
Abstract
C-C chemokine receptor type 5 (CCR5) is a major co-receptor for the entry of human immunodeficiency virus type-1 (HIV-1) into target cells. Human hematopoietic stem cells (hHSCs) with naturally occurring CCR5 deletions (Δ32) or artificially disrupted CCR5 have shown potential for curing acquired immunodeficiency syndrome (AIDS). However, Δ32 donors are scarce, heterologous bone marrow transplantation is not exempt of risks, and genetic engineering of autologous hHSCs is not trivial. Here, we have disrupted the CCR5 locus of human embryonic stem cells (hESCs) and induced pluripotent stem cells (hiPSCs) using specific zinc finger nucleases (ZFNs) combined with homologous recombination. The modified hESCs and hiPSCs retained pluripotent characteristics and could be differentiated in vitro into CD34+ cells that formed all types of hematopoietic colonies. Our results suggest the potential of using patient-specific hHSCs derived from ZFN-modified hiPSCs for treating AIDS.
Introduction
Materials and Methods
Cell culture and characterization
H9 hESCs (passage 47–56, WiCell Research Institute) and an hiPSC clone produced from skin fibroblasts from a healthy human male and fully characterized (passage 15–26, Esteban et al., 2010) were maintained in a feeder-free culture system using mTesR1 (Stem Cell Technologies) and Matrigel (BD Bioscience). OP9 cells (ATCC) were cultured on gelatinized six-well dishes (2×104 per well) in OP9 growth medium consisting of α-modified minimum essential media (Invitrogen) supplemented with 20% non–heat-inactivated defined fetal bovine serum (HyClone). Alkaline phosphatase (AP) staining, reverse-transcription polymerase chain reaction (RT-PCR) and karyotyping were performed following standard procedure. For teratomas, 2×106 cells were injected into NOD-SCID mice.
CCR5 gene modification
ZFNs disrupting the human CCR5 locus were previously described (Perez et al., 2008). The corresponding ZFN sequences were cloned in one open reading frame and linked by a 2A sequence (Szymczak et al., 2004); the expression was controlled by a CAG promoter (Alexopoulou et al., 2008). The plasmid for homologous recombination was prepared by using the pUC19 vector. Enhanced green fluorescence protein (EGFP) and a puromycin-resistance cassette were linked by a 2A sequence as well, with the expression controlled by the EF1α promoter, and cloned within two homologous recombination arms. FuGENE HD transfection reagent (Roche Applied Science) was used for delivery. Puromycin (Sigma-Aldrich) was added at a concentration of 1 μg/mL.
Immunofluorescence
Cells were fixed in 4% paraformaldehyde and washed. The coverslips were blocked in 4% goat serum in phosphate-buffered saline before processing. Nuclei were stained with 4′,6-diamidino-2-phenylindole (Sigma). A Leica DMI4000B microscope (Leica Microsystems) was used for detection. All primary and secondary antibodies of this study are listed in Supplementary Table S1 (Supplementary Data are available online at
Hematopoietic differentiation
We adopted a protocol from Choi et al. (2009). Briefly, OP9 cells were grown for 8 days with half-medium changes on day 4. Undifferentiated hESCs and hiPSCs were added (∼2×105) after treatment with 1 mg/mL dispase (Invitrogen) and dispersion of the cultures into small clumps by scraping and pipetting. The co-cultures were incubated for up to 12 days at 37°C in normoxic conditions and 5% CO2, with half-medium changes every other day from day 4. To obtain a single-cell suspension, the cultures were treated with collagenase IV (Sigma), followed by trypsin/EDTA (0.05%). The suspension was further disrupted by pipetting and filtered through a 70-μm strainer before use in fluorescent activated cell sorter (FACS) analysis with FACSArial flow cytometer (BD Bioscience) or clonogenic forming cell (CFC) assays. For CFC assays, CD34+ cells were isolated using magnetic-activated cell sorting with a human CD34 Microbeads kit (Miltenyi Biotec) and added (2×103/mL) to low-adherence six-well plates (Stem Cell) using MethoCult GF+ H4435 semisolid medium (Stem Cell).
Results and Discussion
To disrupt the human CCR5 locus in hiPSCs, we employed an expression vector in which the ZFN sequences were linked by a 2A sequence and a donor vector for homologous recombination that produced puromycin resistance and EGFP (Fig. 1A). We also used hESCs to compare whether the maintenance of pluripotent characteristics after the gene editing approach is similar in both cell types. In this regard, one could argue that the artificial pluripotent state of hiPSCs might become unstable after zinc finger manipulation. Two days post-transfection EGFP was visualized and the medium was changed to selection medium containing puromycin. Resistant colonies emerged after another 4 days and puromycin was maintained for at least six passages. We then performed PCR analysis using primers that amplified the recombination cassette (Fig. 1A), and we found integration in 67% of the hiPSC and hESC colonies (Fig. 1B). We chose a positive hiPSC colony and another one for hESCs, hereafter termed ΔR5 cells, for further study (Fig. 1C). PCR using additional primers (Fig. 1A) confirmed site-specific integration but also demonstrated that the ΔR5 hiPSC and ΔR5 hESC colonies contained cells homozygous or heterozygous for the CCR5 deletion (Fig. 1D). The two colonies (passage 14 for hiPSCs and 19 for hESCs at the moment of analysis) retained a normal karyotype (Supplementary Fig. S1), expressed hESC-specific genes to the level of normal hESC and hiPSC colonies (Fig. 1E), stained positive for AP and other hESC-surface markers (Fig. 1F), and formed teratomas (Fig. 1G). Next, we investigated whether these modified pluripotent cell lines had the potential to be differentiated into CD34+ cells. FACS analysis confirmed the appearance of CD34+ cells with dynamics similar to those of normal hESCs and hiPSCs (Fig. 2A). We also noticed that EGFP expression was divided into high- and low-expressing populations, both in the CD34+ cells and the remaining cells (Fig. 2B). This is in agreement with the observation that the ΔR5 hiPSC colony is a combination of homozygous and heterozygous CCR5-deleted cells; PCR analysis for purified CD34+ cells confirmed this finding (Fig. 2C). Next, we characterized the multi-differentiation potential of ΔR5 CD34+ cells derived from hiPSCs. Addition of a cocktail of cytokines to purified CD34+ cells cultured on semisolid medium produced different types of colonies: CFC macrophage, CFC granulocyte, CFC granulocyte/macrophage, CFC granulocyte/erythrocyte/macrophage/megakaryo-cyte, and CFC erythroid (Fig. 2D).

Disruption of the endogenous C-C chemokine receptor type 5 (CCR5) locus in human induced pluripotent stem cells (hiPSCs) and human embryonic stem cells (hESCs) by zinc-finger nuclease (ZFN)-mediated homologous recombination.
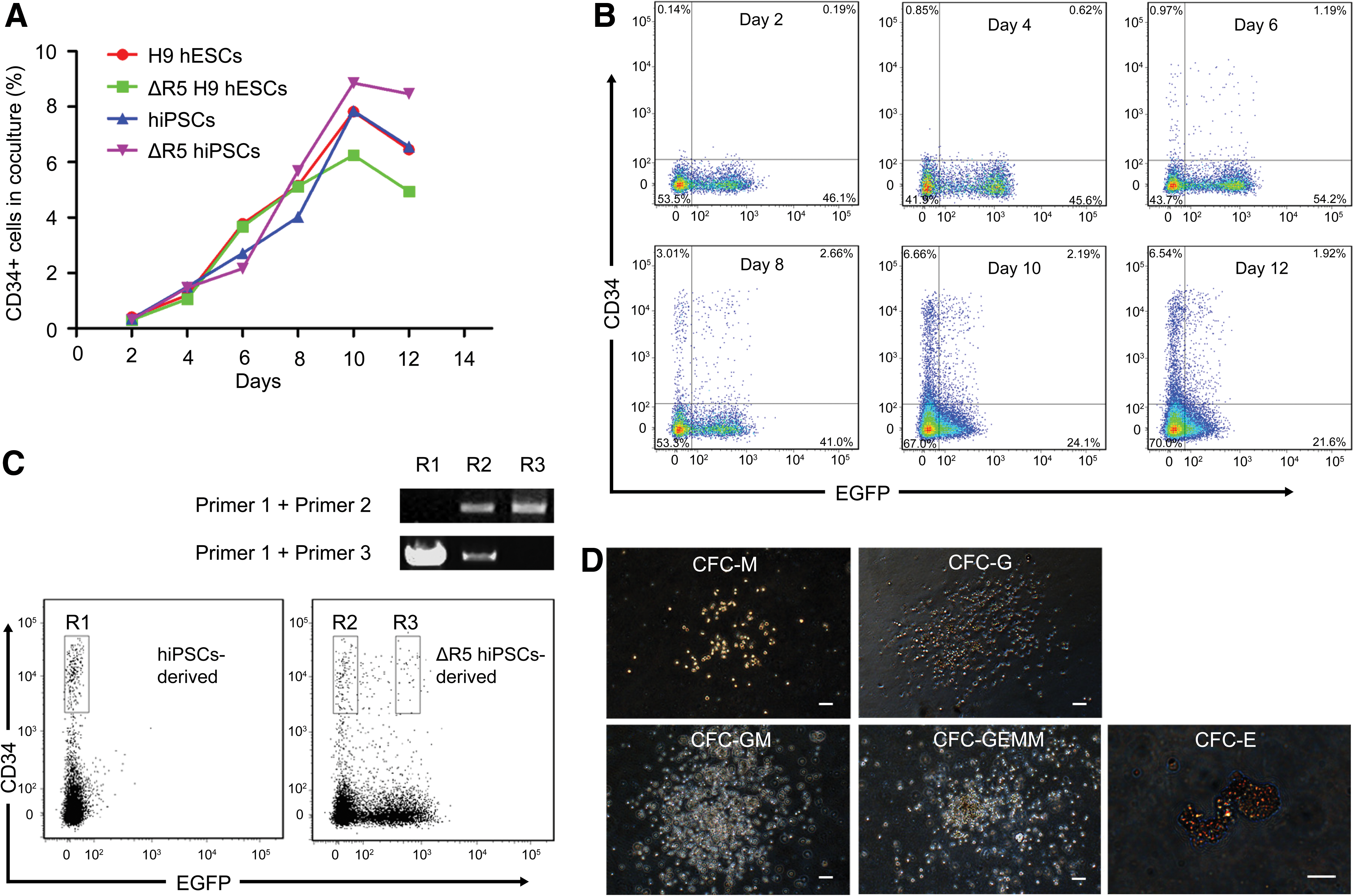
Directed differentiation of CCR5-disrupted hiPSCs and hESCs into CD34+ cells.
In summary, here we have shown the generation of CD34+ cells from CCR5-deleted hiPSCs and hESCs. These cells had the potential to be differentiated into different types of blood cell types. In the future, a similar approach could provide a stable supply of autologous CCR5-disrupted CD34+ cells that might be useful for transplantation in AIDS patients. Before this possibility can be considered, several relevant concerns would need to be addressed. Notably, hiPSCs need to be generated without exogenous factor integration (Yu et al., 2009; Warren et al., 2010) and tested for exclusion of somatic point mutations or epigenetic modifications (Bock et al., 2011; Gore et al., 2011). Generation of hiPSCs in a feeder-free and chemically defined medium (Yu et al., 2011) is also important for guaranteeing the exclusion of xeno-contaminants. Likewise, a feeder-free hematopoietic differentiation system, based on embryoid body formation (Galić et al., 2009), would be necessary to eliminate the need for murine cocultures. On the other hand, the combination of homozygous and heterozygous CCR5-deleted cells within the same colony may not be a critical issue because once these cells have reached macrophage and lymphoid lineages, only those cells bearing homozygous deletion would survive HIV-1 selective pressure in vivo (Holt et al., 2010). Besides, single-cell cloning using the Rho-associated kinase inhibitor could solve this problem (Watanabe et al., 2007). Despite the current limitations, the model presented here may be sufficient for creating humanized mice models for the study of HIV-1 infection (Galić et al., 2006; Holt et al., 2010; Zhang et al., 2007). Such study could be a first step towards considering clinical application of induced pluripotent stem cells in AIDS therapy.
Footnotes
Acknowledgements
The authors would like to thank Dajiang Qin, Dr. Duanqing Pei, Yongxiang Yan, Cuizhu Huang, Hui Zhang, Dr. Huaqiang Yang, and the staff in the Public Instrument Center and Animal Center for discussions and experimental/technical support. This work was supported by a research grant from the Chinese Academy of Sciences (KSCX1-YW-10).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.