
Abstract
Oncolytic viruses represent a novel therapeutic approach for aggressive tumors, such as glioblastoma multiforme, which are resistant to available treatments. Autophagy has been observed in cells infected with oncolytic viruses; however, its role in cell death/survival is unclear. To elucidate the potential therapeutic use of autophagy modulators in association with viral therapy, we analyzed autophagy induction in human glioma cell lines U373MG and U87MG infected with the oncolytic adenovirus dl922–947. dl922–947 infection triggered an autophagic cellular response, as shown by the development of acidic vesicular organelles, LC3-I→LC3-II conversion, and reduction of p62 levels. However, on infection, the Akt/mTOR/p70s6k pathway, which negatively regulates autophagy, was activated, whereas the ERK1/2 pathway, a positive regulator of autophagy, was inhibited. Accordingly, MEK inhibition by PD98059 sensitized glioma cells to dl922–947 effects, whereas autophagy induction by rapamycin protected cells from dl922–947-induced death. Treatment with two inhibitors of autophagy, chloroquine and 3-methyladenine, increased the cytotoxic effects of dl922–947 in vitro. In vivo, the growth of U87MG-induced xenografts was further reduced by adding chloroquine to the dl922–947 treatment. In conclusion, autophagy acts as a survival response in glioma cells infected with dl922–947, thus suggesting autophagy inhibitors as adjuvant/neoadjuvant drugs in oncolytic virus-based treatments.
Introduction
Conditionally replicating oncolytic viruses (OVs) represent a promising approach for the treatment of cancer, and the feasibility and safety of this therapeutic strategy in glioblastoma patients have been demonstrated in previous studies (Haseley et al., 2009). dl922–947 is a replication-selective oncolytic adenoviral mutant, harboring a 24-bp deletion in E1A conserved region-2 (CR2), that is responsible for binding and inactivation of the retinoblastoma protein pRb (Heise et al., 2000), which regulates the G1-to-S phase transition. Thus, dl922–947 virus is unable to induce G1-to-S phase transition in normal cells, but can efficiently replicate in cells with an abnormal G1–S checkpoint. Previous studies have confirmed the efficacy of E1A CR2-deleted adenoviruses, such as Δ24 (Fueyo et al., 2000; Jiang et al., 2007), for glioma cells, and we have demonstrated that dl922–947 exerts antineoplastic activity against the glioma cell lines U373MG and U343MG (Botta et al., 2010). A phase I clinical trial for the use of an E1A CR2-deleted mutant (Ad5-Δ24RGD) in recurrent malignant gliomas has been completed (
Although the efficacy and safety of OVs have been clearly demonstrated in preclinical and clinical studies, most of the molecular and/or biochemical pathways activated in cancer cells, including the cell death mechanisms, are not fully understood. A better understanding of such mechanisms would be advantageous for future selection of novel drugs or treatments to further enhance the efficacy of oncolytic viruses. In various studies it has been shown that OVs activate the autophagic machinery in some cancer cells (Ito et al., 2006; Jiang et al., 2007; Baird et al., 2008; Tyler et al., 2009). Autophagy is a catabolic process whereby cytoplasm and cellular organelles are degraded through lysosomal machinery, allowing amino acid and energy recycling (Mizushima, 2007). It is well known that autophagy provides an alternative energy source, thus representing a temporary survival mechanism under stressful conditions. However, it has also been reported that autophagy plays an active role in cell death (Baehrecke, 2005; Loos and Engelbrecht, 2009).
Here we show that, on infection with the oncolytic virus dl922–947, formation of acidic vesicular organelles and the conversion of the cytosolic form of microtubule-associated protein-1 light chain-3 to the autophagosomal membrane-associated form (LC3-I→LC3-II) occur, indicative of a cellular autophagic response to viral infection.
Regulation of autophagy requires multiple signaling pathways (Meijer and Codogno, 2004). Akt/mTOR/p70s6k is the main pathway involved in the negative regulation of autophagy (Blommaart et al., 1995; Yang et al., 2005). Class I phosphatidylinositol-3-phosphate kinase (PI3K) is activated by ligand binding to growth factor receptors. PI3K activates the downstream target Akt, leading to activation of mammalian target of rapamycin (mTOR) (Díaz-Troya et al., 2008). TOR is a serine/threonine kinase that negatively affects the autophagic process, which is achieved by controlling translation and transcription or by either directly or indirectly affecting the Atg (autophagy-related genes) proteins (Díaz-Troya et al., 2008). p70 S6 kinase (p70s6k) is considered a candidate among the substrates of mTOR and in the control of autophagy downstream of mTOR (Blommaart et al., 1995). In the presence of amino acids, mTOR promotes phosphorylation of S6 through activation of p70s6k, thus facilitating the translation initiation of mRNAs (Díaz-Troya et al., 2008). On the other hand, the extracellular-signal regulated kinase-1/2 (ERK1/2) pathway has been described as a positive regulator pathway of autophagy (Pattingre et al., 2003).
Surprisingly, we found that, despite the autophagy activation in dl922–947-infected cells, viral infection activates the main negative regulator of autophagy—the Akt/mTOR/p70s6k pathway—and inhibits the positive regulator, the ERK1/2 pathway. In line with this, viral cytotoxicity is decreased by rapamycin (an mTOR inhibitor) and increased by ERK1/2 inhibitor PD98059, as well as by specific inhibitors of autophagy (3-methyladenine and chloroquine) or genetic tools such as Atg5 small hairpin RNA (shRNA). Although these data seem to contrast with previous results suggesting autophagy as a cell death mechanism induced by oncolytic viruses in glioma cells (Ito et al., 2006), our results led us to speculate that autophagy might play a survival/defensive role in glioma cells infected with dl922–947. Consistently, we demonstrated that chloroquine treatment of athymic mice bearing glioma tumor xenografts enhances the effects of dl922–947 in reducing tumor growth.
Materials and Methods
Cell lines and reagents
Human glioma cell lines U373MG and U87MG were purchased from the American Type Culture Collection (Manassas, VA) and grown as described (Botta et al., 2010).
Acridine orange, 3-methyladenine (3-MA), PD98059, and rapamycin were purchased from Sigma-Aldrich (St. Louis, MO). Chloroquine (CQ) was purchased from Fluka (Buchs, Switzerland).
Preparation of adenoviruses, infection, and viability assay
dl922–947 is a second-generation adenoviral mutant that has a 24-bp deletion in E1A conserved region-2 (CR2). AdGFP is a nonreplicating E1A-deleted adenovirus encoding green fluorescent protein. dl312 is a nonreplicating adenovirus (ΔE1A, ΔE3B) (Cheong et al., 2008). Viral stocks were expanded, purified, and stored as previously reported (Botta et al., 2010), and a plaque-forming units (PFU) assay was performed on HEK-293 cells to determine viral titer.
To evaluate the dl922–947 cytotoxic effect, 1×103 cells were seeded in 96-well plates. After 24 hr, the cells were treated with 3-MA (1 mM) or CQ (10 μM) for 2 hr and then infected. After 7 days, the cells were fixed with 10% trichloroacetic acid (TCA), stained with 0.4% sulforhodamine B in 1% acetic acid, and solubilized as previously reported (Libertini et al., 2011). Cell death rates, expressed as a percentage of treated cells, were calculated. Dose–response curves were generated to calculate the concentration of each agent required to kill 50% of cells (median lethal dose, LD50); untreated cells or cells treated with single agents were used as a control.
Quantification of acidic vesicular organelles with acridine orange
The development of acidic vesicular organelles is a particular feature of autophagy. The cytoplasm and nucleoli of acridine orange-stained cells fluoresce bright green and dim red, respectively, whereas acidic compartments fluoresce bright red (Paglin et al., 2001). To assess autophagy in U373MG and U87MG cells, the acidic compartment was quantified by supravital acridine orange staining. Treated cells were trypsinized and stained with acridine orange (1.0 μg/ml) for 15 min at 37°C. Data were collected on a CyAn flow cytometer and analyzed using Summit software version 4.3 (both from Dako Cytomation, now Beckman Coulter, Miami, FL).
Generation of stable clones
To investigate autophagy induction, overexpression of GFP-tagged LC3 was induced in U373MG cells (Kabeya et al., 2000). The LC3 expression vector (pCAG-LC3) was kindly provided by N. Mizushima and T. Yoshimori (Osaka University, Suita, Japan). To obtain pGFP-LC3, LC3 cDNA was inserted into the BglII and EcoRI sites of pEGFP-C1, a GFP fusion protein expression vector (Clontech, Mountain View, CA) (Kabeya et al., 2000). Stable expression of GFP-LC3 cDNA in U373MG cells (U373 GFP-LC3) was accomplished by the Lipofectamine (Invitrogen, Carlsbad, CA) method, in accordance with the manufacturer's instructions. Cells were cultured in 60-mm dishes (Corning, Corning, NY) and incubated in serum-free Dulbecco's modified Eagle's medium (DMEM) supplemented with 5 μg of cDNA and 15 μl of Lipofectamine reagent. After 5 hr, DMEM supplemented with 20% fetal bovine serum was added and 1 day later was replaced with DMEM–10% serum. At 48 hr after the medium change, cells were selected in medium containing GIBCO G418 (400 mg/ml; Invitrogen) and G418-resistant clones were obtained. Expression of GFP-LC3 in the selected clones was evaluated by Western blot analysis using anti-GFP antibody.
To stably inhibit the expression of the Atg5 gene, APG5 shRNA (sc-41445-sh) and control shRNA (sc-108060) plasmids (Santa Cruz Biotechnology, Santa Cruz, CA) were transfected/selected in U373MG and U87MG cell lines as described previously.
Analysis of LC3 localization by immunofluorescence staining
U373 GFP-LC3 cells were seeded in 24-well dishes. After treatment, cells were fixed with 4% paraformaldehyde for 15 min at room temperature, washed three times with phosphate-buffered saline (PBS), permeabilized for 10 min with PBS containing 0.5% Triton X-100, and then washed three times with PBS. The cells were blocked with bovine serum albumin (BSA) blotting buffer (1% BSA in PBS) for 30 min and then incubated with BSA blotting buffer plus primary antibody overnight at 4°C (mouse monoclonal anti-hexon, diluted 1:50; US Biological, Swampscott, MA). After washing three times with PBS, cells were incubated with rhodamine-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch, West Grove, PA). Images were acquired with an LSM510 inverted confocal microscope (Zeiss, Oberkochen, Germany), using a ×40 oil objective and processed with LSM software (Zeiss).
Western blot analysis and antibodies
Protein extraction and tumor tissue homogenization were performed as previously described (Botta et al., 2010; Oriente et al., 2011).
Total homogenates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) under reducing conditions. Membranes were incubated overnight with the following primary antibodies: LC3-I/II (ab51520, diluted 1:3000) and caspase-3 (ab13585, diluted 1:500) from Abcam (Cambridge, UK); p62 (#5114, diluted 1:1000), p-ERK1/2 (#9101S, diluted 1:1000), p-Akt (#9271, diluted 1:1000), and p-p70s6k (#9205S, diluted 1:500), from Cell Signaling (Danvers, MA); Atg5 (sc-8666, diluted 1:500), ERK1/2 (sc-93-G, diluted 1:1000), and β-actin (sc-10731, diluted 1:2000) from Santa Cruz Biotechnology; and Akt (#28745, diluted 1:1000) from Upstate Biotechnology (Billerica, MA).
Quantitative real-time PCR of adenovirus
Twenty-four, 48, and 72 hr after infection, viral genomes were collected from the cell medium, using a QIAamp DNA mini kit (Qiagen, Valencia, CA), and then quantified by real-time PCR, using the following primers: 5′-GCC ACC GAG ACG TAC TTC AGC CTG-3′ (upstream primer) and 5′-TTG TAC GAG TAC GCG GTA TCC T-3′ (downstream primer) for the amplification of a 143-bp sequence of the viral hexon gene. Serial dilutions of dl922–947 were used to construct a standard curve for quantification.
In vivo antitumor activity and statistical analysis
CD-I athymic mice were obtained from Charles River (Wilmington, MA), and all experiments were carried out with 6-week-old females. Twenty days postinjection with U87MG cells (5×106), 80 mice with similar tumor size were randomized into four groups: untreated, treated with CQ, treated with dl922–947, or treated with both. CQ (45 mg/kg) was administered intraperitoneally every third day. A viral dose (2×106 PFU) was administered three times a week by intratumoral injection to avoid any first-pass effect (Libertini et al., 2007, 2008, 2011). Saline solution was administered to the control groups. Tumor diameters were measured with calipers and tumor volume (V) was calculated by the formula for a rotational ellipsoid: V=A×B 2/2 (A, axial diameter; B, rotational diameter). The experiment was terminated when tumors reached 1 cm3 in volume and/or symptomatic tumor ulceration occurred. Statistical analysis were done by ANOVA (analysis of variance) and the Bonferroni post hoc test, using commercial software (Prism 4; GraphPad Software, San Diego, CA). Differences in the rate of tumor growth in mice were assessed for each time point of the observation period.
Results
Induction of autophagy in malignant glioma cells by dl922–947
First, we evaluated the cytotoxic effects of dl922–947 in U87MG and U373MG glioma cells, confirming its efficacy. The U373MG cell line is more sensitive to dl922–947, with an LD50 of 0.008 PFU/cell, compared with the U87MG LD50 at 0.09 PFU/cell (Fig. 1A).
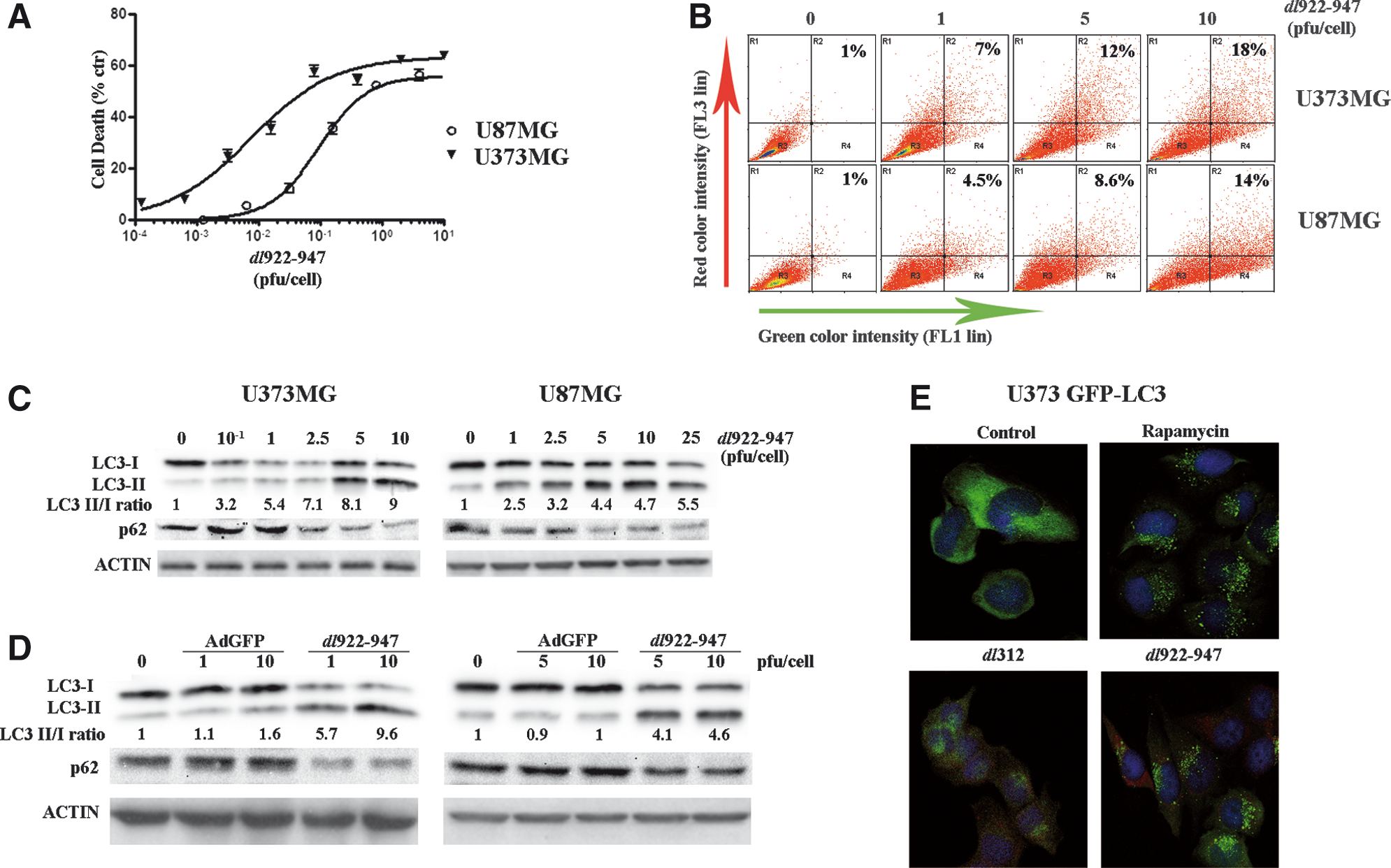
dl922–947 induces cell death and autophagy in malignant glioma cells.
Next, we evaluated the activation of autophagy in glioma cells infected with dl922–947. To this end, we analyzed the formation of acidic vesicular organelles (AVOs), a particular feature of autophagy, after infection with dl922–947. Quantification of AVOs in U373MG and U87MG cells was performed by acridine orange supravital cell staining. U373MG and U87MG cells were infected at 1, 5, and 10 multiplicities of infection (MOIs) with dl922–947 and acridine orange vital staining was performed 72 hr postinfection. FACS analysis showed an increase in bright red fluorescence in infected cells, in a MOI-dependent manner, up to 18% (U373MG) or up to 14% (U87MG). Untreated cells exhibited mainly green fluorescence (Fig. 1B) as the cells were infected with the nonreplicating control adenovirus, dl312 (data not shown). This indicates an accumulation of AVOs in glioma cells infected with dl922–947.
In amino acid starvation-induced autophagy, the microtubule-associated protein-1 light chain-3, LC3-I, is converted to LC3-II through some ubiquitination-like reactions. LC3-I is a cytosolic protein, whereas LC3-II is a protein membrane tightly attached to preautophagosomal (PAS) and autophagosomal membrane structures. The amount of LC3-II reflects the abundance of autophagosomes and variations in the ratio of LC3-II to LC3-I are indicative of autophagy induction (Kabeya et al., 2000). LC3-I→LC3-II conversion was analyzed by Western blotting 72 hr after infection of U373MG and U87MG cells with dl922–947. An increase in LC3-II levels was observed in infected U373MG and U87MG cells, in an MOI-dependent manner (Fig. 1C).
On autophagy activation, LC3-II is recruited to autophagosome membranes; therefore we decided to monitor LC3 localization during dl922–947 infection. U373MG clones expressing LC3 fused to green fluorescent protein were generated (U373 GFP-LC3) by stable transfection and GFP-LC3 tracked by immunofluorescence.
In untreated U373 GFP-LC3 cells or in cells infected with the nonreplicating adenoviral mutant dl312, a diffuse and mostly cytosolic distribution of fluorescence was observed (Fig. 1E). A less diffuse and more punctate pattern was observed 72 hr after infection with 0.1 PFU of dl922–947, indicating that LC3-II is recruited to the autophagosomal membranes; cells were costained with anti-hexon antibody as a control for viral infection. As a positive control, U373 GFP-LC3 cells were treated with the autophagy inducer rapamycin, showing the typical LC3-II localization pattern.
The accumulation of LC3-II could be due to autophagy flux or to interference with autophagosomal/lysosomal function. To discriminate between the two options, we evaluated the levels of the specific autophagy substrate p62/SQSTM1. During autophagy, p62 levels are reduced whereas, in the absence of autophagy, p62 levels are unmodified (Komatsu and Ichimura, 2010). As shown in Fig 1C, a clear decrease in p62 levels was observed, demonstrating that in dl922–947-infected cells the autophagic flux is enhanced.
Glioma cells were infected with a nonreplicating adenovirus expressing green fluorescence protein (AdGFP). AdGFP infection did not induce changes in LC3-II/LC3-I ratio or in p62 levels (Fig. 1D). This observation confirms that autophagy is a specific a feature occurring in infected cells in response to dl922–947 replication.
Taken together, these data demonstrate that glioma cells infected with the dl922–947 virus activate autophagic processes.
dl922–947 infection modulates autophagic signaling pathways and its effects are modified by specific pharmacological inhibitors
We evaluated the effects of dl922–947 infection on the Akt/mTOR/p70s6k and ERK1/2 pathways, which are, respectively, negative and positive regulatory pathways of autophagy.
U373MG and U87MG cells were infected with dl922–947 in a dose–response experiment. Phosphorylation of p70s6k and Akt increased in a dose-dependent manner (Fig. 2A) in both cell lines, indicating activation of the negative regulatory pathway. Conversely, ERK1/2 phosphorylation was reduced on infection with dl922–947 in a dose-dependent manner (Fig. 2A), indicating inhibition of the positive regulatory pathway. AdGFP infection did not induce any of these effects (Fig. 2B). To better evaluate the role of ERK1/2 phosphorylation, a time-course experiment was performed, confirming a dose- and time-dependent reduction of ERK1/2 phosphorylation (Fig. 2C).

Autophagy signaling pathways are modulated in response to dl922–947 infection in glioma cells.
These data suggested to us that dl922–947 acts on autophagic pathways with compensatory mechanisms to counterbalance the cellular response. To clarify the role of the ERK1/2 pathway on the autophagic cellular response in infection, glioma cells were infected with dl922–947 in the presence of MEK-1 (MAPK [mitogen-activated protein kinase]/ERK kinase-1) inhibitor PD98059. An inhibitory dose of 25 μM was used for all the experiments. PD98059 enhanced dl922–947-induced cytotoxicity, leading to a significant reduction of LD50 values in both cell lines: from 0.008 to 0.001 PFU/cell in U373MG cells, and from 0.14 to 0.05 PFU/cell in U87MG cells (Fig. 3A). This observation confirms that blockage of the autophagic positive regulatory pathway favors viral cell-killing activity. Moreover, the combined treatment led to a further reduction of ERK-1/2 phosphorylation (Fig. 3B), along with a reduction on LC3-I→LC3-II conversion; conversely, in uninfected cells, the drug did not modify the LC3-II/LC3-I ratio (Fig. 3B).
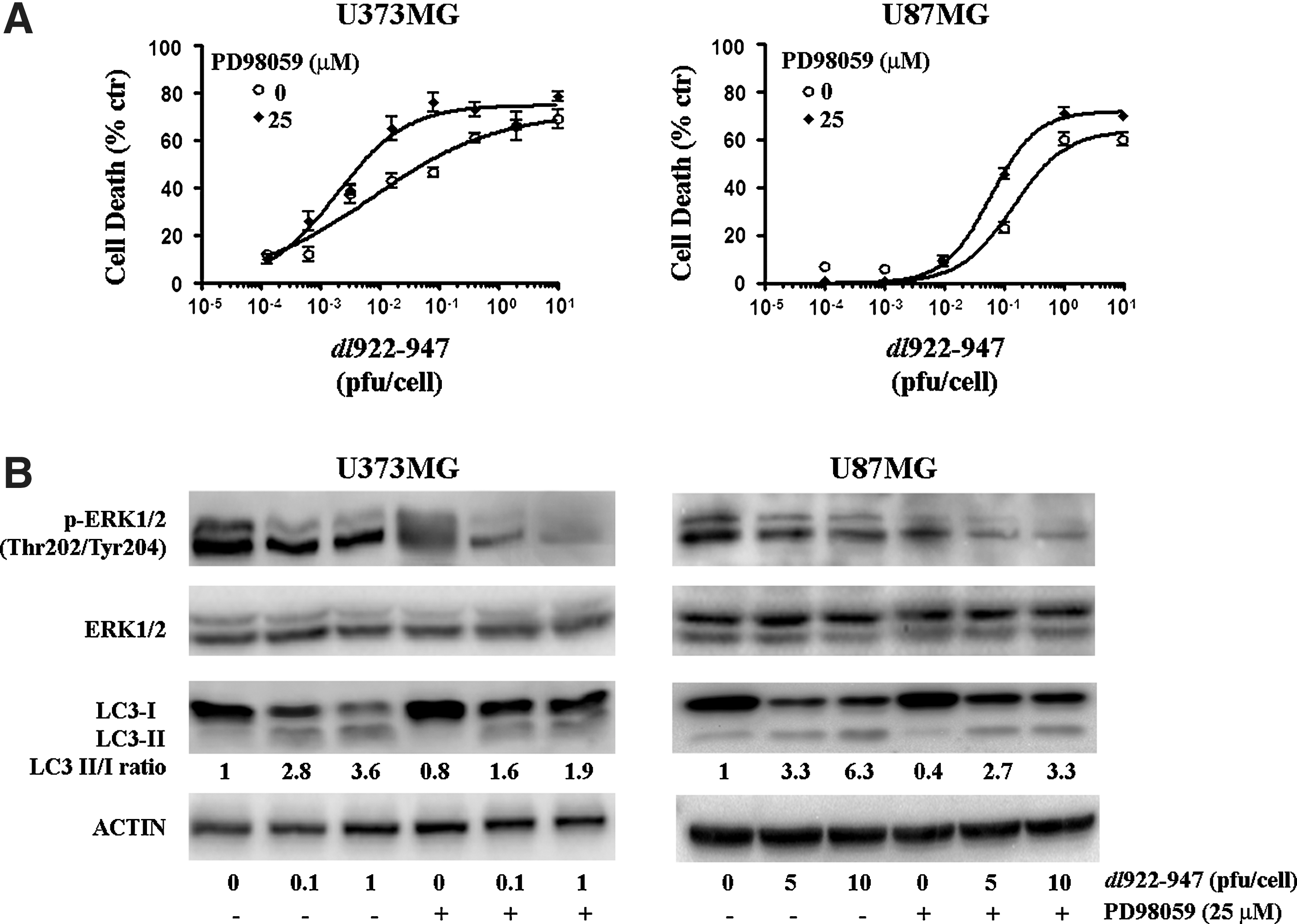
Inhibition of the ERK1/2 pathway inhibits autophagy and enhances dl922–947-induced cytotoxicity.
To investigate the role of the Akt/mTOR/p70s6k pathway in dl922–947 infection, glioma cells were treated with the mTOR inhibitor rapamycin, a well-known autophagy inducer (Meijer and Codogno, 2004), and then infected with dl922–947. Treatment with rapamycin led to a reversion of p70s6k phosphorylation induced by the virus, paralleled by an increase in the LC3-II/LC3-I ratio (Fig. 4A). In both cell lines, rapamycin treatment led to a significant reduction of viral cytotoxicity, with an increase in LD50, 42-fold for U373MG and 23-fold for U87MG (Fig. 4B). These results demonstrate that autophagy acts as a survival mechanism in glioma cells infected with dl922–947 and that the attenuation of autophagic response could increase viral cytotoxicity.
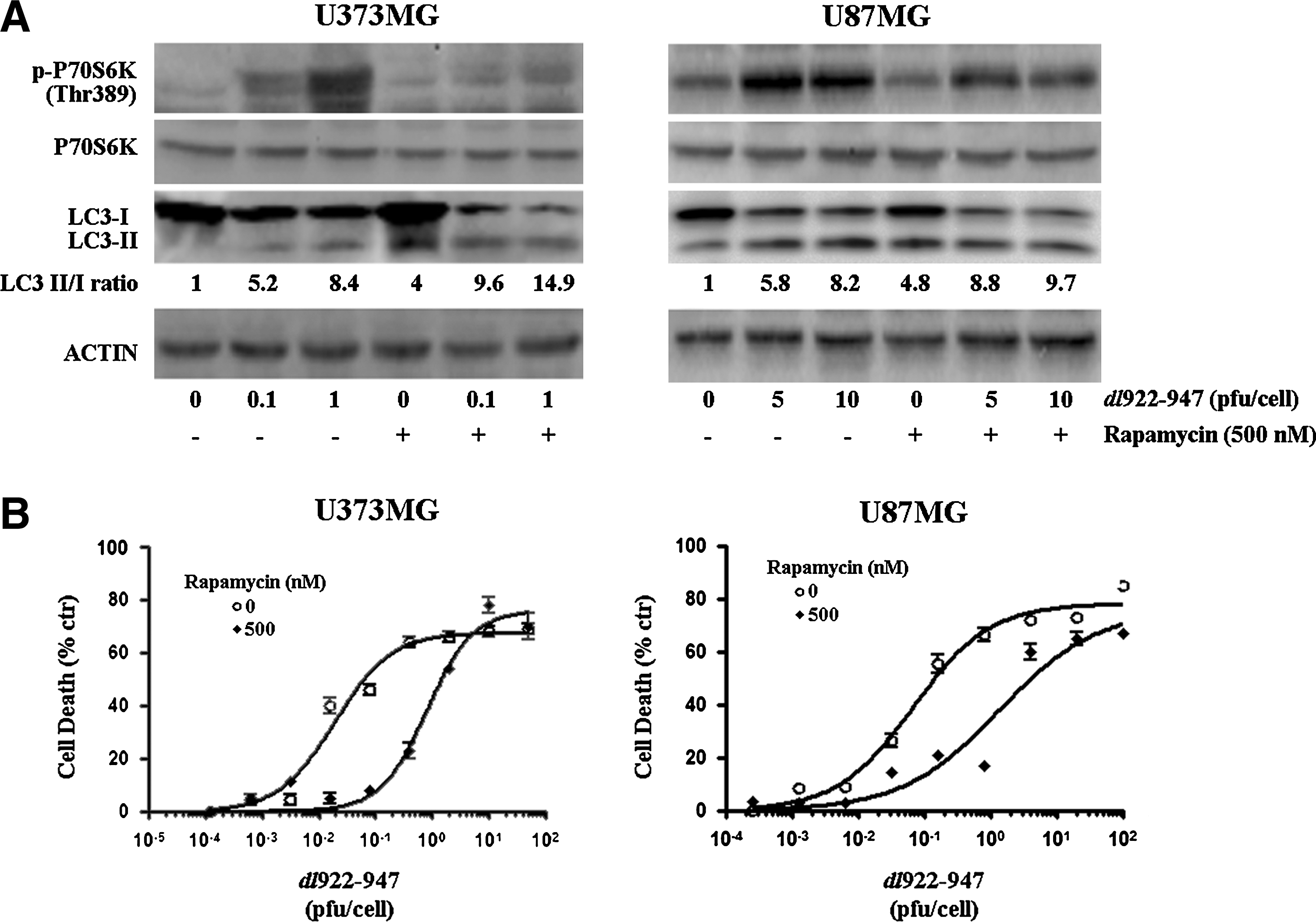
Inhibition of the mammalian target of rapamycin (mTOR) pathway induces autophagy and decreases dl922–947-induced cytotoxicity.
Inhibition of autophagy increases dl922–947 cytotoxicity in glioma cells
Next, we evaluated the effect of pharmacological inhibition of autophagy on dl922–947 infection. To this end, we used two well-known inhibitors of autophagy: 3-methyladenine (3-MA) and hydroxychloroquine (CQ), acting at early and late stages of autophagy, respectively. CQ blocks the formation of autolysosomes, where LC3-II degradation occurs, thus inducing LC3-II accumulation (Amaravadi et al., 2007), whereas 3-MA, an inhibitor of class III phosphatidylinositol-3 kinase (PI3KIII), blocks early autophagic signaling, preventing LC3-I→LC3-II conversion (Kondo et al., 2005).
Accordingly, dl922–947-mediated LC3-I→LC3-II conversion induction and AVO formation were further increased by CQ, whereas they were partially reverted by treatment with 3-MA (Fig. 5A).
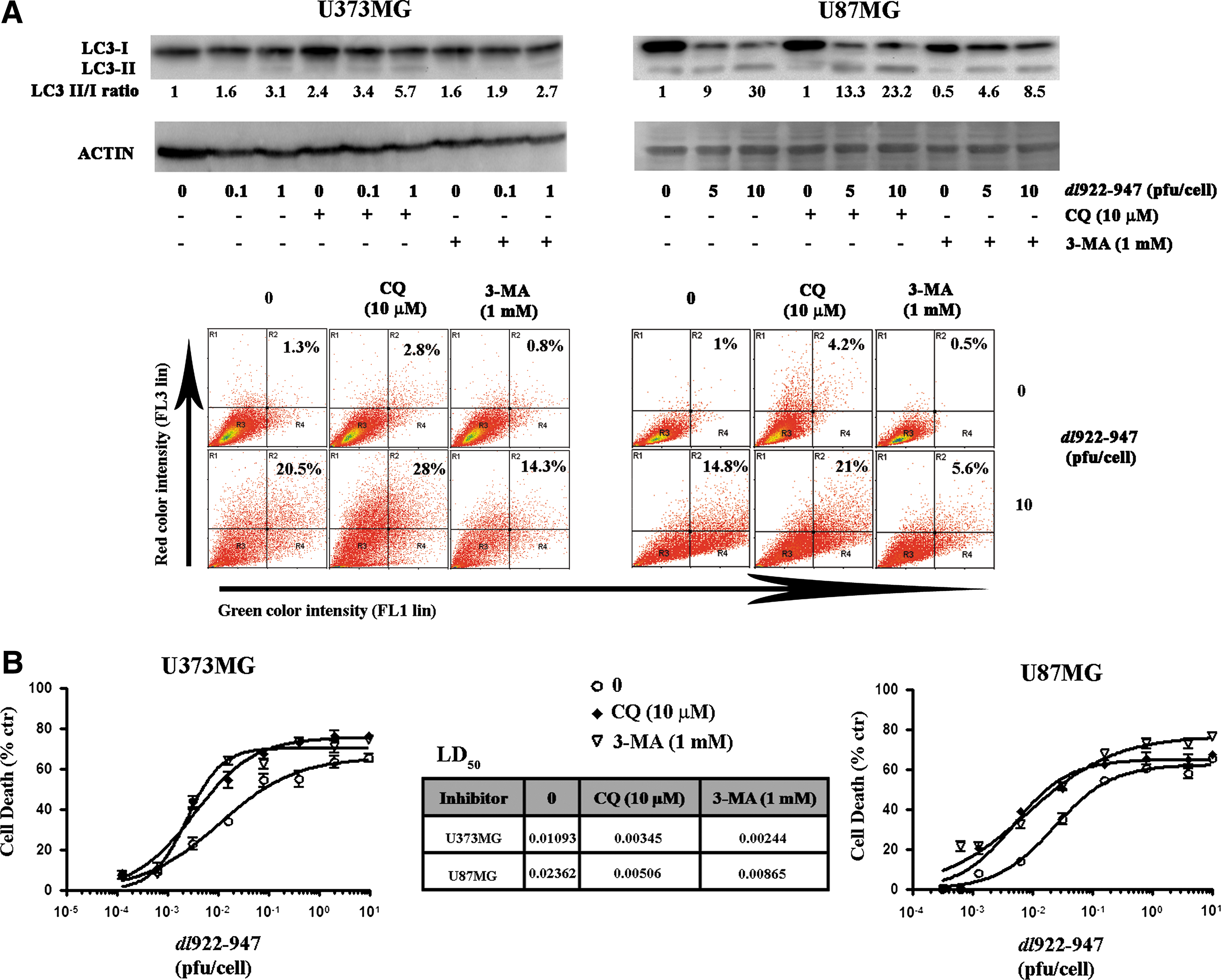
Pharmacological autophagy inhibitors enhance dl922–947-induced cytotoxicity.
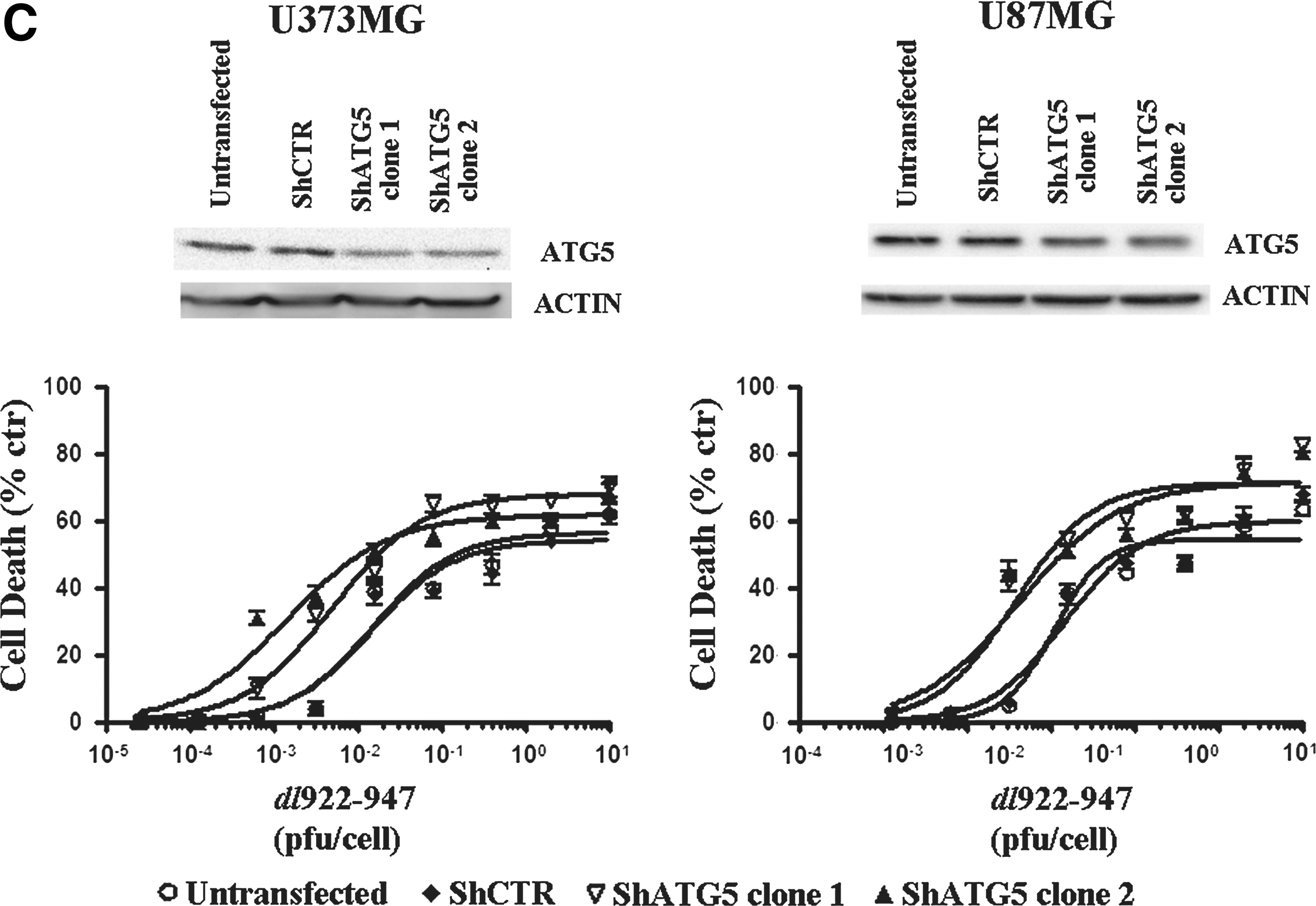
Treatment with a single dose of CQ (10 μM) or 3-MA (1 mM) increased dl922–947-induced cytotoxicity, with a reduction of LD50 values ranging from 2- to 5-fold (Fig. 5B). At the concentrations used, CQ or 3-MA treatment alone did not induce any toxic effect (data not shown).
Because CQ and 3-MA have a wide range of effects, not only specifically linked to autophagy, we have evaluated the effects of autophagy inhibition by generating stable glioma cells clones expressing Atg5 shRNA. We chose Atg5 because this gene is involved in both canonical and noncanonical autophagy (Scarlatti et al., 2008). Although we did not obtain complete gene silencing, in all transfected clones a strong increase in virus-induced cell death was observed (Fig. 5C).
Our data confirm that inhibition of autophagy sensitizes glioma cells to the oncolytic effects of dl922–947.
Viral replication is not increased by autophagy modulation
It has been proposed that the results of oncolytic virotherapy are correlated with the efficacy of viral replication within cancer cells. Therefore, we evaluated viral replication in glioma cells treated with rapamycin, PD98059, and CQ. A slight, albeit not statistically significant, decrease in genome copies number on treatment was observed (Fig. 6A). It has been already demonstrated that inhibition of autophagy with 3-methyladenine (3-MA) resulted in decreased synthesis of adenoviral structural proteins, and thereby poor viral replication (Rodriguez-Rocha et al., 2011).
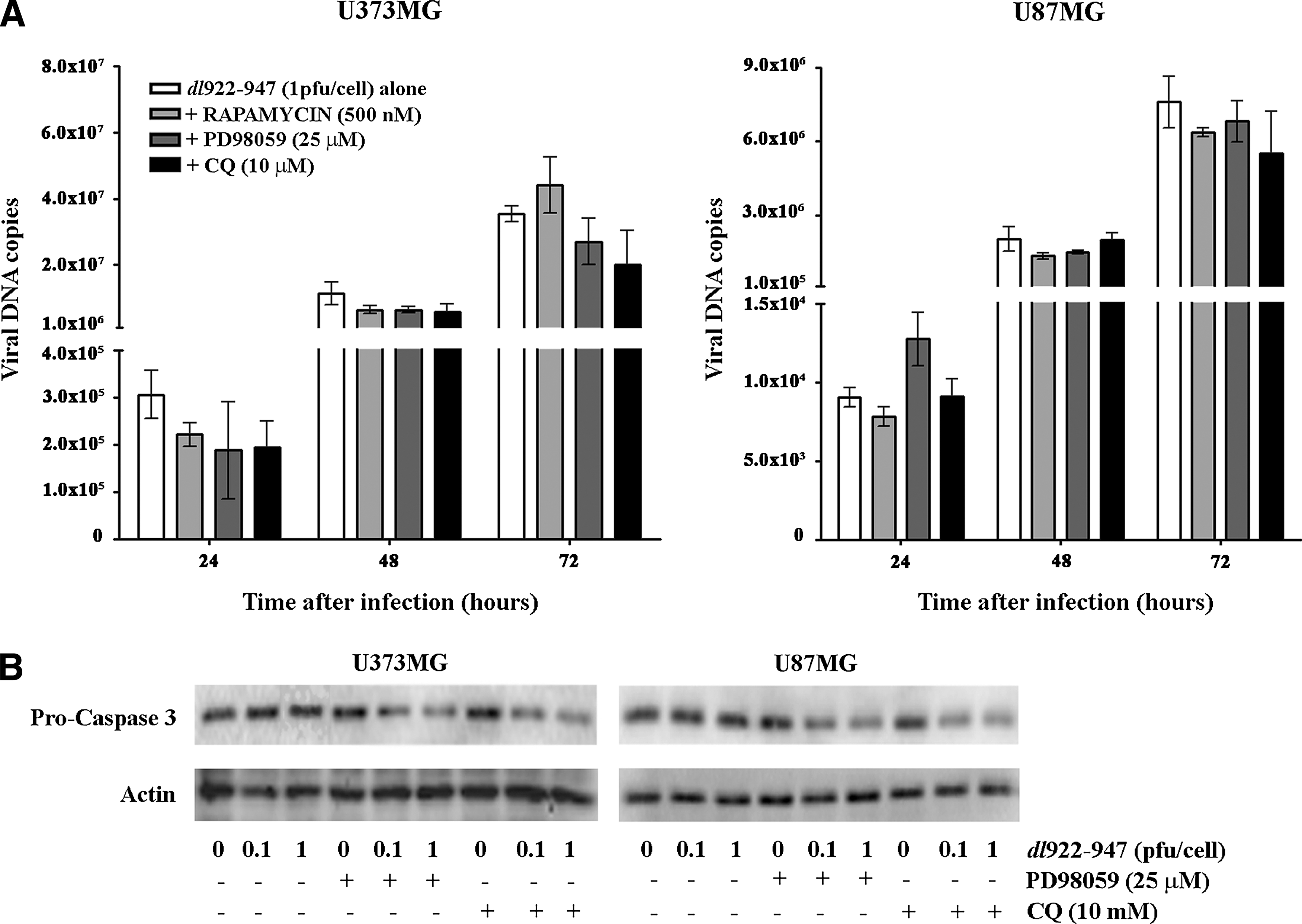
We have previously demonstrated that Aurora B inhibitor AZD1152 enhanced caspase-3-dependent cell death of thyroid carcinoma cells infected with dl922–947, without an increase in viral replication (Libertini et al., 2011). Moreover, several studies have shown that apoptosis and autophagy can be connected by a double switch (Díaz-Troya et al., 2008; Tyler et al., 2009) and that the inhibition of autophagy increased apoptotic cell death in cancer cells (Cardenas et al., 1999; Shigemitsu et al., 1999; Kondo et al., 2005). Analysis of caspase-3 activation in glioma cells infected with dl922–947 showed that the inhibition of autophagy with PD98059 or CQ leads to a reduction of procaspase-3 levels, suggestive of caspase-3 cleavage (Fig. 6B). These results indicate that the increase in viral cytotoxicity in the presence of autophagy inhibitors might be due to the activation of apoptosis, rather than increased viral replication.
Chloroquine enhances dl922–947 oncolytic activity in vivo
To validate the potential therapeutic use of antiautophagic drugs in association with dl922–947, we analyzed the effects of the virus combined with CQ in U87MG xenograft tumors. Athymic mice were injected with U87MG cells and after 20 days the animals were randomized into four groups (T=0 days).
In mice receiving the single treatments (dl922–947 or CQ), a nonsignificant reduction of tumor growth was observed, whereas CQ and dl922–947 combination treatment resulted in highly significant tumor growth inhibition (Fig. 7A). Starting from T=11 days, the differences in tumor volume among the untreated group, the groups receiving the single treatments, and the group receiving the combined treatment were significant (*p<0.05), and became highly significant (**p<0.01) at T=23 days.
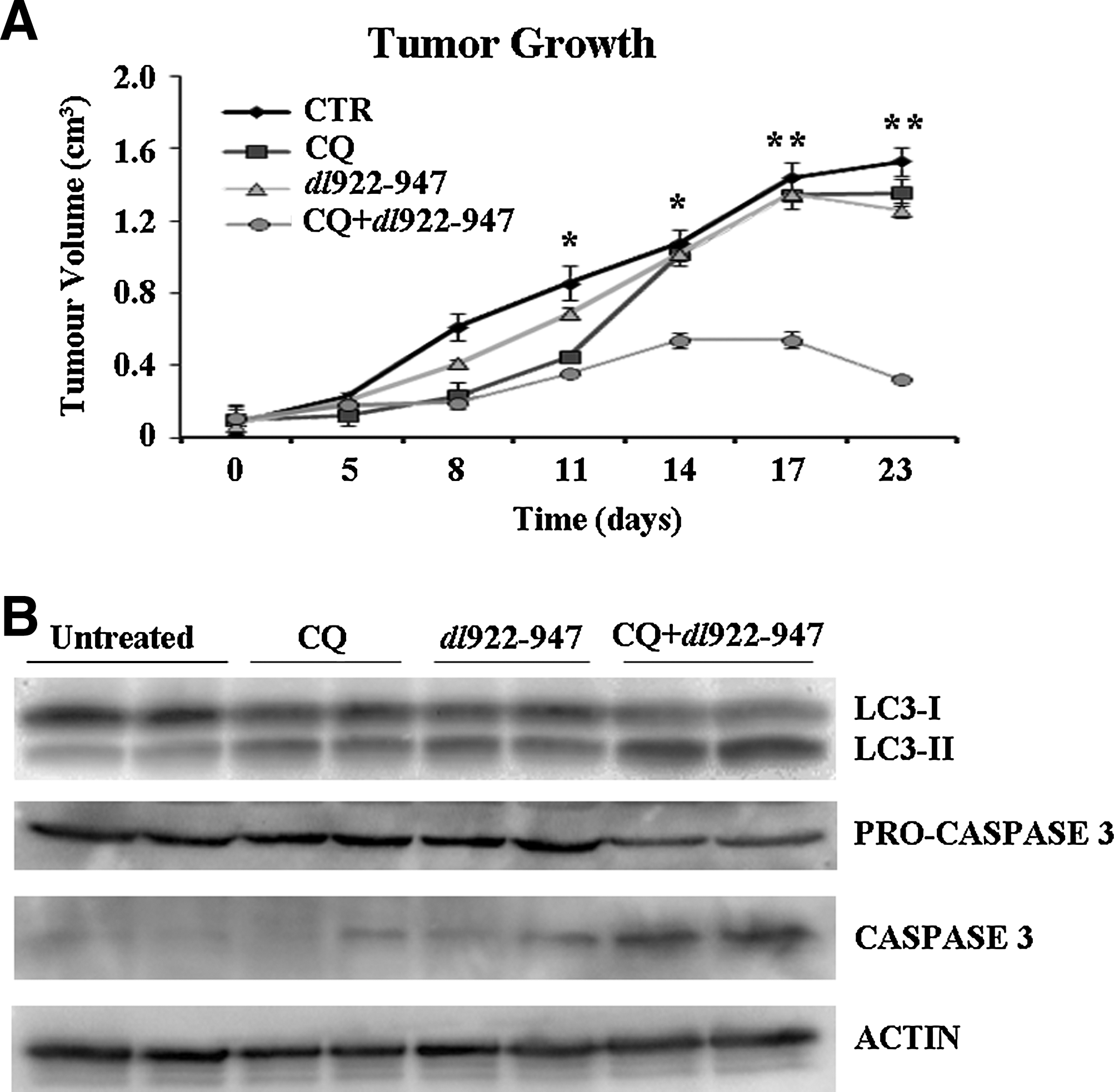
Chloroquine enhances the activity of dl922–947 and delays the growth of glioma tumor xenografts.
CQ treatment and dl922–947 treatment were both able to increase the LC3-II/LC3-I ratio, and this effect was further increased by the combined treatment (Fig. 7B).
Studies have demonstrated that apoptosis-mediated cell death is increased by inhibition of autophagy in cancer cells (Kanzawa et al., 2004; Boya et al., 2005; Dalby et al., 2010; Platini et al., 2010). By analyzing caspase-3 activation in treated tumors, we showed that cleaved caspase-3 levels were undetectable in control tumors and barely detectable in dl922–947-infected tumors. In contrast, in CQ-treated tumors dl922–947 led to a more intense caspase-3 activation, compared with the tumors undergoing the CQ single treatment (Fig. 7B), thus indicating that chloroquine is effective in increasing the antitumor activity of dl922–947 and in inducing apoptosis in experimental glioma in vivo.
Discussion
Glioblastoma multiforme (GBM) is one of the deadliest human cancers (Furnari et al., 2007). The therapy for GBM, which includes surgical resection, radiotherapy, and chemotherapy, has shown limited success. Consequently, the development of novel therapeutic approaches to improve patient survival is urgently required. A number of promising oncolytic viruses have demonstrated antiglioma activity in both preclinical and clinical settings (Brandes et al., 2008).
In the present study, we have analyzed the effects of E1A-mutated adenovirus dl922–947 against glioma cells, confirming its efficacy and showing that glioma cells undergo autophagy in response to dl922–947 infection, as confirmed by the dose-dependent development of acidic vesicular organelles, the increase in LC3-II levels, and the reduction of p62 levels. These effects were not observed on infection with control nonreplicating adenovirus (AdGFP or dl312), indicating that the activation of autophagy is dependent on viral replication.
In contrast to the induction of autophagy observed in infected cells, we show that dl922–947 infection activates the Akt/mTOR/p70s6k pathway, which is considered the main negative regulatory mechanism for autophagy (Blommaart et al., 1995; Yang et al., 2005), and simultaneously inhibits the ERK1/2 pathway, which is known to promote it (Pattingre et al., 2003). The nonreplicating control adenovirus AdGFP did not induce these effects. This apparent discrepancy could be due to compensatory mechanisms selectively operated by glioma cells to escape dl922–947 killing, although it is not possible to exclude that other pathways contribute to the cellular autophagic response to dl922–947 infection.
To address the role of these modulatory pathways in dl922–947-infected cells, we have used pharmacological inhibitors. The MEK-1 inhibitor PD98059 induced a block of autophagy, as shown by a reduction of LC3 conversion. Interestingly, this block was paralleled by an increase in dl922–947 cytotoxicity. Conversely, treatment of infected cells with rapamycin, a well-known autophagy inducer, sharply increased cell survival. All together, our data suggest that autophagy plays a defensive/survival role during the infection with dl922–947, the autophagic machinery probably being involved in the destruction of viral structures. On the other hand, the effects exerted by dl922–947 infection on intracellular signals could likely represent an attempt of the virus to overcome the cellular autophagic response. Indeed, adenoviral infection can trigger an indirect cellular autophagy defense response and specific viral mechanisms have developed to prevent this response. Our data are in agreement with a study showing activation of mTOR and p70s6k pathway on adenoviral infection to ensure translation initiation of viral mRNAs (O'Shea et al., 2005). Moreover, the early-expressed viral E3 gene product RID-α can insert into the autophagosomal membrane and arrest maturation by preventing fusion to the lysosome, thereby decreasing basal autophagy levels (Cianciola and Carlin, 2009). This latter mechanism can be excluded since dl922-947 lacks the E3 gene.
It is known that autophagy and viral infections have complex interconnections. It has been suggested that some viruses could use autophagosomes as sites for replication, and therefore autophagy can support viral replication and assembly (Lin et al., 2010). Moreover, it has been observed that hepatitis C virus (Dreux et al., 2009; Tanida et al., 2009), polio virus (Suhy et al., 2000), and dengue virus serotypes 2 and 3 (Panyasrivanit et al., 2009) are able to sustain their replication by creating membrane alterations through subverting or inducing autophagy. On the other hand, it has been suggested that autophagy can act as a host defense against intracellular pathogens (xenophagy) (Lin et al., 2010), because viral replication may be confined within autophagic vesicles. An antiviral effect of autophagy has been confirmed during neurotropic Sindbis virus infection and in herpes simplex virus (HSV)-1 encephalitis (Lin et al., 2010).
Oncolytic viruses can modulate autophagy in target cells, including glioma cells. Δ24-RGD oncolytic adenovirus has been shown to induce autophagic cell death in brain tumor stem cells (Jiang et al., 2007, 2011). Autophagosome-mediated cell death was also observed in glioma cells infected with an oncolytic adenovirus driven by the survivin promoter (Ulasov et al., 2009). Furthermore, hTERT-Ad, an oncolytic adenovirus regulated by the human telomerase reverse transcriptase promoter, induced autophagic cell death in human malignant glioma through inactivation of the Akt/TOR pathway (Ito et al., 2006), and the inhibition of autophagy reduced viral cytotoxicity (Yokoyama et al., 2008). These results have suggested a role for autophagy as the main pathway of cell death in cells infected with replicating adenoviral vectors (Ito et al., 2006). In line with this hypothesis, in vitro studies showed that autophagy inhibition can decrease the oncolytic activity of Δ24-RGD in glioma cell lines (Jiang et al., 2011) and of the E1B-deleted adenovirus Adhz60 in lung carcinoma cell lines (Rodriguez-Rocha et al., 2011).
Here, we show in vitro and in vivo that inhibition of autophagic pathways increases the activity of the E1A-deleted oncolytic virus dl922–947, indicating that autophagy acts as a cell survival response. Our observation is consistent with Baird and colleagues (2008), who have already shown that 3-MA and chloroquine enhance the effects of dl922–947 in ovarian carcinoma cells, but are in contrast with those of the above-mentioned studies from Jiang and colleagues (2011) and Rodriguez-Rocha and colleagues (2011). The discrepancies could be due to the different oncolytic adenoviruses analyzed. It is also conceivable that different viruses elicit different cellular responses depending on the cancer cell type. In this regard, in anaplastic thyroid carcinoma cells, we did not observe autophagy after dl922–947 infection (Libertini et al., 2011), suggesting that the activation of the autophagic process and the subsequent cellular fate are most likely virus and cell type dependent. Further studies are required to better understand how adenoviruses interact with host cells.
Autophagy inhibition is receiving attention as a novel strategy for cancer treatment. New autophagy inhibitors have been reported to act as potent anticancer drugs and/or to sensitize cancer cells to the activity of anticancer agents (Amaravadi et al., 2007; Carew et al., 2007; Rubinsztein et al., 2007; Shingu et al., 2009). Chloroquine itself is emerging as a potential anticancer agent against various cancers, including gliomas (Degtyarev et al., 2008; Solomon and Lee, 2009). Here, we show that chloroquine strongly enhances the oncolytic effects of dl922–947 both in vitro and in vivo, encouraging the use of antiautophagic drugs for the development of therapies based on the use of dl922–947 oncolytic virus for the treatment of gliomas.
Footnotes
Acknowledgments
The authors thank Dr. Tamotsu Yoshimori (Osaka University) and Prof. Noboru Mizushima (Tokyo Medical and Dental University) for kindly providing the plasmid containing LC3 cDNA. The authors thank Salvatore Sequino for excellent technical assistance and Joanne Smith for proofreading this manuscript. Silvana Libertini was the recipient of an FIRC fellowship and is now the recipient of a Marie Curie fellowship. This study was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC).
Author Disclosure Statement
No competing financial interests exist.