p53VPΔ30 shows potential for improving p53-mediated cancer gene therapy.
Introduction
Proto-oncoprotein murine double minute-2 (Mdm2) is a negative regulator of the p53 protein (Fakharzadeh et al., 1993). Mdm2 directly blocks p53 transcriptional activity and reduces protein levels through ubiquitin-dependent degradation in the proteasome (Michael and Oren, 2003). Amplification of the Mdm2 gene and overexpression of its protein have been reported in a variety of human tumors (Momand et al., 1998; Freedman et al., 1999). Mdm2 overexpression attenuates wild-type p53-mediated antiproliferative and proapoptotic effects, promotes carcinogenesis, and correlates with poor prognosis (Freedman et al., 1999; Onel and Cordon-Cardo, 2004). Hence, overcoming Mdm2-mediated p53 inactivation could be a promising strategy for p53-mediated cancer therapy.
The biochemical activity of p53 is further controlled by C-terminal negative regulation. The C-terminal tail (residues 364–393) of p53 interacts with its core domain, locking the DNA-binding domain in a T state conformation with low affinity for its target sequences (Muller-Tiemann et al., 1998; McKinney and Prives, 2002; Selivanova and Wiman, 2007). This biochemical characteristic implies that modification of the N-terminal Mdm2-binding domain of p53 is not sufficient for potent p53-mediated cancer therapy. Thus, alteration of both the C terminus and N terminus of p53, without abrogating its antitumor actions, could be necessary to improve p53-mediated therapeutic responses.
Adenovirus (Ad) has been extensively used as a vector for cancer gene therapy (Guse and Hemminki, 2009). The clinical benefits of Ad vectors compared with other vector systems have been well documented (Wu et al., 2001; Yun, 2008). However, nonreplicating Ad vectors carrying therapeutic genes have limited clinical success in cancer treatment because of inefficient tumor penetration and lack of long-term transgene expression (Vecil and Lang, 2003). Replication-competent Ad vectors have the potential not only to achieve long-term therapeutic gene expression in more cancer cells but also to enable virus distribution to adjacent cancer cells (Kim et al., 2002, 2007; Choi et al., 2006; Lee et al., 2006; Huang et al., 2010; Kwon et al., 2010). To improve the oncolytic activity and cancer cell-selective therapeutic gene expression of Ad vectors, we previously developed Ad-mTERT-Δ19 (Ad-mΔ19), a conditional replication-competent Ad driven by a modified human telomerase promoter (mTERT) that exerts cytopathic effects in cancer cells only (Kim et al., 2003). However, the E1B 55kD protein of the oncolytic Ad inhibits p53 function in a manner similar to that of Mdm2, thereby suppressing p53-mediated transcriptional activation and apoptosis (Grand et al., 1996; Querido et al., 1997). To improve the efficacy of oncolytic Ad-mediated p53 gene therapy, a p53 protein resistant to the effects of E1B 55kD protein is required to enable p53 accumulation.
In the present study, we developed three p53 variants: p53VP was generated by deleting the N-terminal region of wild-type p53 to confer resistance to Mdm2 and E1B 55kD, replacing the deleted N-terminal region with the transcriptional activation domain of herpes simplex virus (HSV) VP16 to retain transcription activity; p53Δ30 was generated by deleting the wild-type p53 C-terminal region to prevent its negative regulation; and p53VPΔ30 contained both modifications. All three p53 variants were functional transcription factors. We showed that an oncolytic Ad vector expressing p53VPΔ30 (Ad-mΔ19/p53VPΔ30) induced apoptosis in cancer cell lines by enabling accumulation of p53VPΔ30, regardless of endogenous p53 and Mdm2 status. Moreover, we demonstrated that Ad-mΔ19/p53VPΔ30 is an effective p53-mediated cancer gene therapy in mice bearing Mdm2-overexpressing U343 tumors.
Materials and Methods
Cell lines and cell culture
A human embryonic kidney cell line (HEK293), lung cancer cell lines (A549 and H1299), brain cancer cell lines (U343, U373MG, and U251N), osteosarcoma cell line (SJSA), fibrosarcoma cell line (HT1080), and cervical cancer cell line (C33A) were purchased from the American Type Culture Collection (ATCC, Manassas, VA). All cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM; GIBCO-BRL, Grand Island, NY) supplemented with 10% fetal bovine serum (GIBCO-BRL),
Construction of negative regulation-resistant p53 variants
To generate a p53 variant resistant to degradation by Mdm2 and E1B 55kD, we replaced the N-terminal region (amino acids 1–79) of p53 with the transcriptional activation domain of HSV VP16. Briefly, the p53 core domain (amino acids 80–393) was amplified by polymerase chain reaction (PCR) with a sense primer (5′-CCTACACCGGCGGCCCCTGCA-3′) and an antisense primer containing a BamHI site (5′-GATCGGATCCTCAGTCTGAGTCAGGCCCTTC-3′), using the pCEP4-wtp53 plasmid (a gift from B. Vogelstein, Johns Hopkins University, Baltimore, MD) as a template. The VP16 transcriptional activation domain (amino acids 409–420) was amplified by PCR with a sense primer containing an XhoI restriction site (5′-GATCCTCGAGATGAAGCTACTGTCTTCT-3′) and an antisense primer (5′-GAAGCTTCTGCAGACGCGTCG-3′), using the pVP16 plasmid (BD Biosciences, San Jose, CA) as a template. After PCR amplification, p53 core PCR product was ligated with VP16 transcriptional activation domain PCR product, generating a p53VP DNA fragment. The fragment (p53VP) was then inserted into pCEP4 (Invitrogen, Paisley, UK), using the BamHI and XhoI sites, producing pCEP4-p53VP. To generate a p53 variant resistant to C terminus–mediated negative regulation, the p53Δ30 variant (amino acids 364–393 deleted) was amplified by PCR, with a sense primer containing an XhoI site (5′-GATCCTCGAGATGGAGGAGCCGCAGTCA-3′) and an antisense primer containing a BamHI site (5′-GATCGGATCCTCAGTCTGAGTCAGGCCCTTC-3′), using the pCEP4-wtp53 plasmid as a template. The PCR product was inserted into pCEP4 (Invitrogen) to produce pCEP4-p53Δ30. To generate a p53 variant resistant to both Mdm2-mediated/E1B 55kD-mediated degradation and C terminus-mediated negative regulation, the p53Δ30 core domain (amino acids 80–364) was amplified by PCR with a sense primer (5′-CCTACACCGGCGGCCCCTGCA-3′) and an antisense primer containing a BamHI site (5′-GATCGGATCCTCACCTGCTCCCCCCTGGCTC-3′), using the pCEP4-wtp53 plasmid as a template. The PCR-amplified p53Δ30 core domain was ligated with the PCR-amplified VP16 transcriptional activation domain, generating a p53VPΔ30 DNA fragment. The new fragment was then subcloned into the pCEP4 expression vector, using the BamHI and XhoI sites, resulting in pCEP4-p53VPΔ30.
Transcriptional activation assay
To study the transcriptional activity of the p53 variants, we used the PG13-Luc luciferase reporter, in which the luciferase gene is under the control of a promoter consisting of 13 copies of the p53-binding element. H1299, 293, and C33A cell lines were seeded into 6-well plates and transfected with pCEP4-wtp53, pCEP4-p53Δ30, pCEP4-p53VP, or pCEP4-p53VPΔ30 along with p53 reporter plasmid (PG13-Luc), using Lipofectamine and PLUS reagents (Invitrogen, Carlsbad, CA). Forty-eight hours after transfection, both luciferase activities (firefly and Renilla) were determined with a Dual-Luciferase assay kit according to the manufacturer's guidelines (Promega, Madison, WI). Results are expressed as normalized firefly-to-Renilla luminescence intensity ratios.
Construction of replication-competent oncolytic Ads expressing p53 variants
We used E3 shuttle vectors to generate replication-competent oncolytic Ads expressing wild-type p53 and the p53 variants. The coding regions of p53 from pCEP4-wtp53, pCEP4-p53Δ30, pCEP4-p53VP, and pCEP4-p53VPΔ30 were individually subcloned into the BamHI site of the E3 shuttle vector pSP72-E3 (Yun et al., 2005), generating pSP72-E3/p53, pSP72-E3/p53Δ30, pSP72-E3/p53VP, and pSP72-E3/p53VPΔ30, respectively. The E3 shuttle vectors were then digested with XmnI, and transformed into Escherichia coli BJ5183 together with the SpeI-digested oncolytic Ad vector pAd-mΔ19 (Kim et al., 2003) to generate Ad-mΔ19/p53, Ad-mΔ19/p53Δ30, Ad-mΔ19/p53VP, and Ad-mΔ19/p53VPΔ30. The recombinant Ad vectors were purified with a plasmid midi kit according to the manufacturer's specifications (Qiagen, Valencia, CA). Viral particle numbers were estimated from the optical density at 260 nm (OD260), where 1 absorbance unit is equivalent to 1012 viral particles per milliliter, and functional infectious titers (plaque-forming units [PFU]/ml) were determined by limiting dilution titration on 293 cells. In all experiments, infections were normalized on the basis of PFU titers.
MTT assay
To evaluate the cancer cell cytotoxicity of Ads expressing the p53 variants, various cancer cell lines were grown in 24-well plates; at 50–60% confluence, the cells were infected with dE1/lacZ, Ad-mΔ19, Ad-mΔ19/p53, Ad-mΔ19/p53Δ30, Ad-mΔ19/p53VP, or Ad-mΔ19/p53VPΔ30 (A549, U251N, and U373MG, 5 multiplicities of infection [MOI]; U343 and H1299, 10 MOI). Three days postinfection, 200 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich, St. Louis, MO) in phosphate-buffered saline (PBS; 2 mg/ml) was added to each well. After a 4-hr incubation at 37°C, the MTT-formazan formed by metabolically viable cells was solubilized with 1 ml of dimethyl sulfoxide. Absorbance was read on a microplate reader at 540 nm. All assays were carried out in triplicate.
Terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling assay
Apoptosis was analyzed by the terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling (TUNEL) assay, as described previously (Yoon et al., 2006). Briefly, cancer cells were plated onto a chamber slide; the next day they were infected with Ad-mΔ19, Ad-mΔ19/p53, Ad-mΔ19/p53Δ30, Ad-mΔ19/p53VP, or Ad-mΔ19/p53VPΔ30 (MOI, 3). Cells treated with 1 μM camptothecin (CPT) were used as a positive control. Forty-eight hours after treatment with Ad vectors or CPT, cells were washed with PBS, and then DNA single-stranded and double-stranded breaks were visualized with an ApopTag kit (Oncor, Gaithersburg, MD) according to the manufacturer's instructions. Apoptotic cells were counted under×400 magnification in five selected fields. More than 2000 cells were counted to calculate the percentage of TUNEL-positive cells.
Propidium iodide staining
Induction of apoptosis and nuclear damage were also detected by propidium iodide (PI) staining and flow cytometry. Cancer cells were grown in T-25 cell culture flasks and infected with Ads expressing p53 variants (MOI, 5). Forty-eight hours after treatment with Ad vectors or 1 μM CPT, the cells were harvested with PBS containing 0.1% EDTA. Cells were washed twice with PBS, pelleted by centrifugation, resuspended in PBS (containing 3 mM sodium citrate, 0.1% Triton X-100, and PI [50 mg/ml]), and then incubated for 4 hr. Apoptosis and nuclear damage were determined by fluorescence-activated cell sorting (FACS; BD Biosciences); data from 10,000 events were collected for further analysis.
Analysis of cell morphology by transmission electron microscopy
Cells (U343 and H1299) seeded on 10-cm dishes were infected with each Ad vector (MOI, 1). After 36 hr, the cells were harvested and fixed for 1 hr in a cacodylate buffer (0.1 M) containing 3% glutaraldehyde and 2% paraformaldehyde, washed with PBS, and then treated with 0.1% tannic acid for 20 min. The samples were then incubated in 1% buffered osmium tetroxide for 1 hr and stained with 1% aqueous uranyl acetate for 1 hr (Muller et al., 1994). The samples were dehydrated with increasing concentrations of ethanol, and then infiltrated and embedded in Spurr's low-viscosity medium. The blocks were polymerized in a 60°C oven overnight. Thin sections were cut, stained with uranyl acetate and lead citrate, and examined by transmission electron microscopy (TEM) (1200-EX electron microscope; JEOL, Peabody, MA).
Western blot analysis
Cells grown in 6-well plates were infected with Ad-mΔ19/p53, Ad-mΔ19/p53VPΔ30, or control Ad-mΔ19 or dE1/lacZ (MOI, 1). Twenty-four hours postinfection, cells were lysed in lysis buffer (50 mM HEPES containing 0.15 M NaCl, 0.5% Nonidet P-40, and proteinase inhibitors phenylmethylsulfonyl fluoride, tosyl-
Immunoprecipitation analysis
SJSA cells seeded on 10-cm dishes were infected with each oncolytic Ad (MOI, 1). Twenty-four hours postinfection, the cells were harvested and lysed in lysis buffer (50 mM HEPES containing 0.15 M NaCl, 0.5% Nonidet P-40, and proteinase inhibitors phenylmethylsulfonyl fluoride, tosyl-
Animal studies
Male athymic nu/nu mice (6–8 weeks old) were obtained from Charles River Japan (Yokohama, Japan) and maintained in a laminar air-flow cabinet under specific pathogen-free conditions. All facilities were approved by the Association for Assessment and Accreditation of Laboratory Animal Care, and all animal experiments were conducted under the institutional guidelines established for the Animal Core Facility at Yonsei University College of Medicine (Seoul, Korea).
Suppression of tumor growth in vivo
U343 human brain cancer cells (1×107) were implanted in the left abdomen of each mouse by subcutaneous injection. When the tumors reached 70–100 mm3, treatment was initiated by intratumoral injection of Ad vectors (30 μl/tumor; each treatment group, n=6). A total of three intratumoral injections (2×107 PFU) were administered (days 1, 3, and 5). Tumor growth was monitored by measuring the length and width of the tumor with a caliper every 2 days. Tumor volume was calculated as: volume=0.523L×W 2, where L represents tumor length and W represents tumor width.
Statistical analysis
Results are expressed as means±standard error mean (SEM). Statistical analyses of the data were performed by two-tailed Student t test (SPSS 13.0 software; SPSS, Chicago, IL). p<0.05 was considered significant.
Results
Transcriptional activity of p53 variants in vitro
We used the Dual-Luciferase reporter assay system to determine the transcriptional activity of the p53 variants in human cancer cell lines with different p53 status (wild-type p53; 293, mutant p53; C33A, p53-null; H1299). Luciferase activity was assessed 48 hr after transfection with plasmids expressing p53 (pCEP4-wtp53, pCEP4-p53Δ30, pCEP4-p53VP, or pCEP4-p53VPΔ30) along with luciferase reporter plasmid (PG13-Luc). PG13-Luc contains a promoter consisting of 13 copies of consensus p53-binding elements derived from the ribosomal gene cluster upstream of a polyomavirus minimal promoter sequence. As presented in Fig. 1, the transcriptional activities of p53VP, p53Δ30, and p53VPΔ30 variants were not lower than that of wild-type p53, regardless of endogenous p53 status. Further, the C-terminal domain deletions (p53Δ30 and p53VPΔ30) increased activity 3- to 4-fold compared with wild-type p53 (p<0.01). Taken together, these results demonstrate that the modifications to wild-type p53 did not reduce its transcriptional activity.
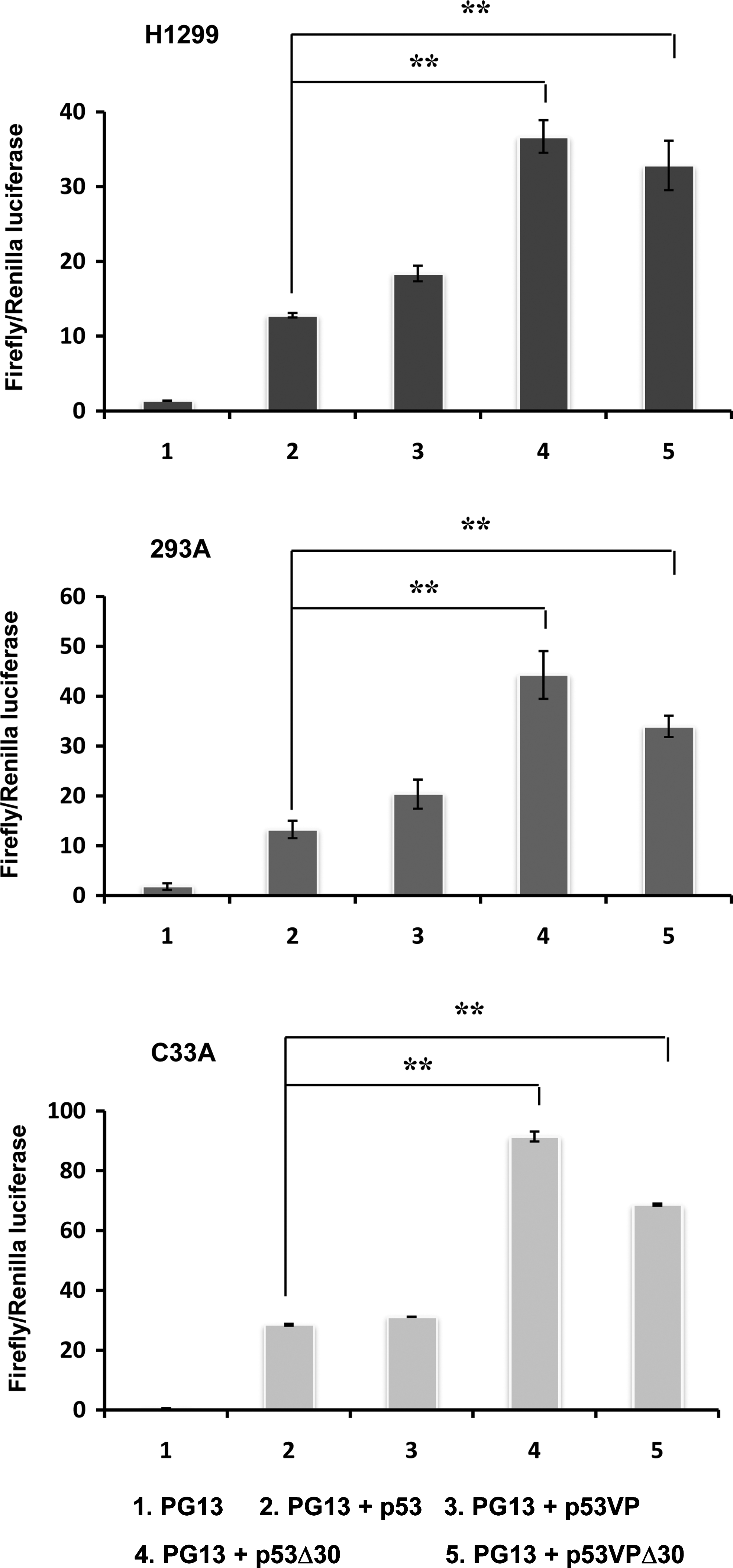
Transcriptional activation by wild-type p53, p53Δ30, p53VP, or p53VPΔ30. H1299, 293, and C33A cells were cotransfected with the luciferase reporter PG13-Luc (PG13) and pCEP4 expressing the p53 variants. Luciferase activity was determined 48 hr after transfection. Data are expressed as the mean (±SEM) firefly-to-Renilla ratio, and are representative of three independent experiments. **p<0.01.
Enhanced oncolytic effect of replication-competent Ad expressing p53VPΔ30
We next evaluated the cytotoxic effect of the p53 variants in human cancer cell lines with different p53 status (wild-type p53, U343 and A549; mutant p53, U251N and U373MG; p53-null, H1299). We used Ad-mΔ19, a replication-competent oncolytic Ad driven by mTERT, for cancer cell-specific expression and amplification of wild-type p53 (Ad-mΔ19/p53) and the p53 variants (Ad-mΔ19/p53Δ30, Ad-mΔ19/p53VP, and Ad-mΔ19/p53VPΔ30; Supplementary Fig. S1 [supplementary data are available online at

Oncolytic effects of Ad-mΔ19 expressing p53 variants. Monolayers of cancer cells were infected with Ad-mΔ19, Ad-mΔ19/p53, Ad-mΔ19/p53Δ30, Ad-mΔ19/p53VP, or Ad-mΔ19/p53VPΔ30 (A549, U251N, and U373MG, 5 MOI; U343 and H1299, 10 MOI). A replication-incompetent Ad, dE1/lacZ, served as a negative control. Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The percentage of viable cells was normalized to that of uninfected cells, and results are expressed as means±SEM (n=3) and are representative of three independent experiments.
Internucleosomal degradation of genomic DNA promoted by p53VPΔ30
To ascertain whether the oncolytic effects of Ad-mΔ19/p53VPΔ30 were due to apoptosis, the internucleosomal degradation of genomic DNA was evaluated by TUNEL assay and expressed as the percentage of TUNEL-positive cells (Fig. 3a and b). Cells treated with CPT (1 μM) to induce apoptosis were used as a positive control. As shown in Fig. 3a, all cancer cell lines infected with the p53VPΔ30-expressing oncolytic Ad exhibited a higher rate of apoptosis than cells infected with Ad-mΔ19, Ad-mΔ19/p53, Ad-mΔ19/p53Δ30, or Ad-mΔ19/p53VP. For example, H1299 cells infected with Ad-mΔ19/p53VPΔ30 had a higher apoptotic fraction (57.7%) than H1299 cells infected with Ad-mΔ19 (2.7%), Ad-mΔ19/p53 (8.9%), Ad-mΔ19/p53Δ30 (29.0%), or Ad-mΔ19/p53VP (40.4%) (p<0.05 compared with Ad-mΔ19/p53, Ad-mΔ19/p53Δ30, or Ad-mΔ19/p53VP; Fig. 3b). The relative effects of Ad-mΔ19/p53VPΔ30 were similar in U343, A549, U251N, and U373MG cells.

Enhanced apoptosis in cancer cells as assessed by the terminal deoxynucleotidyltransferase dUTP nick end-labeling (TUNEL) assay.

The effective promotion of apoptosis by Ad-mΔ19/p53VPΔ30 was further investigated by PI staining in the same cancer cell lines (Fig. 4). FACS analysis showed that the apoptotic effect of Ad-mΔ19/p53VPΔ30 was stronger than that of the other Ad vectors in all cancer cells. In A549 cells, the rate of apoptosis induced by Ad-mΔ19/p53VPΔ30 (60.3% PI-positive cells) was higher than that of Ad-mΔ19 (26.1% PI-positive cells), Ad-mΔ19/p53 (31.3% PI-positive cells), Ad-mΔ19/p53Δ30 (41.3% PI-positive cells), and Ad-mΔ19/p53VP (49.8% PI-positive cells). These results were consistent with TUNEL assay results and indicate that the oncolytic effect of Ad-mΔ19/p53VPΔ30 was due to increased apoptosis.
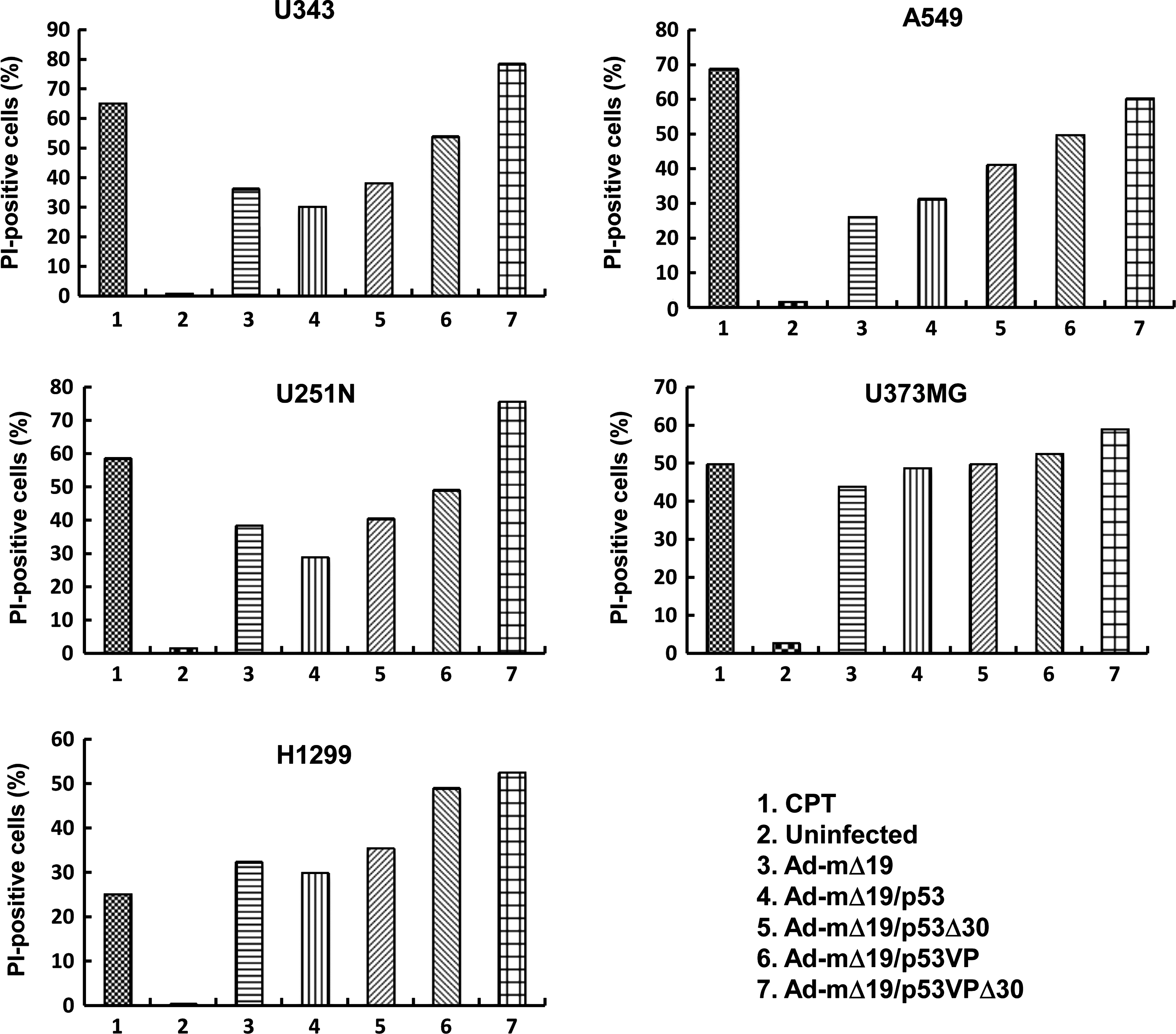
Induction of apoptosis. Cells were infected with Ad-mΔ19, Ad-mΔ19/p53, Ad-mΔ19/p53Δ30, Ad-mΔ19/p53VP, or Ad-mΔ19/p53VPΔ30 (MOI, 5) or treated with camptothecin (CPT, 1 μM). Cells collected 48 hr postinfection were stained with propidium iodide (PI) and analyzed by fluorescence-activated cell sorting. Results are expressed as the percentage of PI-positive cells, and are representative of five independent experiments.
Ultrastructural morphology of cancer cells infected with Ads expressing p53VPΔ30
To confirm the results of the TUNEL assay and FACS analysis, we analyzed apoptosis by TEM in U343 (wild-type p53) and H1299 (p53-null) cells. Thirty-six hours postinfection, Ad-mΔ19/p53VPΔ30-infected cells exhibited nuclear changes (i.e., nuclear membrane blebbing, condensation and segregation of chromatin) as well as cytoplasmic changes (i.e., cytoplasmic vacuolization and mitochondrial swelling) that are consistent with apoptosis (Fig. 5). In contrast, cells infected with Ad-mΔ19/p53Δ30 or Ad-mΔ19/p53VP showed cytoplasmic changes but no detectable nuclear changes. Furthermore, more pronounced morphological changes were observed in Ad-mΔ19/p53VPΔ30-infected cells, and no live cells were detected (Supplementary Fig. S2). Together, these data demonstrate that Ad-mΔ19/p53VPΔ30 induced the strongest apoptotic effects in both p53-positive and p53-negative cancer cells, confirming the results obtained by TUNEL assay and PI–FACS analysis.
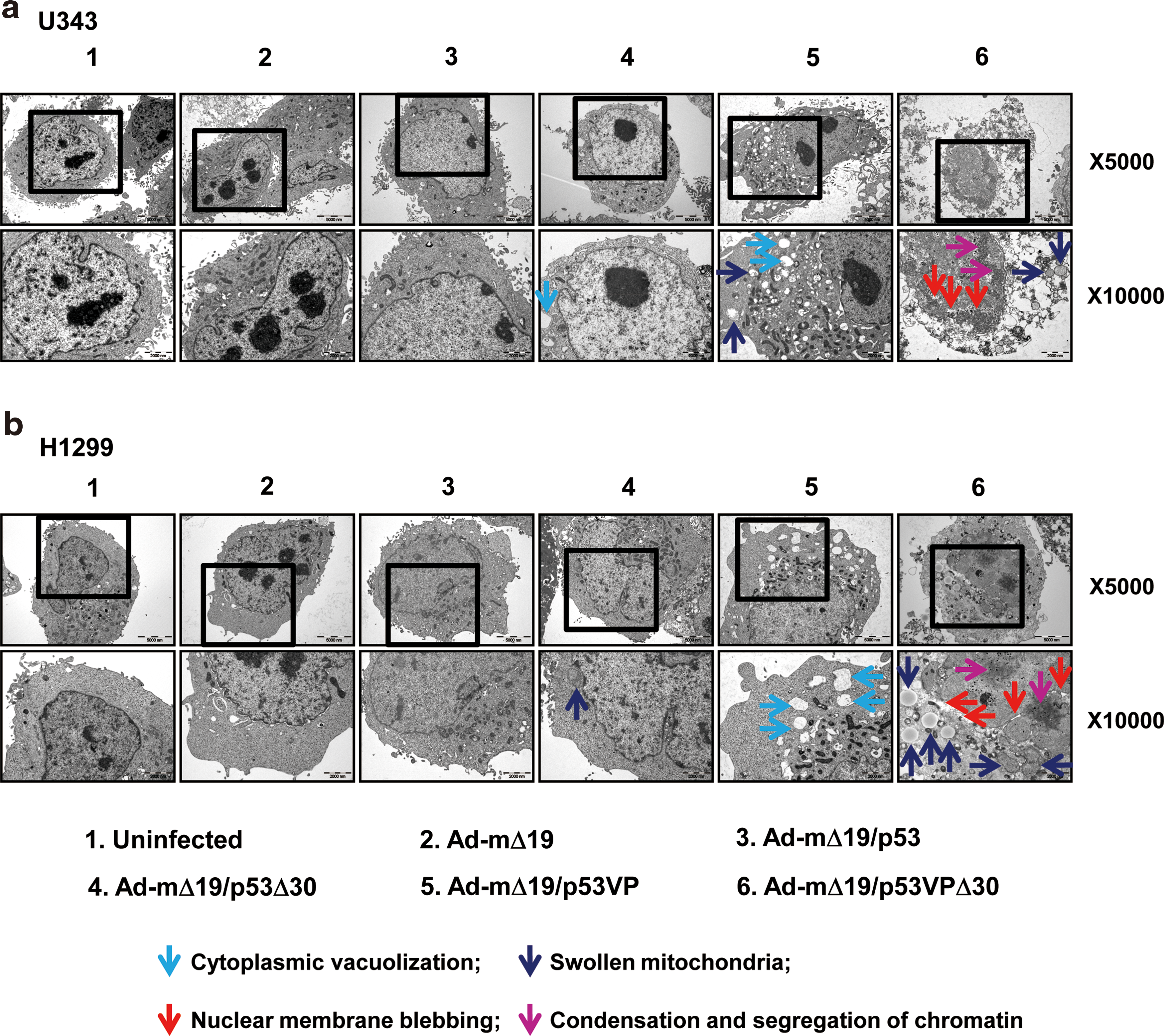
Detection of apoptosis by transmission electron microscopy (TEM).
Enhanced apoptotic signal transduction by Ad-mΔ19/p53VPΔ30 in cancer cells
To analyze the apoptotic signal transduction pathway induced by Ad-mΔ19/p53VPΔ30, the protein levels of p53 and its downstream targets p21, Mdm2, and Bax were determined in wild-type p53-expressing U343, p53-null H1299, high Mdm2-expressing SJSA, and low Mdm2-expressing HT1080 cell lines. In U343 and H1299 cells, protein levels of p53, Mdm2, and Bax were higher in Ad-mΔ19/p53VPΔ30-infected cells than in Ad-mΔ19/p53-infected cells. However, Ad-mΔ19/p53VPΔ30 increased p21 levels only slightly in U343 cells and not at all in the p53-null H1299 cells (Fig. 6a).
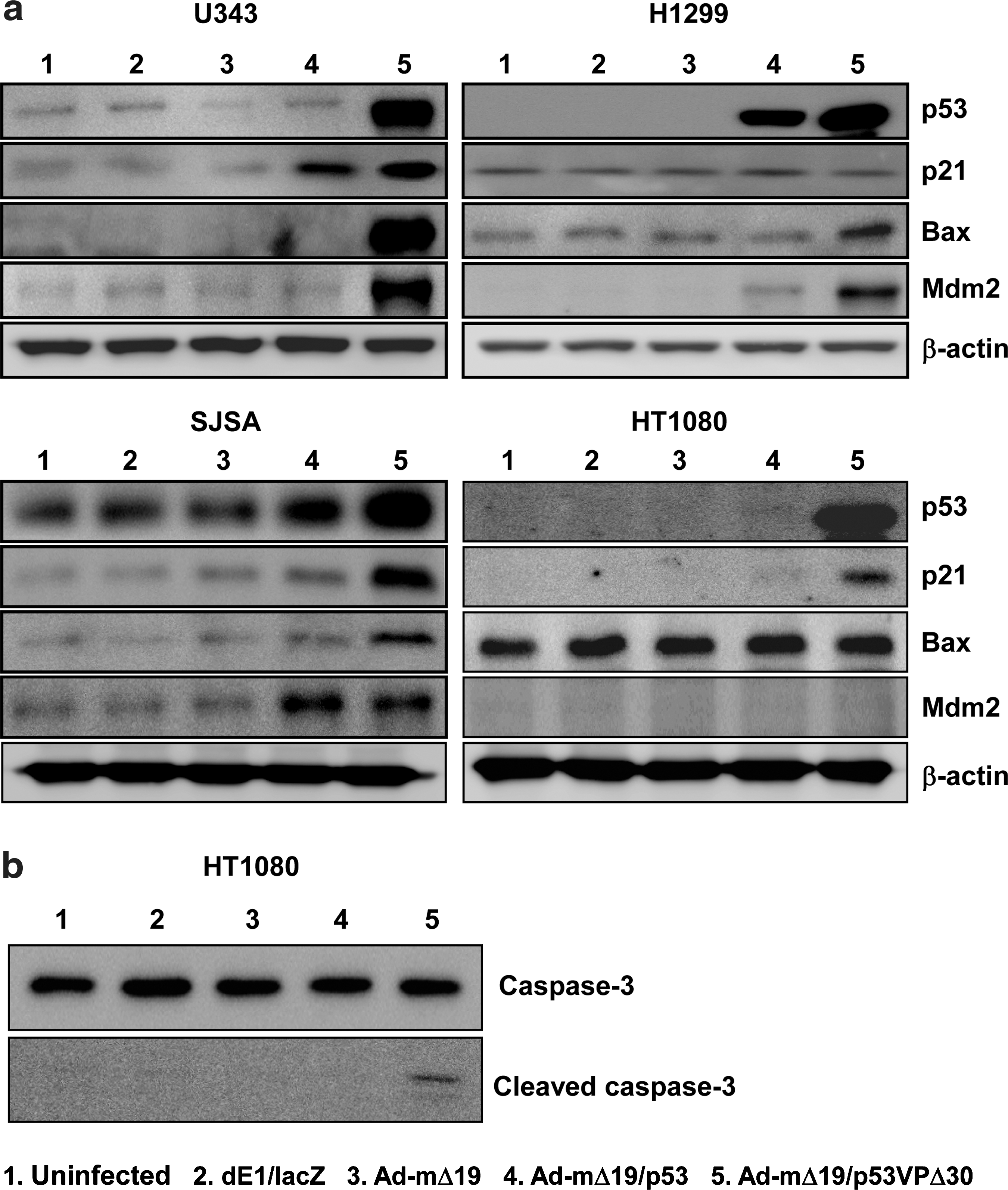
Activation of the apoptotic signal transduction pathway. Cells were infected with dE1/lacZ, Ad-mΔ19, Ad-mΔ19/p53, or Ad-mΔ19/p53VPΔ30 (MOI, 1). Twenty-four hours after infection, cell lysates were analyzed by Western blot with antibodies against p53 and downstream targets p21, Bax, Mdm2, caspase-3, and cleaved caspase-3; β-actin was used as an internal control.
Similarly, the protein level of p53VPΔ30 in Ad-mΔ19/p53VPΔ30-infected SJSA and HT1080 cells was higher than that of wild-type p53 in Ad-mΔ19/p53-infected cells, regardless of Mdm2 expression. Induction of p21 in p53VPΔ30-expressing cells was higher than that of wild-type p53-expressing cells in both cell lines, and Bax expression was increased in SJSA cells infected with Ad-mΔ19/p53VPΔ30. In contrast, Bax levels were not increased in the HT1080 cells infected with Ad-mΔ19/p53VPΔ30, because HT1080 cells undergo apoptosis by activation of caspase-3 in a Bax-independent manner (de Belle et al., 1999) (Fig. 6b).
To further demonstrate the effect of deletion in the N-terminal region of the p53 variant on E1B 55kD-mediated negative regulation, the level of p53 was assessed after infecting p53-null H1299 cells with E1B 55kD-incorporating oncolytic Ads (Ad-mΔ19/p53 and Ad-mΔ19/p53VPΔ30). As shown in Fig. 7a and Supplementary Fig. S3, N-terminal region-deleted p53 variant-expressing oncolytic Ad (Ad-mΔ19/p53VPΔ30) showed a higher expression level of p53. Because p53VPΔ30 was generated by deleting the wild-type p53 C-terminal region (residues 364–393), p53VPΔ30 was easily distinguished from wild-type p53 on the basis of difference in molecular weight. In addition, the E1A expression level in cells infected with oncolytic Ads expressing wild-type p53 or p53 variants was assessed to rule out the possibility of a highly efficient viral infection-mediated increased expression level of p53 in Ad-mΔ19/p53VPΔ30-infected cells. As presented in Supplementary Fig. S4, the expression level of E1A is similar in all cells infected with oncolytic Ads expressing wild-type p53 or p53 variants, verifying that the enhanced expression level of p53 in Ad-mΔ19/p53VPΔ30-infected cells resulted from accumulation of p53VPΔ30. Taken together, these results demonstrate that irrespective of p53 and Mdm2 expression levels, Ad-mΔ19/p53VPΔ30 strongly induces the apoptotic signal transduction pathway through p53VPΔ30 accumulation.
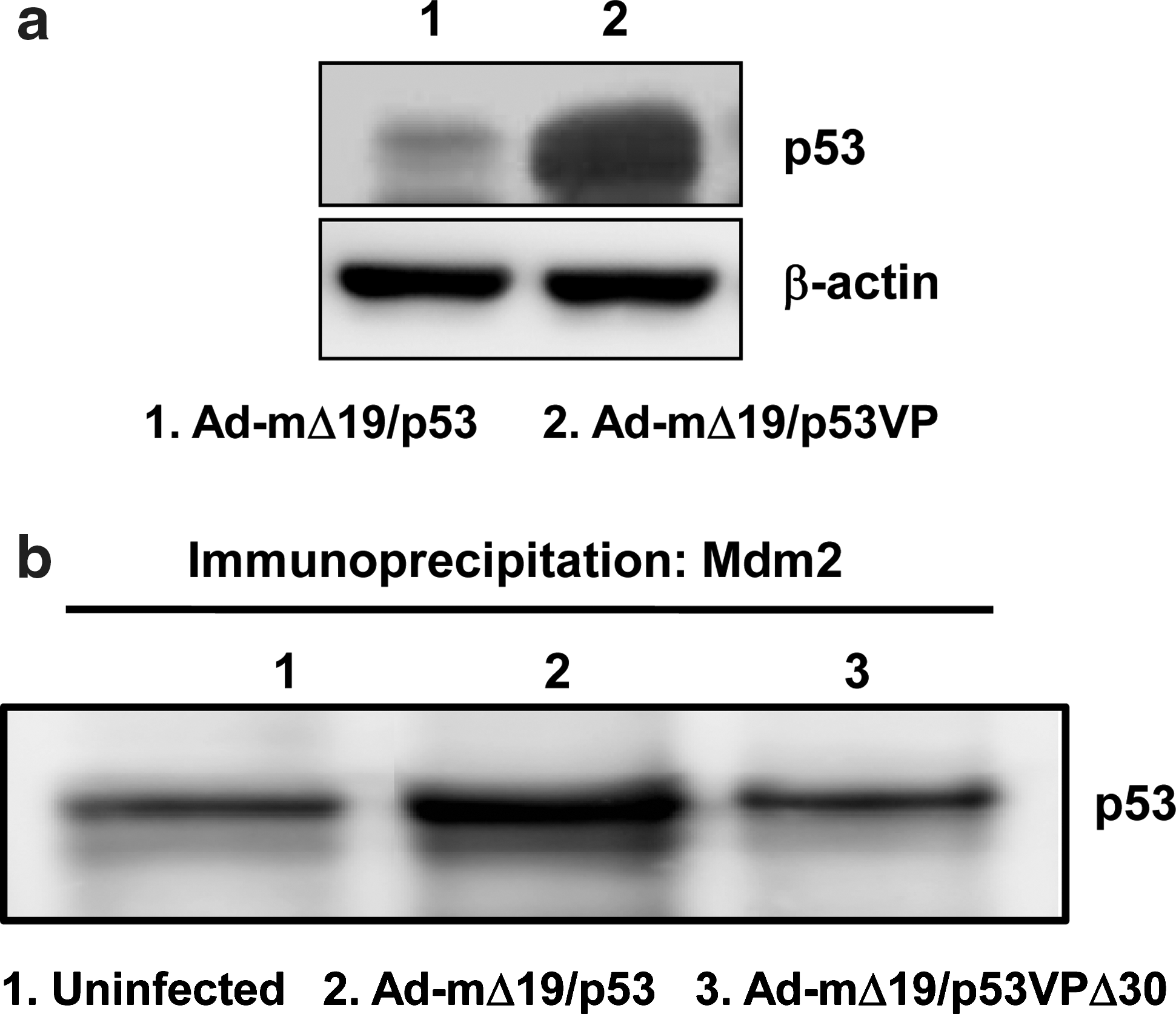
Resistance of oncolytic Ad expressing p53VPΔ30 to E1B 55kD- and Mdm2-mediated negative regulation.
Effect of Ad-mΔ19/p53VPΔ30 on E1B 55kD- and Mdm2-mediated negative regulation
To examine the effect of deletion in the N-terminal domain of the p53 variant on E1B 55kD-mediated negative regulation, the p53 expression level was assessed after infecting u343 cells with E1B 55kD-incorporated oncolytic Ads (Ad-mΔ19/p53 and Ad-mΔ19/p53VP). As seen in Fig. 7a, the protein level of p53VP in Ad-mΔ19/p53VP-infected cells was much higher than that of wild-type p53 in Ad-mΔ19/p53-infected cells, suggesting that N-terminal modification can allow p53 to escape the negative regulation mediated by oncolytic Ad-incorporated E1B 55kD.
We also investigated whether p53VPΔ30 expressed from Ad-mΔ19/p53VPΔ30 can be resistant to Mdm2-mediated degradation. As presented in Fig. 7b, the level of p53VPΔ30 immunoprecipitated by Mdm2 was lower than that of wild-type p53 immunoprecipitated by Mdm2, demonstrating that N- and C-terminal modification of p53 can prevent the interaction with Mdm2, thus endowing resistance to Mdm2-mediated degradation.
Antitumor effects of Ads expressing p53VPΔ30 in U343 human brain cancer xenografts
We next evaluated the ability of Ad-mΔ19/p53VPΔ30 to suppress growth of human glioma xenografts established in nude mice. Tumors were generated by subcutaneous injection of U343 cells, which express wild-type p53 and high levels of Mdm2, into the mouse abdomen. When tumors reached 70–100 mm3, the mice underwent three intratumoral injections of Ad-mΔ19/p53, Ad-mΔ19/p53VPΔ30, or PBS (negative control). PBS-treated tumors grew rapidly, reaching 3704±534 mm3 by day 65 after the first injection, whereas Ad-mΔ19/p53-treated tumors (2589±575 mm3) and Ad-mΔ19/p53VPΔ30-treated tumors (823±228 mm3) were approximately 30% and 78% smaller, respectively (p<0.05 compared with Ad-mΔ19/p53) (Fig. 8a). Treatment with Ad-mΔ19/p53VPΔ30 also increased survival. By day 65, 100% of the mice treated with Ad-mΔ19/p53VPΔ30 were still alive, whereas 0% of the PBS-treated mice and 42.9% of the Ad-mΔ19/p53-treated mice survived (p<0.05 compared with Ad-mΔ19/p53) (Fig. 8b). Throughout the course of the study, no systemic toxicity (e.g., diarrhea, weight loss, and cachexia) was observed. These results demonstrate that Ad-mΔ19/p53VPΔ30 increased survival and tumor suppression in vivo despite high levels of Mdm2.
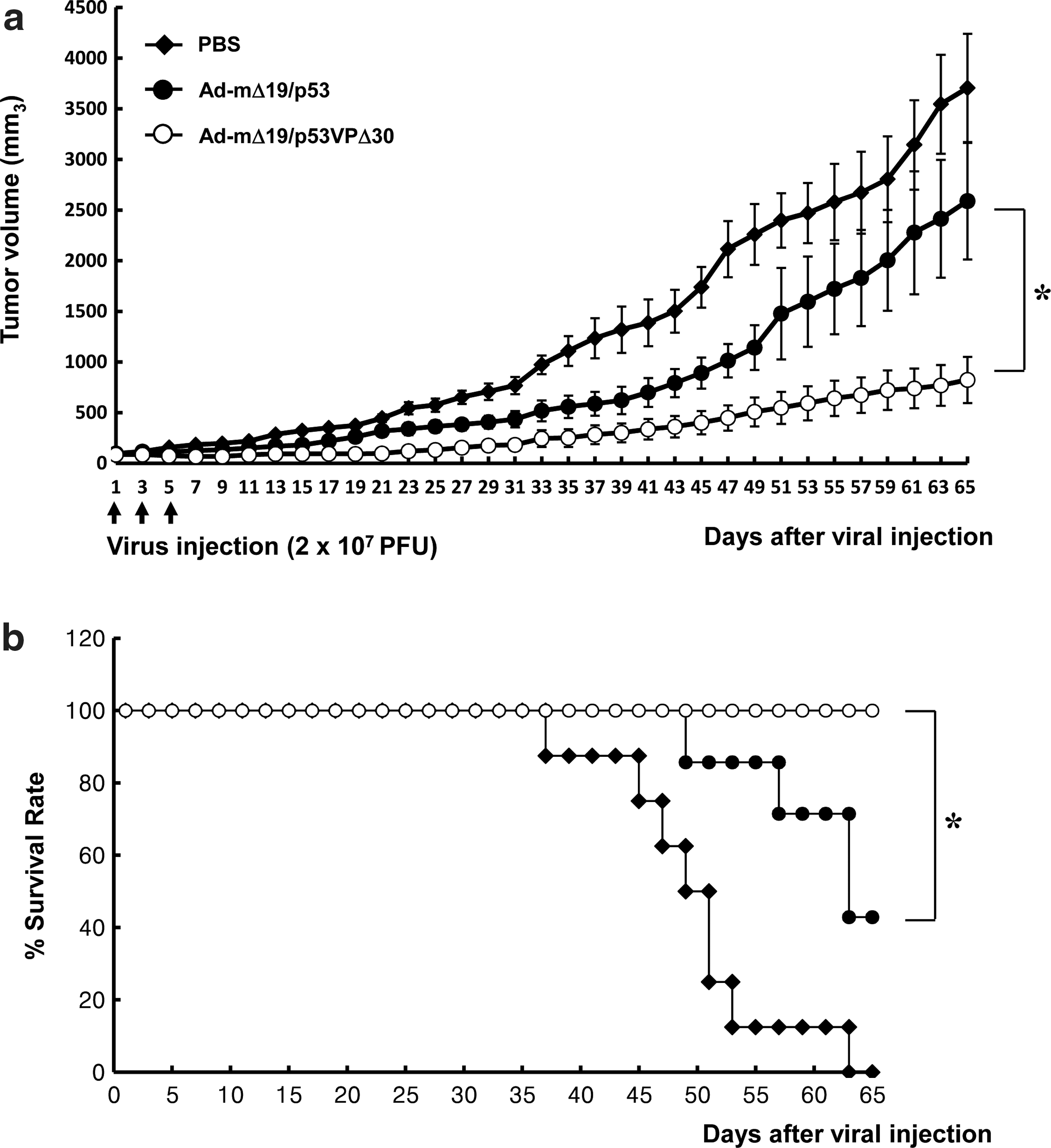
Tumor growth suppression and survival benefit by Ad-mΔ19/p53VPΔ30 in U343 human cancer xenografts established in male athymic nude mice. Subcutaneous tumors derived from U343 cells were treated with Ad-mΔ19/p53 or Ad-mΔ19/p53VPΔ30 (2×107 PFU); negative controls were treated with PBS.
Discussion
The ability of p53 to respond to stress signals is essential to prevent malignant progression and for responsiveness to cancer treatment (Vogelstein et al., 2000). Nearly half of all human tumors express wild-type p53 (Olivier et al., 2002), but these tumors possess alternative mechanisms for disabling p53 function. Overexpression of Mdm2, which degrades p53, is one such mechanism (Chene, 2003); thus, one of the main hurdles of p53-mediated cancer therapy may be to block the action of Mdm2. Indeed, Mdm2 overexpression has been demonstrated in the most human cancers (Momand et al., 1998; Freedman et al., 1999), and these cancers usually retain wild-type p53 (Oliner et al., 1992).
Previous studies have reported that modifying the Mdm2-binding site at the p53 N terminus confers resistance to Mdm2-mediated inactivation, thereby allowing the accumulation of p53 protein (Lin et al., 2000; Tang et al., 2004; Sauthoff et al., 2006). However, the p53 C terminus is also required for Mdm2-targeted degradation (Kubbutat et al., 1998), suggesting that modification of the p53 C terminus is necessary to completely suppress Mdm2-mediated degradation. Moreover, the C terminus of p53 also has a negative regulatory function by binding to the core domain of p53, thereby blocking DNA binding (McKinney and Prives, 2002; Selivanova and Wiman, 2007).
To improve the efficiency of p53-mediated gene therapy against cancer, we therefore explored the therapeutic value of an oncolytic Ad expressing a p53 variant modified at the N-terminal and C-terminal domains (p53VPΔ30). We first assessed the transcriptional activity of three p53 variants containing modifications to the N terminus (p53VP), C terminus (p53Δ30), or both (p53VPΔ30), using a luciferase reporter, and observed that transcriptional activity of the three p53 variants was not lower than that of wild-type p53. This result differed from a previous report that modification of the N-terminal Mdm2-binding region slightly decreased p53 trans-activation activity (Lin et al., 1994; Sauthoff et al., 2006). Our finding indicates that replacing the deleted N-terminal region with the HSV VP16 activation domain prevents loss of function. The VP16 protein is a potent trans-activator of all five HSV-immediate early viral genes, which encode HSV regulatory proteins (Yang et al., 2003).
Moreover, we observed that the two C-terminal domain-deleted variants (p53Δ30 and p53VPΔ30) enhanced the transcriptional activity of p53 three- to four-fold compared with that of wild-type p53. The modification of the Mdm2-binding site at the C terminus of p53 (p53Δ30 and p53VPΔ30) can inhibit Mdm2-mediated p53 inactivation, thus leading to the accumulation of p53 protein (Kubbutat et al., 1998). In addition, the alteration can also enhance transcriptional activity of p53 by escape from the negative regulatory function, that is, the C terminus of p53 binds to the core domain of p53, thereby blocking target DNA binding and its transcriptional activity (McKinney and Prives, 2002; Selivanova and Wiman, 2007). These underlying mechanisms might explain the observed improved transcriptional activity of the C-terminal domain-deleted p53 variants (p53Δ30 and p53VPΔ30) compared with wild-type p53.
For long-term and cancer cell-specific expression of therapeutic genes, we generated Ad-mΔ19 vectors expressing wild-type p53 and the p53 variants. Irrespective of endogenous p53 status, Ad-mΔ19/p53VPΔ30 showed stronger cytotoxicity in cancer cells than Ad-mΔ19/p53, Ad-mΔ19/p53Δ30, or Ad-mΔ19/p53VP (Fig. 2). These results imply that modification of both the N-terminal and C-terminal domains (p53VPΔ30) leads to more effective cancer cell killing than modification of either the N terminus (p53VP) or C terminus (p53Δ30) (Fig. 2). Tumors disrupt the p53 network of signaling pathways through a variety of mechanisms, thereby producing complex heterogeneous phenotypes (Fridman and Lowe, 2003), some of which alter the response to p53-mediated cancer therapy. Thus, it is important to note that oncolysis induced by Ad expressing a p53 variant modified at the N-terminal and C-terminal domains (p53VPΔ30) was not influenced by the endogenous p53 status of cancer cell lines.
To further evaluate Ad-mΔ19/p53VPΔ30-mediated cytotoxicity in cancer cells, apoptosis assays were performed. We found that internucleosomal degradation of genomic DNA was most pronounced in cells infected with Ad-mΔ19/p53VPΔ30, as assessed by TUNEL assay (Fig. 3a and b). This result was supported by PI staining of apoptotic cells (Fig. 4). Visualization of Ad-mΔ19/p53VPΔ30-infected cells by TEM confirmed nuclear changes (nuclear membrane blebbing and chromatin condensation and segregation) and cytoplasmic changes (cytoplasmic vacuolization and mitochondrial swelling) consistent with apoptosis (Fig. 5). Taken together, these findings indicate that Ad-mΔ19/p53VPΔ30 exerts its cytotoxic effects by inducing apoptosis.
To characterize the signal transduction pathway induced by Ad-mΔ19/p53VPΔ30, the protein levels of p53 and downstream targets p21, Mdm2, and Bax were evaluated. Levels of p53 and Mdm2 were higher in cells infected with Ad-mΔ19/p53VPΔ30 than in cells infected with Ad-mΔ19/p53 (Fig. 6a). The transcription factor p53 induces expression of Mdm2, which results in functional inactivation of p53 via protein–protein interactions (Barak et al., 1993; Wu et al., 1993). Thus, increased protein levels of p53 in high Mdm2-expressing cells (U343, H1299, and SJSA) infected with Ad-mΔ19/p53VPΔ30 could be explained by modification of the N-terminal and C-terminal Mdm-2 binding domains of p53, allowing resistance to the negative feedback loop. These results are further supported by the observation that N- and C-terminal modification of p53 (p53VPΔ30) prevents the interaction with Mdm2 (Fig. 7b). Furthermore, the adenoviral E1B 55kD protein of Ad-mΔ19 strongly suppresses p53 activity by binding the same N-terminal binding domain (Grand et al., 1996). Despite Ad E1A-induced p53 stabilization, p53 is rapidly degraded in oncolytic Ad-infected cells in the presence of E1B 55kD (Querido et al., 1997). Thus, resistance to E1B 55kD may contribute to the increased levels of p53VPΔ30 in infected cells (Fig. 7a). Taken together, our results suggest that modification of p53 may prevent negative regulation by Mdm2 and E1B 55kD, thereby allowing accumulation of p53 in Ad-mΔ19/p53VPΔ30-infected cells.
The proapoptotic activity of p53 is mediated by transcription of apoptosis-promoting genes (el-Deiry, 1998). Bax is a p53-regulated proapoptotic Bcl-2 family member (Miyashita and Reed, 1995) that contributes to p53-mediated tumor suppression in vivo (Yin et al., 1997). In the present study, we showed that Bax expression was higher in H1299, U343, and SJSA cancer cells infected with Ad-mΔ19/p53VPΔ30 than in the same cells infected with Ad-mΔ19/p53 (Fig. 6a), demonstrating the strong activity of the accumulated p53VPΔ30. The upregulation of Bax is associated with its translocation from the cytosol to the mitochondria, where it induces mitochondrial permeability transition (MPT), leading to cytochrome c release and eventually induction of apoptosis. Bax-driven MPT was supported by TEM images showing numerous swollen mitochondria in the p53VPΔ30-overexpressing cells (Fig. 5). In HT1080 cells, Bax protein levels induced by wild-type p53 and p53VPΔ30 were similar; however, Ad-mΔ19/p53VPΔ30 increased cleavage of caspase-3 (Fig. 6b), suggesting that in HT1080 cells, Ad-mΔ19/p53VPΔ30 induces apoptosis by activating caspase-3 in a Bax-independent manner. This result is consistent with an earlier report that p53-mediated apoptosis in HT1080 cell lines is associated with caspase-3 activation without a concurrent increase in Bax expression (de Belle et al., 1999). Taken together, these results suggest that irrespective of endogenous p53 and Mdm2 expression, Ad-mΔ19/p53VPΔ30 strongly activated the apoptotic signal transduction pathway in cancer cells.
Excessive Mdm2 expression may result in exaggerated repression of p53, abrogating its protective effects. At least 5–10% of all human tumors exhibit Mdm2 overexpression due to gene amplification or transcriptional and posttranscriptional mechanisms (Michael and Oren, 2003). To closely mimic the environment of these tumors, we used a U343 human glioma xenograft model (wild-type p53 and a high level of Mdm2) to assess the potential therapeutic benefit of an oncolytic Ad expressing p53VPΔ30. Our result showed that Ad-mΔ19/p53VPΔ30 more strongly suppressed tumor growth and increased survival rates compared with Ad-mΔ19/p53 (Fig. 8), suggesting an enhanced antitumor action in vivo despite the high level of Mdm2. These in vivo data are in good agreement with in vitro data (Figs. 2 and 3b), demonstrating that oncolytic Ad expressing p53VPΔ30 elicits a more potent antitumor effect than wild-type p53-expressing oncolytic Ad.
To our knowledge, this is the first study reporting a p53 variant modified at the N-terminal and C-terminal domains that escapes negative regulation by endogenous Mdm2, the Ad protein E1B 55kD, and the p53 C terminus itself, without a decrease of trans-activation activity. We also show that, regardless of endogenous p53 and Mdm2 levels, Ad-mΔ19/p53VPΔ30 induces apoptosis in cancer cell lines through accumulation of p53VPΔ30. Moreover, we demonstrated effective in vivo antitumor effects in Mdm2-overexpressing U343 tumors. Thus Ad-mΔ19/p53VPΔ30 shows potential as an improved p53-mediated gene therapy for cancers.
Footnotes
Acknowledgments
This work was supported by grants from the Ministry of Knowledge Economy (10030051, C.-O.Y.); the National Research Foundation of Korea (R15-2004-024-02001-0, 2010-0029220, 2009K001644, C.-O.Y.); Korea Food and Drug Administration (KFDA-10172-332, C.-O.Y.); and a faculty research grant (6-2010-0052, C.-O.Y.) from the Yonsei University College of Medicine, Seoul, Korea. Taeyoung Koo, Minjung Kim, Jung-Sun Lee, and Eonju Oh are graduate students sponsored by the Brain Korea 21 Project for Medical Sciences, and Il-Kyu Choi is a graduate student sponsored by the National Research Foundation through the National Core Research Center for Nanomedical Technology, Yonsei University, Seoul, Korea.
Author Disclosure Statement
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.