
Abstract
In Duchenne muscular dystrophy (DMD), dystrophin deficiency leading to progressive muscular degeneration is caused by frame-shifting mutations in the DMD gene. Antisense oligonucleotides (AONs) aim to restore the reading frame by skipping of a specific exon(s), thereby allowing the production of a shorter, but semifunctional protein, as is found in the mostly more mildly affected patients with Becker muscular dystrophy. AONs are currently being investigated in phase 3 placebo-controlled clinical trials. Most of the participating patients are treated symptomatically with corticosteroids (mainly predniso[lo]ne) to stabilize the muscle fibers, which might affect the uptake and/or efficiency of AONs. Therefore the effect of prednisolone on 2′-O-methyl phosphorothioate AON efficacy in patient-derived cultured muscle cells and the mdx mouse model (after local and systemic AON treatment) was assessed in this study. Both in vitro and in vivo skip efficiency and biomarker expression were comparable between saline- and prednisolone-cotreated cells and mice. After systemic exon 23-specific AON (23AON) treatment for 8 weeks, dystrophin was detectable in all treated mice. Western blot analyses indicated slightly higher dystrophin levels in prednisolone-treated mice, which might be explained by better muscle condition and consequently more target dystrophin pre-mRNA. In addition, fibrotic and regeneration biomarkers were normalized to some extent in prednisolone- and/or 23AON-treated mice. Overall these results show that the use of prednisone forms no barrier to participation in clinical trials with AONs.
Introduction
DMD is caused by mutations in the DMD gene, located on the short arm of the X chromosome (Xp21), leading to the complete loss of the dystrophin protein it encodes (Hoffman et al., 1987; Koenig et al., 1988). Dystrophin stabilizes muscle fibers by connecting intracellular actin to the extracellular matrix (Blake et al., 2002), through a long repeat domain that intersperses the two essential binding domains. In the absence of dystrophin, muscle fibers will be continuously damaged during contraction, which leads to an influx of inflammatory cells and secretion of cytokines, in particular tumor necrosis factor (TNF)-α (Spencer and Tidball, 2001; Porter et al., 2002; Tidball, 2005). TNF-α activates the IκB kinase (IKK)/NF-κB signaling pathway in macrophages, which in turn increases necrosis and inflammation and reduces regeneration in muscle fibers (Hayden and Ghosh, 2004; Acharyya et al., 2007). Fibrotic tissue is formed by fibroblasts and even by satellite cells, which, once fibrotic tissue has been formed, start to produce collagen type I and no longer take part in regeneration (Alexakis et al., 2007). Fibroblasts from dystrophin-negative mdx mice, a naturally occurring mouse model for DMD (Sicinski et al., 1989), remain activated even when activating factors from immune cells have dissipated (Mezzano et al., 2007). These processes further increase fibrosis, which gradually replaces the damaged muscle fibers. Especially fibrosis in the endomysium is negatively correlated with functional performance in patients (Desguerre et al., 2009). Last, muscle regeneration is reduced through induction of the transforming growth factor (TGF)-β pathway by the fibrotic tissue (Chen et al., 2005).
At present there is no cure for DMD. However, there are pharmacological approaches that try to combat the symptoms caused by the underlying genetic defect. The main treatment is the use of the corticosteroids prednisone, prednisolone (the active form of prednisone), or deflazacort (an oxazolidine derivative of prednisone) (Khan, 1993). Their exact mechanism of action is unknown, but studies have shown that corticosteroid-treated patients have increased muscle strength and remain ambulant for about 3 years longer than untreated patients (Merlini et al., 2003; Angelini, 2007). In mdx mice a positive effect of prednisolone on muscle strength and histology (decrease in centrally located nuclei) was seen (Baltgalvis et al., 2009). This beneficial effect is probably through the antiinflammatory effects and the reduction of muscle necrosis (Gaud et al., 2004; Angelini, 2007). In a dystrophin-deficient Caenorhabditis elegans, the amount of degenerating cells is decreased after treatment with prednisone (Gaud et al., 2004). Because C. elegans has only a simple immune system, this indicates that other mechanisms are involved as well. One study suggests that deflazacort activates the calcineurin/NF-AT (nuclear factor of activated T cell) pathway and thereby increases the expression of NF-AT target genes, among which is the dystrophin homolog utrophin, which can partly take over the function of dystrophin, thereby decreasing the dystrophic muscle fiber pathology (St-Pierre et al., 2004). An alternative possibility is that the anabolic effect of corticosteroids in patients increases muscle regeneration and growth by enhancing proliferation of myogenic precursor stem cells or myoblasts (Angelini, 2007). Furthermore, corticosteroids have a positive effect on calcium homeostasis, which is deregulated in patients with DMD (Khan, 1993). Unfortunately, corticosteroids also have deleterious side effects (Fisher et al., 2005) such as osteoporosis (Lo Cascio et al., 1995), weight gain, growth inhibition (Merlini et al., 2003), delayed puberty, and cataracts (Jobling and Augusteyn, 2002). They have a catabolic effect on muscle in unaffected individuals (Schakman et al., 2009), but in DMD this is generally abrogated by the positive effects. In mdx mice it has even been shown that prednisolone induced fibrosis in the heart (Bauer et al., 2009), but this is not observed in patients with DMD, in whom corticosteroid use prevents/delays ventricular dysfunction (Silversides et al., 2003; Markham et al., 2008).
Approaches aiming to restore the underlying genetic defect of DMD are currently under investigation. At the moment, the most promising strategy is exon skipping, using antisense oligonucleotides (AONs) to restore the disrupted reading frame. These AONs target a specific region in the pre-mRNA involved in appropriate exon inclusion. In this way they prevent the exon from being recognized by the spliceosome and, consequently, it will not be incorporated in the mRNA (Aartsma-Rus et al., 2009). The resulting in-frame mRNA transcript allows translation of a protein that is internally deleted, but contains the essential actin and extracellular matrix binding domains, and therefore it will be partially to largely functional (Aartsma-Rus and van Ommen, 2007). These dystrophins will resemble the internally deleted proteins found in patients with Becker muscular dystrophy (BMD), who have a much milder phenotype and longer life expectancy (Monaco et al., 1988; Koenig et al., 1989). Proof of principle has been obtained in vitro with both healthy and patient-derived cultured primary human myoblasts (Aartsma-Rus et al., 2002, 2003, 2004) and in in vivo studies in mdx mice (Wu et al., 2008; Heemskerk et al., 2009; Yin et al., 2009; Goyenvalle et al., 2010). Exon skipping can be performed using AONs with various backbone chemistries (Heemskerk et al., 2009). For in vivo studies, mainly 2′-O-methyl phosphorothioate (2OMePS) and phosphorodiamidate morpholino oligomer (PMO) AONs are used. These have also been used in two exploratory clinical trials with local injections, which have shown positive results (van Deutekom et al., 2007; Kinali et al., 2009). A phase 1–2a systemic clinical trial with 2OMePS AONs has been completed and a dose-dependent effect was seen after subcutaneous injections of AONs against exon 51. Treatment resulted in exon skipping and dystrophin expression at up to ∼15% of normal expression levels. In an open label extension study, 3 months of weekly injections with the highest dose resulted in an increase in functional performance, without serious adverse events (Goemans et al., 2011). The first systemic clinical trial with PMOs showed promising results as well: 7 of 19 patients, mainly in the higher dose groups, showed dystrophin restoration after treatment, albeit with considerable variability between patients (Cirak et al., 2011).
Larger multicenter placebo-controlled trials with 2OMePS AONs have now been initiated. The majority of the patients involved in these trials will use corticosteroids, primarily predniso(lo)ne. Thus far the effect of corticosteroid treatment on AON biodistribution and skipping efficiency has not been studied. Corticosteroids are thought to stabilize the damaged muscle fiber membrane, whereas the exon-skipping approach actually makes use of the fact that in patients with DMD the membrane is leaky, allowing higher uptake of AONs (Heemskerk et al., 2010). Thus, corticosteroid treatment could result in decreased uptake of AONs and lower levels of exon skipping and dystrophin restoration. On the other hand, the exon-skipping approach requires a sufficient amount of pre-mRNA to be effective, which is expressed only by muscle tissue and not by fibrotic tissue. Therefore, more dystrophin pre-mRNA might be available when the muscle is better preserved, due to corticosteroid treatment. To elucidate this, the effect of cotreatment with 2OMePS AONs and prednisolone on patient-derived muscle cell cultures and in dystrophic mdx mice was assessed.
The results suggest that prednisolone treatment does not interfere with 2OMePS AON uptake and exon-skipping levels in patient-derived muscle cells in vitro and in mdx mice in vivo. Prednisolone might even enhance the dystrophin expression induced by exon 23-specific AONs (23AONs) in mdx mice.
Materials and Methods
Cell cultures and AON transfection
Two patient cell cultures, DL589.2 (deletion exon 51–55) and 53914.1 (deletion exon 52) (previously described by Aartsma-Rus et al., 2003, 2004) were grown to 80% confluency on high-serum medium. Differentiation was induced by switching to low-serum medium. Prednisolone in saline (Leiden University Medical Center [LUMC] Pharmacy, Leiden, The Netherlands) was added in doses ranging from 0.2 to 2.25 μg/ml to determine optimal concentration. For AON transfection experiments prednisolone was added at 0.75 μg/ml to the medium. When differentiation was deemed sufficient (generally after approximately 10 days), h50AON1 and PRO051 (Aartsma-Rus et al., 2002) were transfected in the respective patient cell cultures and the control cells, using polyethylenimine (Exgen 500; MBI Fermentas, Sankt Leon-Rot, Germany), according to the manufacturer's instructions and using 2.5 μl of PEI per microgram of AON (n=12 per condition per cell line). All AONs were 2′-O-methyl RNA oligonucleotides with a full-length phosphorothioate backbone (Eurogentec [Seraing, Belgium] and Prosensa Therapeutics [Leiden, The Netherlands]). Cells were harvested 2 days after transfection.
In vivo 23AON and prednisolone treatment
All experiments were approved by the local animal ethics experimental committees. Mice were housed in individually ventilated cages in the animal facility of the LUMC or Laboratory of Pharmacology and Toxicology (LPT, Hamburg, Germany) and received food and drink ad libitum. Intramuscular experiments were performed at the LUMC and mdx mice (C57Bl/10ScSn-DMD mdx /J) with one or two copies of the utrophin gene (mdx/utrn+/– or mdx/utrn+/+ ) were obtained from our own breeding facilities. The systemic experiments were performed at the LPT and mdx mice from Charles River Laboratories (Sulzfeld, Germany) were used.
For the intramuscular 23AON treatment, mice were subcutaneously (Payne et al., 2006) injected with prednisolone (1 mg/kg; LUMC Pharmacy) (n=4) or saline (n=4) on weekdays from the age of 4 weeks until the end of the experiment. At the age of 8 weeks, the mice were anesthetized with isoflurane and intramuscularly injected via both gastrocnemius muscles on two consecutive days with 2.9 nmol (∼20 μg) of M23D(+2–18), 2′-O-methyl phosphorothioate RNA oligonucleotides with a full-length phosphorothioate backbone, specifically targeting exon 23 (Mann et al., 2002) (produced by Prosensa Therapeutics), in 40 μl of saline. Ten days after the second injection the mice were killed by cervical dislocation and muscles were isolated.
For the systemic 23AON treatment male mdx mice (n=8–10 per group) at the age of 5 weeks (day 1) were anesthetized with ether and underwent surgery to implant a prednisolone pellet (1 mg/kg/day in a 60-day slow-release subcutaneous pellet; Guerron et al., 2010) under the dorsal skin (groups 2 and 4) or underwent mock surgery (groups 1 and 3) at the start of the experiment. Mice were injected subcutaneously with 250 mg of M23D(+2–18)/kg body weight once daily for 5 days in test week 1 and 100 mg of M23D(+2–18)/kg body weight twice per week in test weeks 2 to 8 (groups 3 and 4), or saline (groups 1 and 2). Mice were killed 10 days after the last injections and muscles (gastrocnemius, tibialis anterior, quadriceps, heart, and diaphragm) and organs (liver and kidney) were isolated, snap frozen in liquid nitrogen-cooled 2-methylbutane, and stored at −80°C.
Measurement of creatine kinase levels
Blood samples were taken weekly via the tail vein. Samples were centrifuged at 1700×g for 10 min at 4°C. Serum was stored at 4°C and creatine kinase (CK) levels were measured after diluting the samples 10 times in Dulbecco's phosphate-buffered saline (D-PBS; Invitrogen, Carlsbad, CA) and were measured with a Reflotron system (Roche Diagnostics, Basel, Switzerland) with CK-strips (Roche).
RNA extraction and analysis of exon skipping by RT-PCR
Harvested cells were lysed with RNA-Bee (Campro Scientific, Veenendaal, The Netherlands). Total RNA was extracted and 400 ng of RNA was used for RT-PCR analysis, using Transcriptor reverse transcriptase polymerase (Roche) in 20 μl at 55°C for 30 min with an appropriate primer (primer sequences on request). cDNA was amplified by nested PCR. Three microliters of cDNA was amplified in a 25-μl reaction for 20 cycles of 94°C (40 sec), 60°C (40 sec), and 72°C (80 sec), followed by 32 cycles of 94°C (40 sec), 60°C (40 sec), and 72°C (60 sec), with 1.5 μl of PCR product in a 50-μl reaction.
Muscles were minced in TriPure isolation reagent (Roche), using MagNA Lyser green beads (Roche) or zirconium beads (1.4 mm; OPS Diagnostics, Lebanon, NJ) according to the manufacturer's instructions. Total RNA was extracted and 1 μg of RNA was used for RT-PCR analysis, using Transcriptor reverse transcriptase polymerase (Roche) in 20 μl at 42°C for 45 min with random hexamer primers (20 ng/μl). Then, 1.5 μl of cDNA was amplified in a 50-μl reaction for 30 cycles of 94°C (30 sec), 60°C (30 sec), and 72°C (30 sec), as previously described (Spitali et al., 2010). All PCR products were visualized on 1.5 or 2% agarose gels and quantified with an Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA). PCR products were quantified using a DNA 1000 LabChip on the Agilent 2100 bioanalyzer (Agilent Technologies) according to the manufacturer's protocol. Bioanalyzer analysis has been shown to be an accurate method for exon-skipping quantification in mdx muscle (Spitali et al., 2010).
Hybridization–ligation assay
The assay for measuring the concentration of 23AON in tissue samples is based on a previously published hybridization–ligation assay (Yu et al., 2002). Briefly, a signal probe (containing a peptide for antibody recognition) and a template (complementary to 23AON and the probe) were added to homogenized tissue samples. This was followed by a ligation step that takes place only when both AON and probe are bound to the template. Unbound probe was then washed away and the amount of probe–AON was detected with enzyme-linked antibodies against the probe. Muscles/organs were homogenized in proteinase K buffer (0.1 M Tris-HCl [pH 8.5], 0.2 M NaCl, 0.2% sodium dodecyl sulfate [SDS], and 5 mM EDTA) containing proteinase K (2 mg/ml; Invitrogen), using MagNA Lyser green beads (Roche) or zirconium beads (1.4 mm; OPS Diagnostics), by grinding in a MagNA Lyser (Roche) followed by incubation overnight at 55°C with gentle agitation. Calibration curves of the analyzed 23AON prepared in 60 times pooled control mouse mdx tissue in PBS were included. All tissues were diluted in pooled control mdx mouse tissue. The muscle samples were diluted 500 and 1000 times, and liver and kidney tissue 1000 and 5000 times. All analyses were performed in duplicate.
Protein extraction and Western blot analysis
Western blotting was performed as described (Anderson and Davison, 1999; Aartsma-Rus et al., 2003; Heemskerk et al., 2009). Briefly, muscles were homogenized in 75 mM Tris-HCl (pH 6.8)–15% SDS, using MagNA Lyser green beads (Roche), by grinding in a MagNA Lyser (Roche). Protein concentrations were determined with a Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. Samples containing 75 μg of protein were made in 75 mM Tris-HCl (pH 6.8), 15% SDS, 5% 2-mercaptoethanol, 2% glycerol, and 0.001% bromophenol blue, boiled for 5 min, loaded on a 4–7% gradient polyacrylamide gel, and run overnight at 4°C. Control samples containing 3.75 (5%), 1.5 (2%), 0.75 (1%), and 0.075 (0.1%) μg of protein were used as a reference. Gels were blotted to nitrocellulose BA83 (Whatman/Schleicher & Schuell, Dassel, Germany) for 6 hr at 600 mA at 4°C. Blots were blocked with 5% nonfat dried milk (Campina Melkunie, Zaltbommel, The Netherlands) in Tris-buffered saline (TBS) followed by an overnight incubation at 4°C with NCL-DYS1 (dilution 1:125; NovoCastra, Newcastle-upon-Tyne, UK) in TBS plus 0.05% Tween 20 to detect dystrophin. As secondary antibody the fluorescent IRDye 800CW goat anti-mouse IgG (dilution 1:5000, LI-COR, Lincoln, NE) was used. Blots were visualized and quantified with the Odyssey system and software (LI-COR).
Immunohistochemistry and dystrophin quantification
Sections (thickness, 8 μm) were cut with a Shandon cryotome (Thermo Fisher Scientific) on Superfrost Plus slides (Thermo Fisher Scientific) along the entire length of the gastrocnemius with a minimal interval of 240 μm between the sections. Slides were fixed for 5 min in ice-cold acetone and blocked with PBS–0.05% Tween–5% horse serum. Slides were incubated overnight with dystrophin diluted 1:50 (dystrophin [C-20] sc-7461; Santa Cruz Biotechnology, Heidelberg, Germany) and spectrin diluted 1:200 (anti-spectrin β-3 polyclonal antibody PA1-46007; Thermo Fisher Scientific) as primary antibodies. As secondary antibodies Alexa Fluor 488-conjugated donkey anti-goat IgG diluted 1:1000 (A11055; Invitrogen) for dystrophin and Alexa Fluor 594-conjugated donkey anti-rabbit IgG diluted 1:1000 (A21207; Invitrogen) for spectrin were used. Slides were mounted with VECTASHIELD HardSet mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (H-1550; Vector Laboratories, Burlingame, CA). Images were made with a fluorescence microscope (Leica DM5500; Leica Microsystems, Rijswijk, The Netherlands) at ×20 magnification. Dystrophin levels were assessed with Leica MM Basic Offline software (Leica Microsystems) by calculating the average maximal intensity of 10 randomly placed rounds at the membranes minus the average of the average of 10 randomly placed rounds in the cytoplasm.
Biomarker analysis
Total RNA was purified with a NucleoSpin RNA II kit according to the manufacturer's instructions, including a DNase digestion (Macherey-Nagel, Düren, Germany). The integrity of the purified RNA was checked with an RNA 6000 Nano LabChip on the Agilent 2100 bioanalyzer (Agilent Technologies) according to the manufacturer's protocol.
One microgram of RNA was used for cDNA synthesis, using BioScript (GC biotech, Alphen aan den Rijn, The Netherlands) in 20 μl at 70°C for 10 min and 42°C for 1 hr, with random hexamer primers (20 ng/μl). Gene expression levels were determined for CD68, Lgals3 (lectin, galactoside binding, soluble, 3), biglycan, Lox, MyoD, myogenin, MRF4, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) by real-time qPCR, using SensiMix SYBR (GC biotech) and the Roche LightCycler 480 (Roche) (primer sequences on request), with a program consisting of 45 cycles of 95°C (10 sec), 60°C (30 sec), and 72°C (20 sec).
For microRNA (miRNA) analysis 1 μg of unpurified RNA was used. Input RNA was reverse transcribed, using an miScript reverse transcription kit (Qiagen, Venlo, The Netherlands) according to the manufacturer's protocol. Two microliters of 10 times diluted cDNA was used as input for the real-time qPCR, using SensiMix SYBR (GC biotech) on the Roche LightCycler 480 (Roche), using a program consisting of 55 cycles of 95°C (10 sec), 57°C (30 sec), and 72°C (20 sec). Specific forward primers for miR-31 and 5S were used in combination with a universal reverse primer complementary to the adapter sequence of the RT-primer (primer sequences on request).
Calculations of relative expression were done with LinRegPCR quantitative PCR data analysis software, version 11.3 (Ruijter et al., 2009). GAPDH, stably expressed across all conditions, was used to correct for differences in cDNA input for protein-coding transcripts and 5S was used to correct for differences in starting concentrations for miR-31.
Statistical analysis
Data are represented as means±SD. Exon-skipping percentages, 23AON concentrations, and protein levels between AON and prednisolone plus AON groups were compared using an independent samples, two-tailed, Student t test in Excel 2003 (Microsoft Office Professional Edition 2003; Microsoft, Redmond, WA). Values of p less than 0.05 after correcting for multiple testing were considered significant.
Statistical significance between all four groups for dystrophin staining, plasma creatine kinase levels at the end of the experiment (day 64), and biomarker expression was assessed by one-way analysis of variance (ANOVA), followed by a Bonferroni correction for multiple testing in case of significance (p<0.05) in SPSS 17.0.2 (SPSS, Chicago, IL).
To assess a possible treatment effect on the weight of mice over time a longitudinal analysis was perform in R (R Development Core Team, 2009), using the lme4 package (Bates and Maechler, 2010). A baseline corrected model was used, including fixed linear and quadratic time effects; the mouse effect was considered to be random.
Results
Prednisolone does not interfere with exon skipping in cultured cells and intramuscular 23AON injection
The effect of prednisolone on exon-skipping efficiency was first determined on cell cultures. Various doses of prednisolone were added to culture medium (0.2–2.25 μg/ml) The highest dose did not induce cell death, but impaired differentiation was observed (data not shown), a known effect of corticosteroids (Schakman et al., 2009). The dose of 0.75 μg/ml was selected for further study, as it did not induce cell death, did not affect differentiation, and is comparable to the dose that is used in most patients (recommended dose, 0.75 mg/kg/day) (Mendell et al., 1989; Khan, 1993; Bonifati et al., 2000; Angelini, 2007).
Patient-derived cell cultures were pretreated for 10 days with prednisolone (0.75 μg/ml), or with saline as a control. Subsequently, cells were transfected with a 400 nM concentration of the appropriate AON. Skipping of the targeted exon was seen in all cells (Fig. 1) and no difference was seen between prednisolone- and saline-treated cells. Exon 51 skipping in cells with a deletion of exon 52 (53914.1) averaged 40% for both saline- and prednisolone-treated cells. For exon 50 skipping in cells with a deletion of exon 51–55 (DL589.2) the percentages were ∼70%, with relatively small variation (Fig. 1A). Similar exon-skipping levels were also found for other AON concentrations in the presence or absence of prednisolone (data not shown).
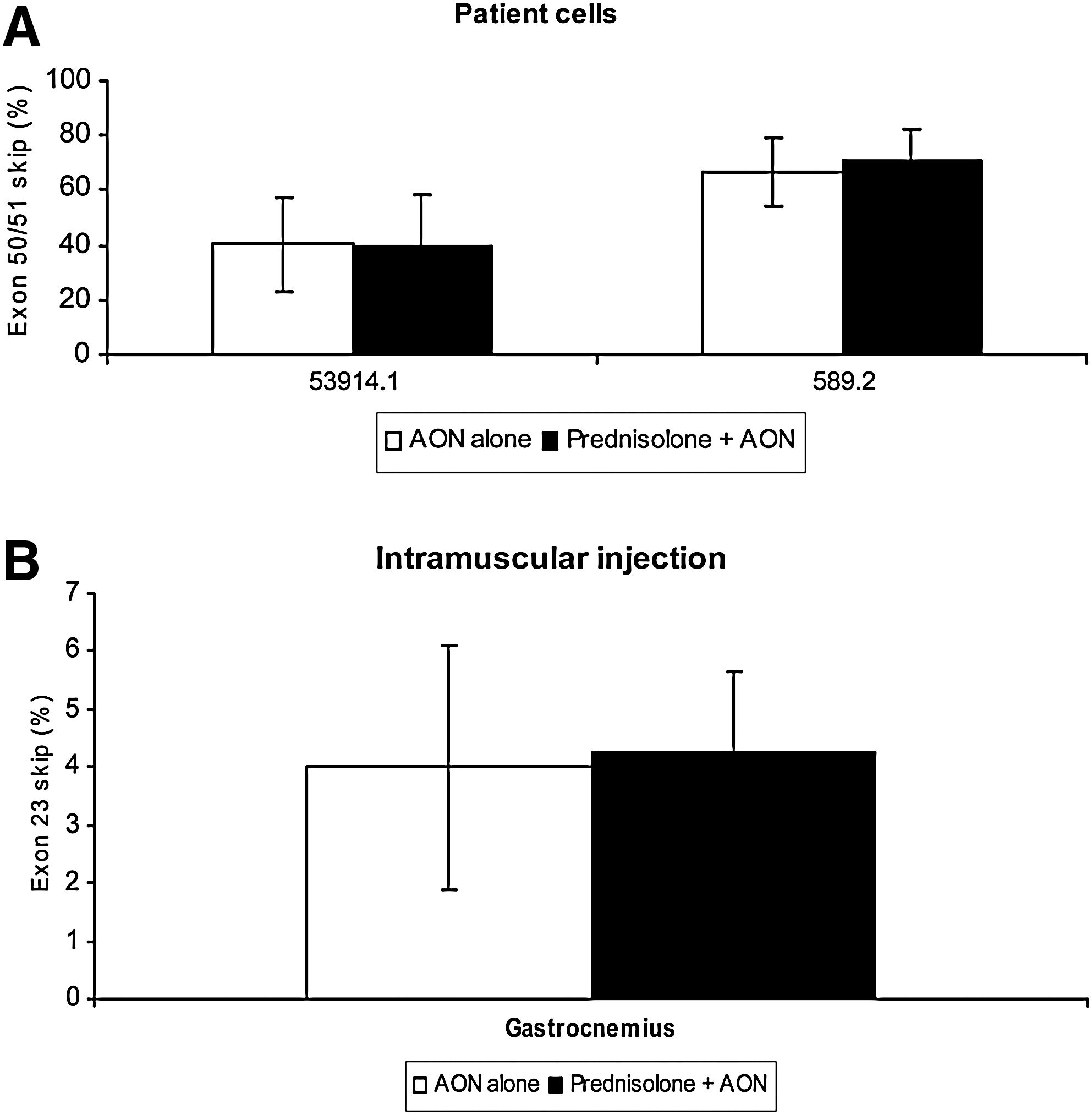
Effect of prednisolone on antisense oligonucleotide-mediated exon skipping in vitro and intramuscularly in the mdx mouse. Means are shown for each group. Error bars represent the standard deviation.
In vivo, the effect of systemic prednisolone treatment was first tested by local, intramuscular, injections with 23AON, a 2′-O-methyl phosphorothioate AON inducing exon 23 skipping [M23D(+2–18)] (Mann et al., 2002). mdx mice were pretreated subcutaneously with saline or 1-mg/kg prednisolone for 4 weeks, starting at the age of 4 weeks (Payne et al., 2006). This age was chosen because the major regeneration and degeneration cycles in mdx mice have been shown to take place at about this age (Tanabe et al., 1986). The prednisolone dose is comparable to doses used by patients with DMD (Khan, 1993). At the age of 8 weeks, mice received two consecutive injections with 2.9 nmol of 23AON locally in the gastrocnemius muscles. Exon-skipping percentages were determined by primary PCR and bioanalyzer analysis (Spitali et al., 2010). No significant differences in exon-skipping levels were observed (Fig. 1B). Nested PCR analysis, the most commonly used method for determining exon skipping at present (Lu et al., 2005; Denti et al., 2008; Ivanova et al., 2008; Yin et al., 2008; Doran et al., 2009; Heemskerk et al., 2009), resulted in higher skipping levels of about 20% (data not shown). However, it has been shown that nested PCR gives an overestimation of absolute exon-skipping percentages (Spitali et al., 2010).
Prednisolone does not affect exon skipping efficiency systemically in mdx mice
To investigate the effect of prednisolone on systemic treatment with 23AON, 5-week-old male mdx mice were treated for 8 weeks simultaneously with prednisolone (1 mg/kg/day via a subcutaneous slow-release pellet, shown to be an effective mode of delivery [Yao et al., 2008; Guerron et al., 2010]) or saline and 23AON (250 mg/kg, five times in week 1; and 100 mg/kg, two times in weeks 2 to 8, subcutaneously), or saline. During the treatment the weight of the mice was monitored. In contrast to the weight increase seen in humans, prednisolone-treated mice (both prednisolone alone and prednisolone and 23AON) weighed significantly less compared with saline- or 23AON alone-treated mice (Fig. 2A). This is in line with results described in the literature (Keeling et al., 2007). In the absence of prednisolone, no difference in weight between 23AON- and saline-treated mice was observed. Plasma creatine kinase (CK) levels (a measure of muscle damage) did not differ significantly for the various groups after treatment (data not shown). Notably, levels were generally much lower than expected (up to 2000 U/liter in all mice, whereas, in general, levels up to 8000 U/liter are found in untreated mdx mice). This may have been due to the time between serum taking and measuring (1–4 weeks).
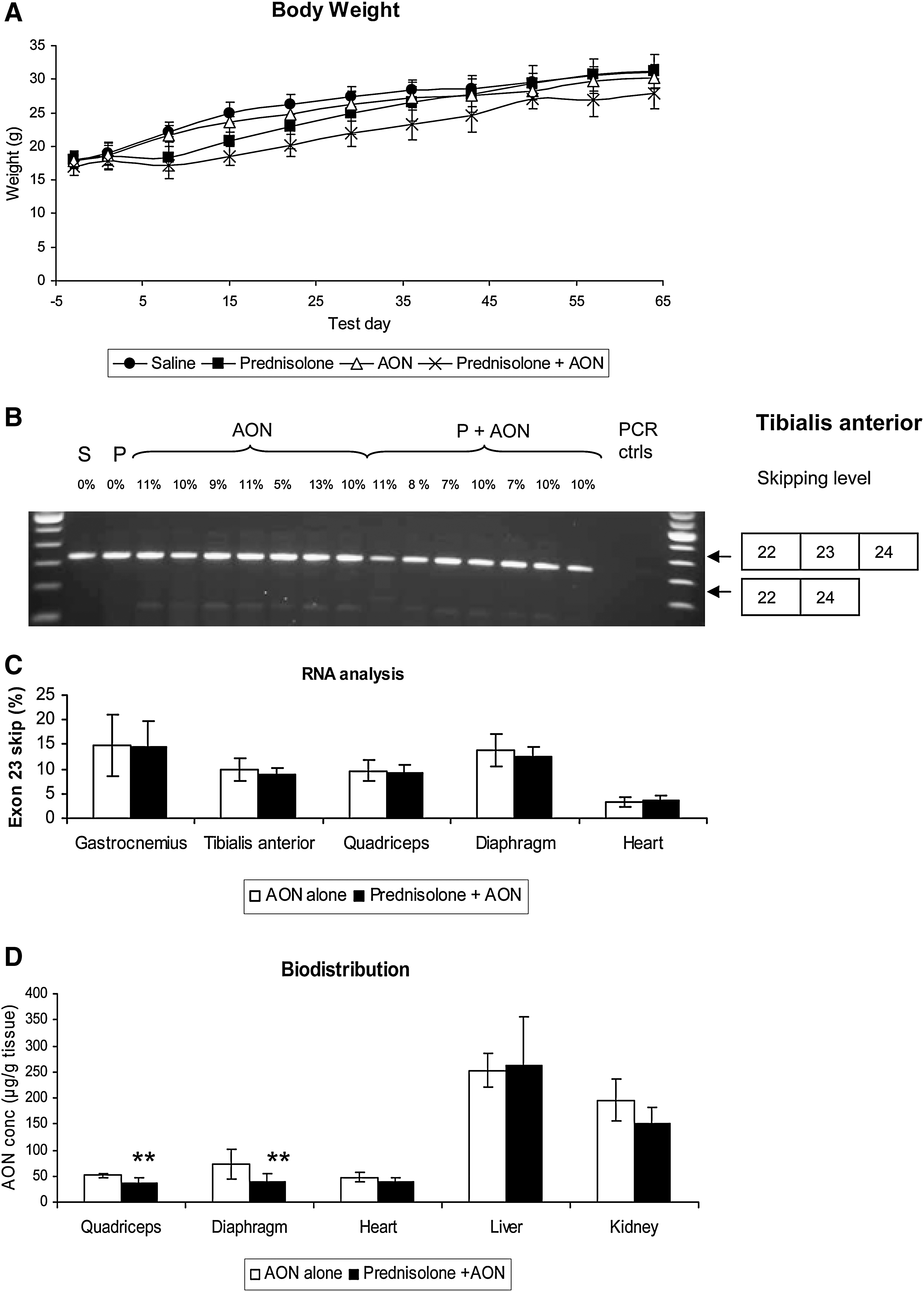
Systemic treatment with prednisolone and/or 23AON. Shown is the effect on body weight of all treatments and antisense oligonucleotide-mediated exon skipping and 23AON biodistribution after cotreatment with prednisolone or saline. Per group, eight mice were analyzed. Means are shown for each group. Error bars represent the standard deviation.
Average skipping levels were comparable for skeletal muscles and diaphragm and skipping levels did not differ between saline- and prednisolone-treated animals (Fig. 2B). In heart, exon-skipping levels were much lower, as described previously (Heemskerk et al., 2009), but detectable in all samples. No skipping was observed without 23AON treatment for any of the muscles (data not shown).
Assessment of the concentrations of 23AON in the various tissues showed in prednisolone-treated mice a small, but significant decrease in 23AON concentration in the quadriceps and diaphragm (Fig. 2D). In the heart the levels were almost comparable to those found in skeletal muscle, without real difference between both treatment groups. The majority of the 23AON ends up in the liver and kidneys. Levels were similar in the liver for prednisolone-treated animals versus control animals, whereas an almost significant decrease in 23AON concentration was seen in the kidneys. Overall, 23AON levels in muscle and organs were slightly lower in the prednisolone-treated mice.
23AON-mediated expression of dystrophin protein is slightly increased in prednisolone-treated animals
Dystrophin protein expression was determined in two ways: by Western blot analysis and by immunofluorescence staining of cross-sections. Assessment of protein levels by Western blot (Fig. 3A and B) revealed restoration of expression in all 23AON-treated mice, albeit at low levels (<5% of wild-type control). Quantification indicated slightly elevated protein levels for mice treated with both prednisolone and 23AON compared with mice treated with 23AON alone; the difference was significant for the gastrocnemius. In the diaphragm the same trend was seen, but this increase was not significant (Fig. 3A and B). However, with these low dystrophin expression levels, differences may be more difficult to observe. Immunofluorescence staining showed dystrophin expression above background levels (saline/prednisolone alone-treated mice) for both 23AON-treated groups, but no difference was observed between saline- and prednisolone-treated animals (Fig. 3C). Untreated mdx mice did not express dystrophin.
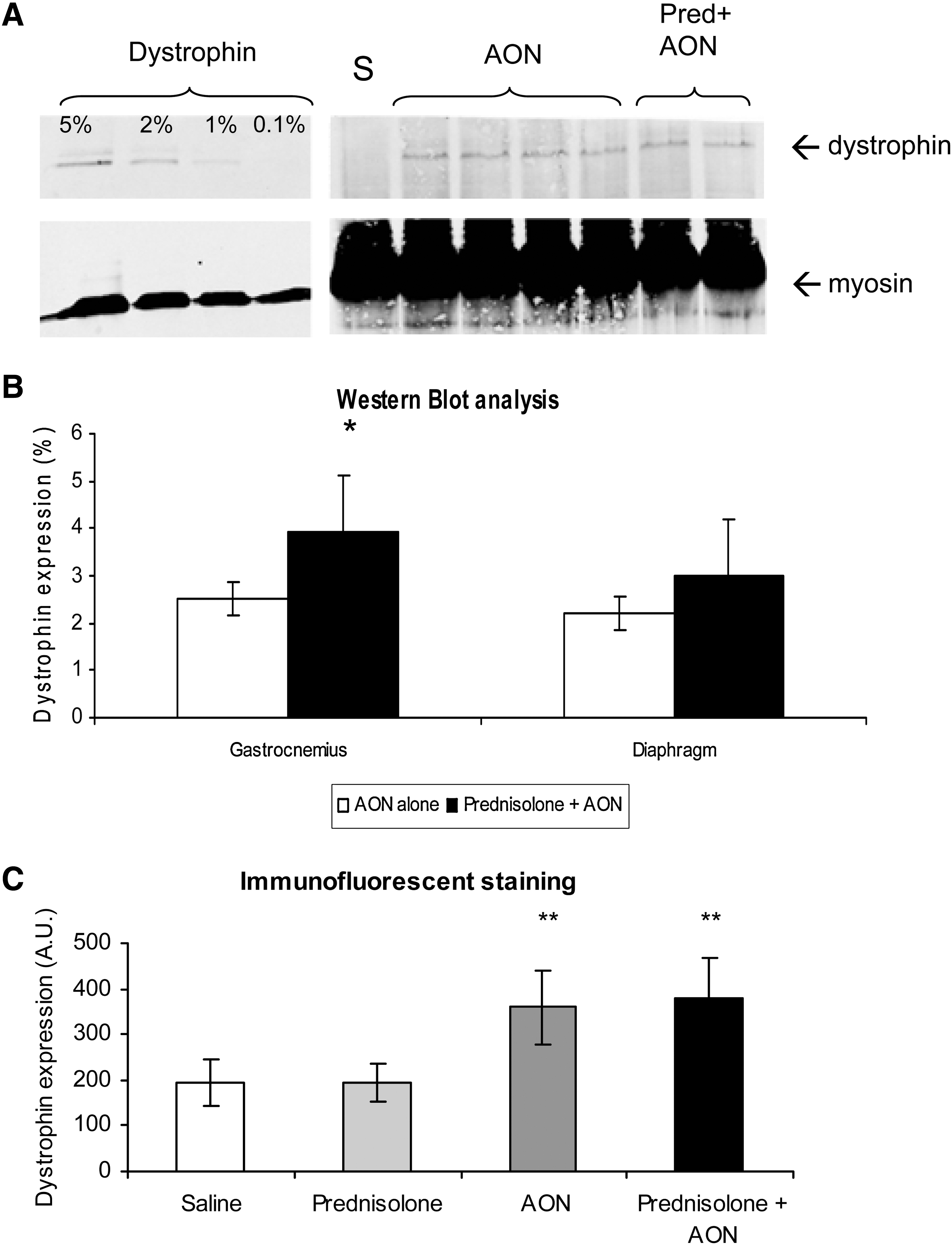
Dystrophin expression detected by immunofluorescence staining and Western blot analysis. Per group, eight mice were analyzed. Means are shown for each group. Error bars represent the standard deviation.
Biomarkers
The expression levels of multiple mRNA transcripts have been shown to be changed in the mdx mouse. These can be used as biomarkers to assess the effect of therapeutic interventions ('t Hoen et al., 2006). Expression levels for several immunological, fibrosis, and early and late regeneration markers for the tibialis anterior and diaphragm were assessed by quantitative PCR. The main changes in expression levels were seen in the tibialis anterior of 23AON- and/or prednisolone-treated mice (Fig. 4; and see Supplementary Table S1 online at

Expression of biomarker levels measured by quantitative PCR. Per group, eight mice were analyzed. Means are shown for each group, corrected for GAPDH or 5S expression. Values are expressed relatively to tibialis anterior levels of saline-treated mice. Error bars represent the standard deviation. Immunological markers CD68
Discussion
AON-mediated exon skipping as a potential therapy for DMD is being tested in placebo-controlled clinical trials and is likely to become clinically applicable in the near future. Most patients currently use corticosteroids, mainly prednisone, to slow disease progression. Prednisolone has been shown to have a positive effect on muscle maintenance and quality. This could affect exon-skipping efficiency either positively or negatively. It has been noticed previously that uptake of AONs and exon-skipping efficiency are higher in dystrophic muscle than in wild-type muscle (Heemskerk et al., 2010). This is thought to be caused by the leakiness of the dystrophic muscle membranes, which facilitates the entry of AONs into the muscle cells (Bremmer-Bout et al., 2004; Heemskerk et al., 2010). In fact, in mice containing only one utrophin allele in the absence of dystrophin (mdx/utrn +/–), which are more severely affected, exon-skipping levels are higher than in the mdx mouse (Tanganyika-de Winter, C.L. et al., unpublished observations). Therefore stabilization of the muscle fibers by prednisolone could have a negative effect on AON uptake and thereby decrease its efficiency. Conversely, improving the quality of the muscle could lead to more muscle fibers, increasing the total amount of dystrophin pre-mRNA, the target of AONs, and thereby increase the therapeutic potential of AONs. In this study the effect of prednisolone on AON uptake and biodistribution, exon-skipping levels, and dystrophin restoration was examined.
In two patient cell cultures and one healthy control cell culture no differences were seen in 2OMePS AON-induced exon skipping between prednisolone and saline cotreatment. Thus, the effect prednisolone has on muscle cells does not seem to influence skipping efficiency in vitro. The same appears to be true in vivo, where muscle cells are in their natural environment. After local injections of 23AON, skipping efficiency was the same between saline- and prednisolone-treated mice. The same holds for systemic 23AON treatment, where exon skipping percentages were similar between both treatment groups for skeletal muscle, diaphragm, and heart.
The 2OMePS dose used in this study is much higher than that used in clinical trials using the same chemistry (Goemans et al., 2011) (200 vs. 6 mg/kg/week). However, for most drugs a correction factor applies when translating doses between small and larger animals, based on normalization to body surface area (see Guidance for Industry,
The exon-skipping levels in vivo after 2OMePS AON treatment (both locally and systemically) are relatively low. Vivo morpholinos (i.e., modified morpholinos, conjugated with a dendrimeric octaguanidine) have been shown to lead to much higher skip levels (Wu et al., 2009, 2011). This may partly be due to differences in analysis method, primary versus nested PCR (Spitali et al., 2010). In either case, at the moment the clinical relevance of these high levels is limited, as exon-skipping levels in the currently ongoing systemic trials are low (Cirak et al., 2011; Goemans et al., 2011). Higher exon skip efficiency would obviously be desired, but for now only nonconjugated AONs are being tested in clinical trials. Nevertheless, low levels of dystrophin protein have already led to histological and functional improvement in mice (van Putten and Aartsma-Rus, 2011; Goemans et al., 2011), and the low exon-skipping levels in our study did result in dystrophin restoration, which was slightly increased in prednisolone-treated mice.
As seen in previous studies (Heemskerk et al., 2009, 2010) the majority of the 23AON ends up in the liver and the kidneys after systemic treatment. A small decrease in 23AON concentration was seen in the quadriceps and diaphragm in prednisolone-treated mice compared with saline-treated mice, but this did not lead to a decrease in exon-skipping percentages. This might be explained by the localization of the AON within the tissue. The 23AON concentration is measured in whole muscle and therefore reflects AONs present both in the fibers (where exon skipping can be induced) and in the extracellular matrix and interstitial spaces (where no exon skipping can be induced). The lower levels observed in muscle may be due to less sequestration in the extracellular matrix caused by an improvement in muscle quality. Therefore, the higher levels in the AON-treated animals do not necessarily have to result in higher exon-skipping levels, because part may be sequestered in the extracellular matrix and interstitial spaces. This is supported by the finding that more dystrophin was expressed in prednisolone-treated mice, which might be another indication of a positive effect on muscle quality (more dystrophin pre-mRNA) by prednisolone, because the same percentage of skipping in more transcripts leads to higher protein production. In this study an AON with 2′-O-methyl phosphorothioate chemistry was used; however, it is anticipated that the results also hold for the other chemistry that is approaching use in large clinical trials (morpholino phosphorodiamidate AONs).
The best known effect of prednisolone is its suppressive effect on the immune system. However, no clear decrease in the immunological markers CD68 and Lgals3 was seen in prednisolone-treated mice. By contrast, the levels of myogenic transcription factors MyoD and myogenin (regeneration markers) were decreased, although not significantly everywhere, for all treated groups, suggesting that prednisolone treatment did have an effect on muscle. This concurs with previous findings that prednisolone treatment leads to a general reduction in proliferation, and MyoD and myogenin are downregulated in methylprednisolone-treated adrenalectomized rats (Almon et al., 2007). Because AONs interact only with their target sequence, a general effect as observed for prednisolone is unlikely. In this case regeneration might be reduced because of reduction in muscle damage, inflammation, and fibrosis as a consequence of the dystrophin restoration. The less pronounced reduction in MRF-4 levels might be explained by the timing. MyoD and myogenin are elevated early in regeneration (Marotta et al., 2007), peaking 72 hr after induction of regeneration (Jin et al., 2000), whereas MRF-4 is increased in maturing myofibers (Marotta et al., 2007). The combination of prednisolone and 23AON showed mixed effects. The absence of a decrease in immunological markers, as seen in 23AON-treated mice, is probably due to conflicting results in one or two mice, which abolish the small differences observed. Fibrotic and regeneration markers followed roughly the same pattern as prednisolone and/or 23AON treatment alone. miR-31 is a microRNA that, amongst other effects, targets the 3′ untranslated region of the dystrophin mRNA, thereby repressing its translation and expression. It has been shown to be localized in regenerating myoblasts and is almost absent in wild-type muscle fibers, but is upregulated in mdx mice. Furthermore, repression of miR-31 led to an increase in dystrophin expression in AON-treated human DMD myoblasts (Cacchiarelli et al., 2011). In all treated mice a trend was observed toward normalization, which seemed to be more pronounced after combinational treatment. The downregulation of miR-31 might be another explanation for the improved dystrophin levels after prednisolone treatment.
It is surprising that even the limited levels of dystrophin found in this short-term study resulted in improved muscle quality to some extent. However, the long-term presence of low amounts of dystrophin has been reported to result in beneficial effects before. The mdx3cv mouse model, which has approximately 5% of dystrophin since birth, performs significantly better in the grip strength test compared with mdx mice (Li et al., 2008). Our own results in mice with low levels of dystrophin (mdx-Xist Δhs) confirm these results; here, low levels of dystrophin (<15%) were sufficient to improve histology and muscle function and to normalize biomarkers (van Putten, M., et al., manuscript submitted). Notably, in clinical trials low dystrophin levels after AON treatment appear to result in functional improvement (Goemans et al., 2011).
In conclusion, this work shows that there is no negative effect of prednisolone and 23AON on each other's therapeutic outcome in any of the tests, suggesting that patients can continue using prednisone during exon-skipping trials. Prednisolone might even have a positive effect on AON treatment.
Footnotes
Acknowledgments
The authors are grateful to Rick Vermue and Ruurd Verheul (Prosensa Therapeutics) for assistance with the hybridization–ligation assay, and to Maarten van Iterson (LUMC) for assistance with the statistical analysis. This work made use of the infrastructure of the Center for Medical System Biology (CMSB) and the Center for Biomedical Genetics (CBG) in which the LUMC participates. LUMC is a partner in the FP6 TREAT-NMD network of excellence (LSHM-CT-2006–036825). This work is supported by grants from the Prinses Beatrix Fonds, The Netherlands (grant W-OR09-12) and the Dutch Duchenne Parent Project.
Author Disclosure Statement
Tatyana G. Karnaoukh, Ingrid G.M. Kolfschoten, Anne Vroon, and Judith C.T. van Deutekom report being employed by Prosensa Therapeutics. LUMC has patents on the exon-skipping applications. Hans Heemskerk, Gert-Jan B. van Ommen, Judith C.T. van Deutekom, and Annemieke Aartsma-Rus report being coinventors on some of these patents, and as such are entitled to a share of royalties.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.